
Abstract
In this study, the analytical and developed method was successfully validated for the simultaneous determination of benzoic acid (BA) and Sorbic acid (SA) by natural deep eutectic solvent (DES) composed of thymol/water and dispersive liquid–liquid microextraction (DLLME) followed by HPLC/UV in orange juice samples. A response surface methodology (RSM) based on a central composite design (CCD) was applied for optimization of the main variables. The significant parameters including DES volume (600 µL), dispersive solvent volume (THF) (709 µL), sample volume (11.1 mL), salt amount (5% w/v), and vortex time (10 min) were optimized. Under the optimal conditions, the limits of detection (LOD) and limits of quantifications (LOQ) were obtained 0.468 and 1.419 mg L−1 for BA and 2.517 and 7.628 mg L−1 for SA, respectively. The recovery values were between 94 and 97%. R2 of BA and SA were 0.9940 and 0.9921, respectively.
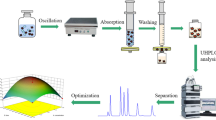
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Preservatives are chemical compounds used in foodstuffs in order to extend the shelf life based on their antimicrobial properties [1, 2]. One of the most commonly food preservatives is benzoic acid (BA), often utilized in combination with sorbic acid (SA) [1, 3]. Due to the optimal antibacterial activity of aforementioned preservatives at pH levels (pH < 4.5), they are well-suited in order to control the microorganisms in acidic foods. Fruit juices are highly susceptible to microbial growth because of high moisture and sugar content [4]. Several toxicological studies have shown that excessive levels of the preservatives (like BA and SA) may cause adverse health effects for human such as allergic responses, metabolic acidosis, asthma, urticaria, seizures, and hyperpnea [5]. It was indicated that BA can convert into benzene, which is a chemical hazard and has carcinogenic effects [6]. Furthermore, SA may change to mutagenic products through reaction with nitrite at high temperature and concentration [7]. As regards, acceptable daily intake of 5 and 25 mg Kg−1 body wight per day has been defined for BA and SA [8]. Therefore, it is important to determine and monitor the amount of preservatives in food samples, especially in drinks to protect consumers’ health. In different countries, it has been approved specific laws for food additives based on the nutritional pattern of society. For example, the usage of preservatives including BA and SA is not permitted in some foods in Iran. Nevertheless, the existence of the aforementioned preservatives is reported in drink-based products in some studies [9,10,11]. In this regards, simultaneous quantification of food preservatives in beverages is in high importance.
Due to the complexity of orange juice matrices, the application of sample preparation methods before instrumental analysis is required. Different preparation procedures such as liquid-phase and solid-phase extraction methods have been introduced for the extraction of BA and SA in beverages. These methods are time-consuming, complex, low efficiency, and expensive. Furthermore, it is required high amounts of sample and organic solvents. So, they cannot fulfill the concept of green chemistry, which has recently become a rising topic globally, due to eco-environmental benefits [12]. Hence, microextraction methods have been developed as new sample preparation techniques to increase the sensitivity of the analysis method [13,14,15,16].
In this context, DLLME is developed by using a mixture of extractive and dispersive solvents [4]. On the other hand, the discovery of environmentally friendly solvents, DES has become a major breakthrough in green chemistry for the replacement or reduction of the conventional toxic organic solvents [12,13,14,15, 17, 18]. Nowadays, various studies investigated the application of green DES [19,20,21,22]. These types of solvents have some superior properties compared to ionic liquids, including environmentally friendly, easy preparation, biodegradability, high purity, low cost, non-toxicity, excellent solubility, and low volatility [15, 16, 21]. DES is generally referred to binary or ternary mixtures of compounds at a certain molar ratio linked to each other mainly via hydrogen bonds, which causes a significant decrease in the melting point of such a combination. DES is mainly comprised of a hydrogen bond donor and a hydrogen bond acceptor [12, 22]. Given that hydrophilic DES such as choline chloride or tetrabutylammonium bromide are decomposed upon contact with water. Hence, hydrophobic DES like natural terpenoids (i.e., thymol and menthol) are better choices in the analysis and effective separation of various analytes from aqueous samples (i.e., fruit juices) with the reason of higher water stability [23, 24]. Therefore, it was established that hydrophobic natural DES based on thymol and water and DLLME method coupled with HPLC/ UV provide effective separation of BA and SA from orange juice at current study. To the best of our knowledge, the application of DES-based extraction systems for BA and SA microextraction has not been presented in literature.
Materials and methods
Materials and reagents
In this study, all used chemicals were analytical reagent grade and purchased from Merck (Darmstadt, Germany). BA and SA standards were purchased from Sigma Aldrich (Steinheim, Germany). KCl (salt) was obtained from Neutron Chemicals (Tehran, Iran).
Preparation of solutions
The mixed 1000 mg L−1 standard solution of BA and SA were prepared with the suitable dilutions in deionized distilled water and maintained in the refrigerator at 4°C until use. An intermediate standard solution of both standards (100 mg L−1) was also prepared; meanwhile, the working solutions was prepared at 10 and 50 mg L−1 by diluting stock solution with the deionized distilled water and were refrigerated at 4 °C. Solutions were applied for the preparation of calibration curves, and validation standards (Fig. Supp. 1).
Sampling and sample preparation
In this study, a total of 5 bottles of pulp-free and non-carbonated orange juice were randomly purchased from a local store at Isfahan, Iran. All the samples were stored at cold place before analysis.
DES preparation and selection solvent for DLLME
Initially, to synthesis of an appropriate DES, different green compounds with the following combinations; combination 1: thymol + phenylalanine (molar rate 1:1, 1:2, and 2:1), combination 2: thymol + urea (molar rate 1:1, 1:2, 3:1), combination 3: thymol + acid citric (molar rate 1:1), combination 4: thymol + water (molar rate 10:1 and 5:1), combination 5: eugenol + water (molar rate 10:1), combination 6: acid citric + urea (molar rate 1:1), combination 7: acid citric + phenylalanine (molar rate 1:1, 1:2, and 2:1), combination 8: phenylalanine + water (molar rate 10:1), combination 9: phenylalanine + urea, were selected to be examined as an extractive solvent (Table 1). To prepare DES, the above-mentioned compounds at the mentioned specified molarity ratios were mixed in a screw cap-glass test tubes and placed in an oil bath. Then the mixtures were stirred until clear, homogenous, and colorless liquids formed with magnetic stirrer at a temperature of 80–90 °C for 1–2 h. The acquired liquid DESs (if acquired) were kept at ambient temperature. Afterward, different compounds like acetone, ethyl acetate, acetonitrile, ethanol, methanol, propanol, and tetrahydrofuran (THF) were selected to be examined as the dispersive solvents. In the current research, a univariate approach was used to examine different combinations of extraction and dispersive solvents to find the best solvent pair for the DLLME of BA and SA in water samples under the same conditions (i.e., 5 mL water sample spiked with 10 mg L−1 BA and SA standards, 200 µL DES or extractive solvent, 800 µL dispersive solvent, 10 min vortex, 10 min centrifuge). Finally, the best solvent pair was selected according to the analytical performances. In order to confirm the formation of the hydrophobic DES, the fourier transform infrared (FTIR) results were obtained in the 300 to 4000 cm−1 range by FTIR spectrophotometer (Jasco, 6300, Tokyo, Japan).
Methods
DES-based DLLME procedure
To perform the DLLME procedure, an aliquot of 10 mL of orange juice sample spiked with 10 ppm of BA and SA standards, was inserted into a 15 mL falcon having 200 μL DES (i.e., thymol + water, 10:1 molarity rate). Then, 800 μL THF was inserted swiftly into the solution. The falcon was lightly agitated and vortexed for 10 min. In this step, the targeted analytes were extracted into tiny droplets of DES (extractive solvent). After that, each falcon was centrifuged at 5000 rpm for 10 min at 25 °C. Subsequently, DES in the upper layer and concentrated analytes was transferred to a microtube (1.5 mL). Eventually, 20 μL of solution injected into the HPLC apparatus.
HPLC–UV apparatus
The extracted phase was inserted into the HPLC–UV system. A Hitachi (Santa Clara, CA, USA) Elite LaChrom system was equipped with a vacuum degasser, an LC-20AT quaternary solvent delivery system (pump), L-2400 UV detector, a L-2200 auto sampler with a 20 μL loop. HPLC separations were carried out on an analytical C18 Nucleosil Macherey Nagel (Germany) column (250 × 4.6 mm id, 5-μm particle size). An optimized mobile phase (HPLC grade water; acetonitrile purity HPLC) (60: 40, v/v) was used at a flow rate of 1 mL min−1. The separation was performed at room temperature in an isocratic elution mode within 20 min. The injection volume was 20 μL and the detection wavelength was 270 nm.
Optimization of DLLME method and statistical analysis
It the current study, it was used the Design Expert 12 Software to minimize the number of testes and investigate the impact of the variables on the response, along with optimizing the DLLME method. In this context, a central composite design (CCD) was used for regression and mathematical modeling of the practical experimental findings. Influential factors on the performance of DLLME method such as the selection of the extracting and dispersive solvents were performed through univariate approach. Other effective variables on the sensitivity of DLLME procedure, including extractive solvent volume (A), dispersive solvent volume (B), sample volume (C), salt amount (D), and vortex time (E), were selected according to previous literature review and initial experiments, and then optimized by the CCD. It provides a response surface modelling (RSM) of the data in a rotatable form of a multi-level factorial design to evaluate the impacts and the interactions between included factors and optimize the proposed DLLME method. The optimal conditions were finally determined by performing the analyses of variance (ANOVA) and the graphical optimizations together with the desirability function. According to the design, five variables (A, B, C, D, and E) at five levels were studied. In order to construct an orthogonal design, each of the five factors have comprised of a high and low values. The design consisted of six replicates of the central point. Therefore, the applied design included 48 random experiments (Table 2, and Supplementary Table 1). Deionized distilled water was used for the optimization process. The extraction analytical performance of BA and SA was evaluated. The response surface graphs explaining the design and the modelled CCD information were drawn by Design Expert 12 Software.
Results and discussion
DES preparation and selection of suitable solvent pair for DLLME
The selection of appropriate extractive and dispersive solvents is one of the most relevant factors in order to develop an effective and applicable DLLME method. A suitable extractive solvent has particular specifications including low solvability in water, high water miscibility (to fully dissolve in each other at any concentration and forming a homogeneous mixture), appropriate density (for simplifying the recovery), low boiling point (for speeding up its evaporation), good chromatographic performance, and high extraction efficiency as well as sensitivity to analyte [4, 14]. Based on the aforementioned characteristics, different combinations of green compounds were selected to be examined as the extractive solvents at the current study (Table 1). The highest extraction recovery was referred to combination 4 (thymol + water, molar rate 10:1) (Table 1). The presence of water in DES can decrease viscosity. Regarding with highly efficient extraction, combination 4 was eventually nominated as the best green extractive solvent or DES for the extraction of BA and SA from orange juice. Those combinations containing phenylalanine did not form any DES, even at high temperature (100 °C) (combination 1, 7, 8, and 9). In case of eugenol, DES like liquid was formed but the extraction peaks were not stable. Besides, the dispersive solvent must have high miscibility with both water and the extractive solvent to favor the creation of a stable cloudy solution [4]. Therefore, different compounds were selected to be examined as the dispersive solvents (Table 1). Afterward, the mixture of 200 µL of DES with 800 µL of the dispersive solvents were examined in order to choose the most appropriate dispersive solvent. The best suitable dispersive solvent was THF after checking the visual cloudy appearance of the tested tubes. Consequently, THF was nominated as the best solvent pair for the DLLME in sample based on the acquired recovery data of BA and SA. This may be due the fact that both thymol and water (as DES compounds) have hydroxyl functional groups in their structure, which can form hydrogen bonds with each other. Subsequently, this situation can lead to the formation of a stable and effective DES [20]. Moreover, the formation of the π–π interaction type of Van der Waals forces between aromatic cycle of thymol and conjugated bonds of BA and SA is the reason of their interaction and subsequently acids separation by thymol-based DES [21].
Characterization of DES
The FT-IR spectra of pure thymol and DES were shown in Fig. 1. Accordingly, the bands relating to the tension and stretching –OH (3397 and 1456 cm−1) in the spectrum of pure thymol as well as the absorption relating to the tension C–O (1029 cm−1) were recognized. After the incorporation of water and thymol, some alterations appeared in the band position and their intensity. Particularly, the obvious reductions was observed in band intensity at 1876 cm−1 in spectrum of DES compared to thymol spectrum. This band relates to the position of (C–O) bond. This change confirms the dipole–dipole interaction between H2O with OH group which result in change of stretching vibrations of C–O bond. In addition, the acquired spectra illustrates that the location of the absorption band of the phenol group of the thymol alters (from 3397 cm−1 to 3456 cm−1), which shows the interaction of the water molecules with the OH group of thymol. Thus, this shift confirms the existence of hydrogen bonding between water and thymol based on the formation of DES [25]. The position and shape of the band at 2962 cm−1 remained unchanged. The obtained data suggested that there was no chemical interaction between aromatic group and water molecules [26].

FT-IR spectra of pure thymol (A) and DES (B), respectively
In addition, HPLC was used to compare the retention time of thymol with the synthesized DES. Fig. Suppl. 2 shows the chromatogram of DES and thymol. It was observed that retention time of DES is more than pure thymol. This condition can be explained by this fact that DES is more polar than thymol (because of the presence of water in DES molecules). Furthermore, the interaction between nonpolar stationary phase (C18) and nonpolar compound is much higher than polar compounds Fig. 2.

HPLC partial chromatogram of a juice sample contained benzoic (1) and sorbic (2) acids
Optimization of DES-DLLME method
Regarding to the concept of green chemistry, a multivariate optimization approach that significantly reduced the number of experiments and subsequently the consumption of samples and reagents was used to optimize the DLLME conditions [13]. In the current study, the multivariate optimization design of the DES-DLLME method was performed using CCD. By applying this design, the best model of the relationship between the influential factors were identified to obtain the optimum values [13, 27]. CCD provides RSM of the data in a rotatable form of a multi-level factorial design. It describes the interaction and quadratic effects of the variables along with the linear effects of each factor on the response. This modelling design estimates the regression coefficients between different factors by specifying equal variance to each component [13]. The present study was performed with five factors at five levels, which were displayed in Table Supplementary 2. The five factors were comprised of extraction solvent volume (DES, µL), dispersive solvent volume (THF, µL), sample volume (orange juice, mL), salt amount (%W/V), and vortex time (min). Due to the rotatory inclination of CCD, five levels were specified to each variable from low to high values comprising a center point. The validation of the model was examined using ANOVA. The significancy of the model, the variables, and the interaction affinity between variables was evaluated by F-value and the p-value. Table Supplementary 3 depicts the ANOVA summary of the included variables and their interaction terms for response surface quadratic model of BA and SA. As shown in Table Supplementary 3, the model was significant and applicable with a p-value less than 0.0001 and F-value of 158.92. F-values is an indicator of the most contributing factor to the CCD model. A higher F-value shows a greater contribution of the factor to the CCD model. Accordingly, as represents in Table Supplementary 3, the most contributing factors to the CCD model are AD (F-value: 554.15), AB (F-value: 535.78), and vortex time (F-value: 256.05). A Lack of Fit P value is applied to express the repeatability of the CCD model. It is preferred to be greater than 0.05 at the 95% confidence level. As shown in Table Supplementary 3, the Lack of Fit P value of 0.3654 shows that the regression equation model was not lack of fit and it is not significantly attributed to the pure error. The data in Table Supplementary 3 is an indication of the efficient compatibility of the operating conditions with the criteria of all the included responses, using as an alternative for concurrent optimization of multiple responses [13].
To predict response at any point, even those not involved in the design, a quadratic regression model was fitted on acquired findings, which is shown in Eq. 1:
R is the predicted response (average recoveries of BA and SA) (Eq. 1). The quality and reliability of model fitting was evaluated by the coefficient of determination (i.e., R2 and adjusted-R2), which represents the potential of the quadratic model for predicting the response and demonstrates if the model can effectively predict the response. R2 indicates the credibility of analytical information acquired from the CCD model, which is preferred to be close to 1.0. Moreover, the difference between the adjusted R2 and predicted R2 values should be less than 0.2. In the present study, the R2 value for tested analytes was 0.9922, which is so close to 1.0. Besides, the adjusted R2 and predicted R2 values for tested analytes were 0.9860 and 0.9726 which indicated a good agreement between the experimental results and the proposed model. The low coefficient of variation (CV % = 0.0252) illustrates that the precision of the acquired findings was very good.
Verification of regression model on diagnostic plots
The verification of regression model was examined on diagnostic plots, as shown in Fig. Suppl. 3. It was indicated the plot of the normal probability i.e., residual for the recoveries (Fig. Suppl. 3a). The normal probability plot of the residuals gives useful information about normality of distribution of errors and their independence from each other. It can be seen that the plotted points are distributed around the regression line, which suggests that the data set is approximately normally distributed. Based on the visual examination of the findings, it could be concluded that the residuals have a normal distribution. To further investigate the validity of the model, residuals versus run plot was given in Fig. Suppl. 3b. The random distribution of residuals against run number indicates the accuracy of the model and the efficiency of obtained model for the observed data.
Three-dimensional response surface plots
The three-dimensional graphs were actually the graphical descriptions of the regression equation (Fig. Suppl. 4). The effect of DES and dispersive volume on the recovery of BA and SA was shown in Fig. Suppl. 4a. The DES volume was changed from 400 to 600 μL and the dispersive volume was changed from 600 to 900 μL. As it was displayed the recovery percentage enhanced by the concurrent elevation of the DES volume up to 600 µL along with decrease of the dispersive solvent volume to 600 µL. It is evident that the extraction efficiency of acids increased when the DES volume increased and the dispersive solvent volume decreased and vice versa (Fig. Suppl. 4a). The reason might be for a better cloudy state produced in a higher volume of DES and lower volume of dispersive solvent leading to higher recovery [14]. Moreover, THF (as dispersive solvent) promotes phase separation by decreasing the interactive strength between DES and the water molecule [19]. Water solubility of BA and SA enhanced at higher volume of the dispersive solvent. It can lead to lower extraction efficiency [14]. Furthermore, it was demonstrated that the low DES volume (50–100 μL) would result in difficult withdrawal of the upper extract phase, leading to decreased extraction recovery [21]. Therefore, dispersive and extractive solvents was chosen in the amount of 709 and 600 μL. It was selected as the best solvent pair volume.
The effect of salt amount and DES volume on the extraction efficacy of BA and SA was represented in Fig. Suppl. 4b. In the current study, 5–8%w/v salt was used to estimate the extraction efficiency. It was observed that concurrent increase of the DES volume to 600 μL along with decreasing the salt amount to 5%w/v, enhances the extraction efficiency and vice versa. It is obvious that the recovery values increase with higher DES volume. The increased efficiency corresponding to the increased volume of the DES to 600 μL, was due to a rise in the concentration of the extracted analytes [4, 21]. Generally, increasing the DES volume significantly affects the DES-DLLME procedure by increasing the enrichment factor of analytes [27]. Moreover, it was examined the DES volume in the range of 50 to 500 μL. It was hard in volume of 50 μL to repeatedly withdraw the upper layer, so the extraction efficacy was low [21]. This situation was comparable with extraction recoveries provided at the DES volume between 100 to 500 μL. However, the maximum enrichment factor was acquired at the DES volume of 100 μL. Based on our results, the concurrent increase of the salt amount higher than 6.2 w/v% and the DES volume up to 600 μL, has not a positive effect on the response. Similar to our results, it was indicated that the extraction efficiency would improve by increasing the salt amount (up to 0.75 g) [14]. It seems that higher amounts of salt (more than 0.75 g) would decrease the extraction efficiency. The best explanation to this phenomenon might be due to the salting out effect. A reduction in solubility of analyses in sample solutions can assist their movement into the organic phase and increases the extraction efficiency since the added salt promotes a rise in ionic strength. On the other hand, it was demonstrated that solutions with high amounts of salt (more than 0.75 g) have low extraction performance. Because they are heavier than the water solution, which inhibits the sedimentation of the extraction solvent. It is confirmed that the extraction efficiency reduced by increasing the amount of salt from 0% to 8.0% (w/v). Alshana, Ertaş et al. 2015 illustrated that an appropriate cloudy state did not produce, when the amount of salt was above 8.0%, which might be as a result of the lower miscibility of acetonitrile with water [28]. Therefore, a salt amount of 5 W/V% and a DES volume of 600 μL were selected as the best solvent pair volume.
The effect of vortex time and DES volume on the recovery of acids was depicts in Fig. Suppl. 4c. The response surface acquired by plotting vortex time versus DES volume with the volume of aqueous sample fixed at 10 mL. It is critical to select the most appropriate DES volume in liquid phase microextraction techniques in order to provide high enrichment factors, low toxicity, high extraction of target analytes, and practical possibility for the injection into an analytical instrument [29]. Fig. Suppl. 4c shows that the extraction efficiency enhanced with concurrent elevation of the vortex time up to 15 min together with decreasing the DES volume solvent to 400 μL. The maximum response was obtained at 15 min for vortex time and 400 μL for DES volume. But the extraction efficiency remained almost constant when DES volume was 600 μL and vortex time was increased from 10 to 15 min. It seems that vortex provides effective and rapid dispersion of DES molecules into the sample solution by producing the microspheres of DES and, accordingly, effective phase separation, and consequently maximum extraction efficacies were achieved [19].
The effect of salt amount and dispersive volume on the recovery of BA and SA was represented in Fig. Suppl. 4d. As it is demonstrated, the extraction efficiency enhanced with simultaneously elevation of the amount of salt to 8%w/v and the dispersive volume solvent to 900 μL. As previously mentioned, the best explanation to this would be the salting out effect.
The effect of vortex time and dispersive volume on the recovery of BA and SA was shown in Fig. Suppl. 4e. It is obvious that concurrently increasing the vortex time and decreasing the dispersive solvent volume improved the performance of the proposed method. The maximum recoveries were achieved when the vortex time was 15 min and the THF volume was 600 μL. The main reason would be the effect of vortex on the solution, which lead to elevate the interaction between the DES solvent and the dispersive solvent (THF). In this way, both phase separation is accelerated and the extraction time is decreased [19]. An adequate vortex time is essential to maintain an equilibrium in the extraction system [21].
The effect of salt amount and sample volume on the recovery of BA and SA was depicted in Fig. Suppl. 4f. As it is displayed, it is evident that concurrently increasing of the salt amount and the sample volume increased the performance of the proposed method. The maximum response was acquired at a salt amount of 8 g and a 15 mL volume of sample. It was observed that the extraction efficiencies were almost constant when the salt amount was at the minimum amount (e.g. 5% w/v) and the sample volume increased from 10 to 15 mL, which might be due to the dilution effect [21]. However, maximum recoveries were obtained by increasing the sample volume in the presence of 8% w/v salt, which might be due to the increased number of analytes and the salting out effect.
Optimum conditions and confirmation test
Briefly, the optimized values by CCD for the five factors studied in simultaneous DES-DLLME of BA and SA, with a desirability (DF value) of 1, were as followed: DES volume (600 µL), dispersive solvent volume (709 µL), sample volume (11.1 mL), salt amount (5%W/V), and vortex time (10 min). The real samples were analyzed under optimized conditions in order to verify the efficiency of the developed model. The samples were spiked with BA and SA at 20 ppm. The predicted recovery values (96%) were close to the obtained experimental recovery values (95 ± 3%). The good responses (mean recoveries of BA and SA) were obtained in real samples which ranged from 94 to 97%. These results showed the good reliability, accuracy and repeatability of the proposed method, so that matrix constituents of the samples particularly carbohydrate compounds had no significant effects on the identification of BA and SA. This compliance between the CCD predicted data and the experimental findings approved that these extraction conditions were optimal. Hence, these optimized data can be applied for validation studies of the proposed method.
Evaluation of analytical performance of DES-DLLME-HPLC–UV method
To examine the analytical performance of DES-DLLME-HPLC–UV technique, different figures of merits such as limit of quantification (LOQ), limit of detection (LOD), repeatability, and linearity were verified under optimal conditions. LOD and LOQ were 0.468 and 1.419 mg L−1 for BA and 2.517 and 7.628 mg L−1 for SA, respectively. The calibration curves of BA and SA were linear over the range of 10–25 mg L−1 (4 point) and 1–35 mg L−1 (8 point), respectively. For SA two calibration curves were used, including 1, 5, 10, 15 and 20, 25, 60, 35. The square of correlation coefficient (R2) was 0.9940 for BA and 0.9921 for SA. To the best of our knowledge, the developed method, in the current study, is applied for the first time for quantification of BA and SA in food products. So, a comparative table of the analytical performance of recent literature [1, 13, 30,31,32] with our proposed method is provided in Table Supplementary 4. Compared to the data presented in literature, the proposed method offers comparable linear range and recoveries in recently reported methods in the literature for extraction and determination of the BA and SA. The permissible level of BA and SA in beverages is 150 mg L−1 based on FAO/WHO expert committee recommendation which is higher than LOQ and in the linear range of this method. The proposed method has intrinsic advantages for detecting simultaneously BA and SA, including rapidity, simplicity and low consumption of solvents and reagents compared to other methods. All of these results of present study reveal that the developed, optimized, and validated DES-DLLME-HPLC/UV method could efficiently and simultaneously assess BA and SA in orange juice within the concept of green chemistry.
Conclusions
In the current study, the DES-DLLME procedure was applied for preconception of BA and SA from orange juice prior to the simultaneous quantification of these preservative in HPLC/UV. Since the volume of a toxic extractant solvent decreased by substitution of an environmentally-ecofriendly solvent (i.e., DES: thymol and water, molarity rate: 10:1), the method fulfills the concept of green chemistry. Moreover, a multivariate optimization approach (i.e., CCD) was used in the current study to optimize the influential factors on the performance of DES-DLLME method, which also meets the green chemistry principals. The optimized conditions for DLLME were obtained at 600 μL DES volume and 709 μL dispersive solvent. The analytical figures of merits showed that the developed method had appropriate LOD, high recoveries, good precision linearity, and sensitivity. So, it can efficiently use for concurrent determination of BA and SA in orange juice.
References
N. Tungkijanansin, W. Alahmad, T. Nhujak, P. Varanusupakul, Simultaneous determination of benzoic acid, sorbic acid, and propionic acid in fermented food by headspace solid-phase microextraction followed by GC-FID. Food Chem. 329, 127161 (2020). https://doi.org/10.1016/j.foodchem.2020.127161
M. Shoeb, M.M. Islam, M.S. Reza, N. Nahar, M.M. Islam, HPLC analysis of artificial preservatives, stimulants and sweeteners in carbonated beverages in Bangladesh. Curr. Res. Biosci. Biotech. 3(2), 215–221 (2022)
Z. Esfandiari, M. Saraji, R.A. Madani, E. Jahanmard, Status of benzoic acid amount during processing from Yoghurt to its by-product drink (Doogh). Ital. J. Food Sci. 28(3), 536–541 (2016)
T. Abderrahmane, M. Marzouk, A. Youcef, Optimized DLLME method coupled to HPLC for simultaneous analyses of benzoates, sorbates and methyl p-hydroxyl benzoates in fruits products. J. Food Eng. Technol. 8(1), 39–49 (2019). https://doi.org/10.32732/jfet.2019.8.1.39
K. Danchana, P. Jitthiang, K. Uraisin, V. Cred, WinMLR program for the determination of sorbic and benzoic acids in food samples. Food Chem. 361, 130086 (2021). https://doi.org/10.1016/j.foodchem.2021.130086
V. P. Salviano dos Santos, A. Medeiros Salgado, A. Guedes Torres, K. Signori Pereira. Benzene as a Chemical Hazard in Processed Foods. Int J Food Sci 545640, 1–7 (2015). https://doi.org/10.1155/2015/545640
N.K. Merugu, S. Manapuram, T. Chakraborty, S.K. Karanam, S.B. Imandi, Mutagens in commercial food processing and its microbial transformation. Food Sci. Biotech. 32, 599–620 (2023). https://doi.org/10.1007/s10068-022-01240-7
Z. Esfandiari, M. Badiey, P. Mahmoodian, R. Sarhangpour, E. Yazdani, M. Mirlohi, Simultaneous determination of sodium benzoate, potassium sorbate and natamycin content in Iranian yoghurt drink (Doogh) and the associated risk of their intake through Doogh consumption. Iran. J. Public Health 42(8), 915–920 (2013)
H. Hosseini, M. Shabazaz, S. Asadinejad, Iran Food Additives, 1st edn. (Food and Drug Administration Publication, Iran, 2008), pp.209–220
M. Kamankesh, A. Mohammadi, Z. Modarres Tehrani, R. Ferdowsi, H. Hosseini, Dispersive liquid–liquid microextraction followed by high-performance liquid chromatography for determination of benzoate and sorbate in yogurt drinks and method optimization by central composite design. Talanta 109, 46–51 (2013). https://doi.org/10.1016/j.talanta.2013.01.052
B. Akbari-Adergani, M. Poorasad, Z. Esfandiari, Sunset yellow, tartrazine and sodium benzoate in orange juice distributed in Iranian market and subsequent exposure assessment. Int. Food Res. J. 25(3), 975–981 (2018)
T. El Achkar, H. Greige-Gerges, S. Fourmentin, Basics and properties of deep eutectic solvents: a review. Environ. Chem. Lett. 19, 3397–3408 (2021). https://doi.org/10.1007/s10311-021-01225-8
A.S. Abedi, A. Mohammadi, E. Azadniya, A.M. Mortazavian, R. Khaksar, Simultaneous determination of sorbic and benzoic acids in milk products using an optimized microextraction technique followed by gas chromatography. Food Addit. Contam. Part A. 31(1), 21–28 (2014). https://doi.org/10.1080/19440049.2013.859742
F. Javanmardi, S.R. Arefhosseini, M. Ansarin, M. Nemati, Optimized dispersive liquid–liquid microextraction method and high performance liquid chromatography with ultraviolet detection for simultaneous determination of sorbic and benzoic acids and evaluation of contamination of these preservatives in Iranian foods. J. AOAC Int. 98(4), 962–970 (2015). https://doi.org/10.5740/jaoacint.14-260
J. Cao, Y. He, X. Zhang, L. Shi, B. Zeng, F. Zhao, Development of a tributyl phosphate-based hydrophobic deep eutectic solvent for liquid-liquid microextraction of total parabens in beverages. J. Mol. Lipid 387, 122593 (2023). https://doi.org/10.1016/j.molliq.2023.122593
J. Cao, C. Wang, L. Shi, Y. Cheng, H. Hu, B. Zeng, F. Zhao, Water based-deep eutectic solvent for ultrasound-assisted liquid–liquid microextraction of parabens in edible oil. Food Chem. 383, 132586 (2022). https://doi.org/10.1016/j.foodchem.2022.132586
S.S. Özak, A vortex-assisted microextraction based on deep eutectic solvents for determination of four parabens from cosmetic baby oils and optimization by Box-Behnken design. Düzce Univ. J. Sci. Technol. 8, 1936–1947 (2020). https://doi.org/10.29130/dubited.721252
B.B. Kefi, S. Baccouri, R. Torkhani, S. Koumba, P. Martin, N. M’Hamdi, Application of response surface methodology to optimize solid-phase extraction of benzoic acid and sorbic acid from food drinks. Foods 11, 1257 (2022). https://doi.org/10.3390/foods11091257
A.Ö. Altunay, A. Elik, A novel sonication assisted dispersive liquid-liquid microextraction method for methylparaben in cosmetic samples using deep eutectic solvent. Sustain. Chem. Pharm. 29, 100781 (2022). https://doi.org/10.1016/j.scp.2022.100781
M. Tiecco, A. Grillo, E. Mosconi, W. Kaiser, T.D. Giacco, R. Germani, Advances in the development of novel green liquids: thymol/water, thymol/urea and thymol/phenylacetic acid as innovative hydrophobic natural deep eutectic solvents. J. Mol. Liquids 364, 120043 (2022). https://doi.org/10.1016/j.molliq.2022.120043
A. Shishov, E. Nizov, A. Bulatov, Microextraction of melamine from dairy products by thymol-nonanoic acid deep eutectic solvent for high-performance liquid chromatography-ultraviolet determination. J. Food Comp. Anal. 116, 105083 (2023). https://doi.org/10.1016/j.jfca.2022.105083
M.S. Taghavi Dehaghani, Z. Esfandiari, H. Rostamabadi, H. Rashidi Nodeh, Application of amino acid-based natural deep eutectic solvents in extraction of different analyses: a review study. Trends Food Sci. Technol. 147, 104448 (2024). https://doi.org/10.1016/j.tifs.2024.104448
P. Makoś, E. Słupek, J. Gębicki, Hydrophobic deep eutectic solvents in microextraction techniques–a review. Microchem. J. 152, 10434 (2020). https://doi.org/10.1016/j.microc.2019.104384
M. Devi, R. Moral, S. Thakuria, A. Mitra, S. Paul, Hydrophobic deep eutectic solvents as greener substitutes for conventional extraction media: examples and techniques. ACS Omega 8, 9702–9728 (2023). https://doi.org/10.1021/acsomega.2c07684
T. Khezeli, A. Daneshfar, R. Sahraei, Emulsification liquid–liquid microextraction based on deep eutectic solvent: an extraction method for the determination of benzene, toluene, ethylbenzene and seven polycyclic aromatic hydrocarbons from water samples. J. Chromatogr. A 1425, 25–33 (2015). https://doi.org/10.1016/j.chroma.2015.11.007
C. Xing, W.Q. Cui, Y. Zhang, X.S. Zou, J.Y. Hao, S.D. Zheng, T.T. Wang, X.Z. Wang, T. Wu, Y. Liu, X.Y. Chen, S.G. Yuan, Z.Y. Zhang, Y.H. Li, Ultrasound-assisted deep eutectic solvents extraction of glabridin and isoliquiritigenin from Glycyrrhiza glabra: optimization, extraction mechanism and in vitro bioactivities. Ultrasonics Sonochem. 83, 105946 (2022). https://doi.org/10.1016/j.ultsonch.2022.105946
F.C. Pinheiro, M.Á. Aguirre, J.A. Nóbrega, N. González-Gallardo, D.J. Ramón, A. Canals, A dispersive liquid-liquid microextraction based on deep eutectic solvent for elemental impurities determination in oral and parenteral drugs by inductively coupled plasma optical emission spectrometry. Anal. Chim. Acta 1185, 339052 (2021). https://doi.org/10.1016/j.aca.2021.339052
U. Alshana, N. Ertas, N.G. Goger, Determination of parabens in human milk and other food samples by capillary electrophoresis after dispersive liquid–liquid microextraction with back-extraction. Food Chem. 181, 1–8 (2015). https://doi.org/10.1016/j.foodchem.2015.02.074
H. Farahani, Screening of parabens in natural water by salting-out based centrifugeless dispersive liquid-liquid microextraction combined with HPLC-UV. Acta Chim. Slov. 66(3), 576–653 (2019). https://doi.org/10.17344/acsi.2018.4889
I. Timofeeva, D. Kanashina, D. Kirsanov, A. Bulatov, A heating-assisted liquid-liquid microextraction approach using menthol: separation of benzoic acid in juice samples followed by HPLC-UV determination. J. Mol. Liquids 261, 265–270 (2018). https://doi.org/10.1016/j.molliq.2018.04.040
T. D’Amore, A.D. Taranto, G. Berardi, V. Vita, M. Iammarino, Going green in food analysis: a rapid and accurate method for the determination of sorbic acid and benzoic acid in foods by capillary ion chromatography with conductivity detection. LWT (2021). https://doi.org/10.1016/j.lwt.2020.110841
S. Das, M.N. Uddin, A.S.M. Khaled, M.R.O.K. Noyon, D. Chakraborty, M. Mostafa, S.M.M. Islam, S.C. Bhattacharjee, S.K. Das, M. Uddin, Health risk assessment of three preservatives in beverage, cake, ketchup, and therapeutic products available in Bangladesh using the new validated HPLC-PDA method. J. Food Compos. Anal. 2024(126), 105907 (2024). https://doi.org/10.1016/j.jfca.2023.105907
Acknowledgements
The authors thank Isfahan University of Medical Sciences for facilitating and financial support. This work was supported by the Vice-Chancellor of Research and Technology of Isfahan University of Medical Sciences under Grant number 2401234 with ethics code IR.MUI. RESEARCH.REC.1401.321.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mohamadi, S., Esfandiari, Z., Khodadadi, M. et al. Simultaneous analysis of benzoic and sorbic acids in orange juice using thymol/ water natural deep eutectic solvent and DLLME method followed by high performance liquid chromatography. Food Measure 18, 6285–6294 (2024). https://doi.org/10.1007/s11694-024-02647-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11694-024-02647-y