
Abstract
Sheep primary epithelial cells are short-lived in cell culture systems. For long-term in vitro studies, primary cells need to be immortalized. This study aims to establish and characterize T immortalized sheep embryo kidney cells (TISEKC). In this study, we used fetal lamb kidneys to derive primary cultures of epithelial cells. We subsequently immortalized these cells using the large T SV40 antigen to generate crude TISEKC and isolate TISEKC clones. Among numerous clones of immortalized cells, the selected TISEKC-5 maintained active division and cell growth over 20 passages but lacked expression of the oncogenic large T SV40 antigen. Morphologically, TISEKC-5 maintained their epithelial aspect similar to the parental primary epithelial cells. However, their growth properties showed quite different patterns. Crude TISEKC, as well as the clones of TISEKC proliferated highly in culture compared to the parental primary cells. In the early passages, immortalized cells showed heterogeneous polyploidy but in the late passages the karyotype of immortalized cells became progressively stable, identical to that of the primary cells, because the TISEKC-5 cell line has lost the large SV40 T antigen expression, this cell line is a valuable tool for veterinary sciences and biotechnological productions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Kidney-derived epithelial primary cells and cell lines like the human embryonic kidney (HEK-293) (Graham et al. 1977), the monkey Vero (Simizu et al. 1967), the bovine Madin-Darby bovine kidney (MDBK) (Madin and Darby 1958), or canine MDCK (Gaush et al. 1966) cell lines have been extensively used in various studies. Primary lamb fetal kidney cells originated from the kidney cortex of animal fetuses less than 2 mo of age have been widely used for a long time (De Lange 1959) for isolation of viruses such as “peste des petits ruminants” (PPR), sheep and goat pox (SGP), and bluetongue (BT) viruses (Taylor and Abegunde 1979). The cells were also used for capripoxvirus propagation and laboratory diagnosis, such as titration and serological detection (Babiuk et al. 2007).
Fetal ovine kidney cells, traditionally used for ovine-poxvirus propagation, require a stock of primary cells freshly isolated from kidneys of fetal animals. In this situation, these cells do not lend themselves to proper control of their health status, so ensuring that the cells are free of adventitious agents such as DNA and RNA viruses is difficult. In the absence of continuous cell lines sensitive to those viruses, their isolation and detection prevent the development, validation, and widespread use of viruses for disease confirmation.
Primary cells are useful within the first few passages, usually within two to four, and their uses present several drawbacks. Primary cells undergo a limited, predetermined number of cell divisions before entering an irreversible growth arrest. The risk of endogenous contamination is higher than for cell lines. In general, access to primary cultures is difficult and expensive for large-scale production. In contrast to primary cell cultures, which require optimized growth conditions, immortalized or permanent cell lines offer several advantages such as being able to undergo unlimited cell divisions and having an infinite lifespan. They are cost-effective, easy to use, provide an endless supply of material, and bypass ethical concerns associated with the use of animal and human tissues. Cell lines also provide a homogeneous population of cells, which is invaluable since it offers a consistent supply and reproducible results. They are consistently being used in vaccine production, testing drug metabolism and cytotoxicity, antibody production, the study of gene function, and synthesis of therapeutic compounds (Gómez-Lechón et al. 2003).
Stable cell lines are preferred in most diagnostic laboratories over cultured primary cells due to their longevity, rapidity of multiplication, and ease of maintenance. Immortalized cell lines are handy tools for the study of biological processes and may also provide useful systems for cellular regulation and differentiation. In biotechnology, stable cell lines are fundamental for the production of viral vaccines or viral vectors for therapeutic gene transfer. The immortalization approach could avoid animal embryo collections from the slaughterhouse, every time primary cells are needed. Also, with the new regulation and development of technologies to limit pregnant animals in the slaughterhouses, access to fetuses and embryos became more difficult (EFSA 2018).
The purpose of the current study is to describe the immortalization of primary kidney sheep cells using a plasmid harboring the sequence encoding the SV40 large T antigen. The resulting immortalized cells were characterized according to their morphology, cell growth, and expression of the SV40 large T antigen. The results of this study suggested SV40 large T immortalized sheep cells could be considered as a valuable tool for a long-time period of SECK cell study as they manifest multiple essential properties in the cell culture system.
Materials and Methods
Cells
Placenta containing a sheep embryo at 8 to 10 weeks of gestation was obtained from the slaughterhouse and transferred to the laboratory on ice to extract the fetus in aseptic conditions. Both kidneys were collected and transferred into cold Dulbecco’s modified Eagle medium (DMEM) supplemented with 12.5 mg/ml of fungizone, 0.6 mg/ml L-glutamine, 200 U/ml penicillin, and 200 mg/ml streptomycin (Eurobio, Les Ulis, France) and kept in ice till dissection. Each of the kidneys was transferred into a 100-mm petri dish containing 5 ml DMEM and then aseptically dissected. The outer cortex was excised, scrapped, and then minced with a scalpel blade. The minced tissue suspension was adjusted to 50 ml with DMEM, 0.1% trypsin (Eurobio, France), and then incubated 2 h at 37°C under gentle agitation. The suspension was decanted for 2 min, and the supernatant transferred into a 50-ml conical tube, centrifuged 5 min, 1500 rpm at 4°C. The cell pellet was re-suspended into 20 ml DMEM supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cell suspension was seeded into four 25 cm2 surface flasks and then incubated at 37°C, 5% CO2 in a humidified incubator. Cells were maintained until confluency with a medium change every 72 h. Epithelial cell monolayers were dissociated by trypsin treatment and passaged in new flasks in DMEM medium for three or four times. These cells were henceforth called sheep embryo kidney cells (SEKC).
Plasmid and transfection
The 7.3 kb plasmid DNA pMK16-SV40 (ori) (Gluzman et al. 1980) was amplified in E. coli K12 bacteria and purified using a commercial kit and the manufacturer’s protocol (Macheray Nagel, Hoerdt, France). This plasmid expresses the oncogenic large T antigen of the simian virus SV40 constitutively. Transfection of this plasmid DNA into cultures of primary SEKC was performed using the calcium phosphate method. Briefly, SEKCs were seeded at a density of 5.105 cells/well in a 6-well plate and grown 24 h in DMEM supplemented with 10% FCS in a cell culture incubator. Transfection mixtures containing 2.5 μg plasmid DNA with CaCl2 and × 2 Hanks’ balanced salt solution (HBSS) were incubated 30 min at room temperature to allow the precipitate formation and then inoculated drop-wise onto the SEKC monolayers. Cells were incubated overnight in a cell culture incubator. The transfection medium was discarded, and monolayers of transfected cells were rinsed three times with phosphate-buffered saline (PBS) and then maintained in culture in DMEM containing 10% FCS. When they reached confluency, monolayers were dissociated with trypsin and seeded at lower density (15–20%) into new T-25 flasks. DMEM supplemented with 10% FCS was replaced every 3–4 d, and sub-confluent cell monolayers were passaged following trypsin treatment as described above. As a positive control for transfection, we used the 5.5 kb expression plasmid containing the coding sequences of GFP, pCG-GFP plasmid DNA (Matsuda and Cepko 2004).
Cloning of TISEKCs
Transfected SEKCs were dissociated by trypsin, counted, and diluted in DMEM containing 10% FCS to obtain about four cells/ml. The diluted cell suspension was aliquoted into 24-well tissue culture plates (250 μl/well) to seed approximately 1 cell/well. Cells were incubated at 37°C in 5% CO2. Each well was regularly monitored for cell growth by observation under light microscopy to detect wells with monoclonal development resulting from a single cell per well. These clones were dissociated by trypsin and allowed to grow in the same well till they reached 90–100% confluency. They were then dissociated with trypsin and transferred into 25-cm2 tissue culture flasks. Subsequent passages (over 25 passages) were performed when monolayers reached confluency. Crude immortalized cells were named TISEKC.
Immunocytochemistry
Crude TISEKC and cloned TISEKC cultures were stained by immunocytochemistry to detect large SV40 T antigen using a specific monoclonal antibody (sc-53,448, Santa Cruz Biotechnology, Nanterre, France). Expression of cytokeratin and vimentin proteins was analyzed using monoclonal antibodies anti-cytokeratin 5/6 (CM048A, BioCare Medical, France) and anti-vimentin (CM105A, BioCare Medical, Les Ulis, France), respectively. Cell monolayers were fixed 15 min at room temperature with 100% acetone. Fixed cells were rinsed three times with PBS and then incubated in 0.3% hydrogen peroxide for 30 min at RT to block endogenous peroxidase. Goat serum (1/10 dilution in PBS) was used to incubate the monolayers 30 min at RT for blocking. Monolayers were then incubated 30 min at RT with the primary monoclonal antibody Pab419 diluted 1/50 in PBS, 1% BSA to detect SV40 T antigen. For cytokeratin and vimentin detection, mouse mAb (CK5/6.007, BIOCARE Medical, France) and mouse mAb (V9, BIOCARE Medical, France) were used diluted at 1/100 and 1/50 respectively in PBS, 1% BSA. Fixed cells were rinsed twice with PBS and then incubated 30 min at room temperature with the respective diluted mAbs and then rinsed twice with PBS. For SV 40 T antigen detection, we used a purified biotinylated goat anti-mouse IgG polyclonal antibody (Dako Kit ref.: K0377, Les Ulis, France), diluted 1/50 in × 1 PBS, 1% BSA. After 30 min incubation at 37°C, cells were rinsed with × 1 PBS and then incubated for 15–30 min at RT in a solution containing extravidin–peroxydase diluted 1/100 in PBS as recommended by the supplier (Vector-NovaRED TM Substrate Kit, Les Ulis, France). Cells were rinsed and observed under light microscopy. For cytokeratin and vimentin labeling, monolayers treated with the primary mAbs were then incubated 30 min with the secondary fluorescent highly cross-adsorbed purified goat anti-mouse IgG antibody, Alexa Fluor ® 568, (Fisher/Invitrogen ref.: A11029, Illkirch, France) at 5 μg/ml in PBS, 1% BSA as recommended by the supplier. Cells were rinsed with PBS and directly observed under a fluorescent microscope, and the picture was taken.
Kinetics of cell growth and dependence of fetal calf serum
To evaluate the proliferation and persistence capacity of primary SEKC, cells were seeded in triplicates at a density of 105 cells/well in 6-well plates in DMEM 10% FCS. Every 3 d, cells were dissociated with trypsin and enumerated. Data were used to plot the kinetics growth according to the passage number. To compare the proliferative capacity of SEKC, TISEKC, and TISEKC-5, cells were seeded at a density of 0.25 × 105 cells/well in 6-well plates in DMEM 10% FBS. Triplicates of wells were dissociated with trypsin daily 1–5 and every 2 d (7, 9, and 11) and the last point at 14 d after cell seeding and enumerated. Cell counts were used to plot the kinetics proliferation curves. For evaluation of serum dependence of cultured SEKC, crude TISEKC, and the TISEKC-5 clone, cells were seeded at a density of 0.125 × 105 cells/well in 6-well plates in DMEM supplemented with 10%, 5%, 2.5%, 1%, 0.5%, or 0.25% FCS. Triplicate wells of each culture were dissociated and counted every day for 10 d after cell seeding. Average cell counts were used to plot proliferation kinetics.
PCR detection of SV40 T antigen coding gene
Cellular DNA was isolated using the genomic DNA kit (Nucleospin tissue, Machery-Nagel, France), according to the manufacturer’s instructions. Detection of SV40 T antigen gene was performed by PCR using the primers, SV40 T antigen-F: 5′-CTGACTTTGGAGGCTTCTGGG-3′ and SV40 T antigen-R: 5′-ACATCCCAAGCAATAACAACACATC-3′. PCR reaction in 25 μl was conducted by an initial denaturation step of 94°C for 5 min, followed by 30 cycles of 94°C for 10 s, 60°C for 30 s, 72°C for 30 s, and a final extension of 72°C for 10 min. After amplification, 3 μl of PCR products were analyzed by 1% agarose gel and visualized under UV light.
Examination of TISEKC karyotypes
TISEKC cultures from selected clones were grown in on eight-chamber Labtek slides in DMEM till reaching approximately 60% of confluency. The cells were then stopped in metaphase by addition of 20 μL of Colcemid (N-Deacetyl-N-methylcolchicine, 10 μg/mL, Gibco, Illkirch, France) to the culture for 30 min at 37°C, incubate at 37°C for 20 min in a hypotonic solution to induce plasma membrane destruction. Monolayers were then fixed for 30 min using (3:1 methanol:acetic acid). G-banding coloration was performed with Giemsa after trypsin exposition and R-banding coloration was performed with Giemsa and BrdU (5-bromodésoxyuridine). Chromosome numeration analyses were performed on at least 100 mitoses per sample. Finally, chromosome morphology was evaluated on at least 3 mitoses per sample.
Evaluation of susceptibility of TISEKC to transfection
TISEKC were seeded at a density of 105 cells/well in a 6-well plate and grown for 24 h in a tissue culture incubator. Transfection was performed either with calcium phosphate or with TransIT-LT1 methods (Mirus-Euromedex, Souffelweyersheim, France). Cells at a density of 80–90% of confluency were transfected with calcium phosphate as here above described and with TransIT®-LT1 Transfection Reagent according to manufacturer’s recommendations. The expression plasmid (2.5 μg) containing the coding sequence of GFP, pCG-GFP plasmid DNA (Matsuda and Cepko 2004) was used as an indicator of transfection efficiency. After 13 d, expression of GFP was observed in cell monolayers by fluorescence microscopy.
Results
Immortalization of primary cells
The replication-defective plasmid pMK16-SV40 (ori-) expressing the immortalizing SV40 T antigen was used to immortalize primary cell cultures of sheep embryo kidney cells by transfection (Fig. 1a). Greater than 50% of transfected SEKC with the control plasmid pCG-GFP were found to express the green fluorescent protein. This indicated that the transfection conditions of these primary cells were efficient. Serial passages of crude transfected SEKC with the immortalizing plasmid revealed that these cells had acquired the capacity to undergo greater than six passages without senescence or decreasing their capacity of proliferation. While the non-transfected SEKC undergone cell division arrest and could not be kept for more than six passages. Indeed, SEKC from the fifth passage grew slower than at passage three, and at the end of the sixth passage, the cells showed signs of senescence with a minimal number of cells actively dividing (Fig. 1b). These results indicated that transfected cells acquired immortalization properties and, therefore, were named TISEKC.

Evaluation of cell growth kinetics. (a) Cartoon of cell immortalization pipeline. (b) Cell growth of primary SEKC following serial cell culture passages. Triplicates of cells were seeded in 6-well plates at a concentration of 105 cells per well, and then dissociated with trypsin and counted every 3–4 d for each passage during seven passages. The standard deviations of cell counts are represented by the black bars. (c) Comparative growth kinetics of SEKC, TISEKC, and TISEKC-5. Cells were seeded at a density of 0.25 × 105 cells/well in 6-well plates in DMEM 10% FBS, and triplicates of wells were dissociated with trypsin and enumerated at each indicated time point. The mean of cell numbers is illustrated with standard deviations.
Derivation and selection of TISEKC clones
We used the classical limiting dilution method with TISEKC to generate single cell–derived colonies that were subsequently amplified to generate cloned cell lines. Twenty-one clonal cell lines numbered as TISEKC-1 to -21 were selected for further studies.
Kinetics of cell growth and dependence of fetal calf serum
The growth kinetics was determined by chronological counting of total viable cells in wells at selected time points. The values of cell counts were used to plot the graphs shown in Fig. 1c. The growth kinetics of SEKC showed the absence of a lag phase, indicating that the cells had all their proliferative capacities. The exponential growth phase that followed the initial phase was quite long, lasting about 5 and 6 d. This latter was followed by the slowdown phase, observed between days 7 and 9, beyond which SEKC was in a stationary phase, probably due to a balance between growth and cell death. TISEKC showed an accelerated rate of division, reaching the confluency within 2 and 3 d while actively dividing SEKC seeded at the same density required at least 5 and 6 d to reach confluency (Fig. 1c). At confluency, SEKC stopped growing as a result of contact inhibition, while TISEKC continued to overgrow in layers on top of the monolayer cells (data not shown). TISEKC were serially passaged over 20 times and have been successfully frozen, stored at − 80°C, and thawed with a high rate of viability (> 80%) before storing a large stock in liquid nitrogen.
However, the growth kinetics of TISEKC and TISEKC-5 clone was characterized by their coevolution, although TISEKC cells grew less rapidly than TISEKC-5. Indeed, the shape of the curves is similar for the first 3 d with a strong increase for the next 4 d to finally reach a plateau. Moreover, comparing these two kinetics with that of SEKC, it is clear that the proliferation phase was shorter, and the stationary phase, represented on the curve by a plateau, was rapidly reached for SEKC. Overall, TISEKC-5 proliferated exponentially, at a faster growth rate than the primary cells, with a doubling time of 17 h.
Next, we examined the dependency of TISEKC on fetal bovine serum by decreasing the concentration in their culture medium. Cell counts of triplicate wells grown in various concentrations of fetal bovine serum in the medium were determined every day, and value numbers were used to plot growth kinetics shown in Fig. 2. With all cell cultures, the highest serum concentration (5%) correlated with the highest growth kinetics with the highest cell numbers at all examined time points post-seeding. However, the concentrations of TISEKC and TISEKC-5 reached higher values in almost all time points compared to those obtained for SEKC samples. The decrease of serum concentration resulted in decreased growth kinetics for all three cultured cell types. However, curiously this decrease in serum concentration seems to have a higher effect on TISEKC and TISEKC-5 than the parental SEKC. This is mainly seen for the level of serum about 0.5% (Fig. 2a, b, and c).

Longitudinal evaluation of serum dependence for cell growth. SEKC (a), TISEKC (b), and TISEKC-5 (c) cultures were seeded at a density of 0.125 × 105 cells per well in 6-well plates in DMEM supplemented with 10%, 5%, 2.5%, 1%, 0.5%, or 0.25% FCS. Triplicate wells of each culture were dissociated and counted at indicated time points after seeding. The mean of cell numbers is represented with standard deviations.
Detection of SV40 T antigen, cytokeratin, and vimentin protein expression in TISEKCs
Monolayers of crude TISEKC and clonal TISEKC-5 cultures at P14 passage were used to detect SV40 T antigen expression by immunocytochemistry with anti-T SV40 mAb. The T immortalized goat embryo fibroblasts (TIGEF), previously immortalized in our Lab., (Da Silva Teixeira et al., 1997), were used as a control to assess large T antigen expression. Monolayers of both crude and cloned cell cultures displayed uniform nuclear staining typical of the SV40 T antigen. This indicated that even following serial passages, the cells were still expressing the immortalizing antigen. Figure 3a shows the detection of SV40 T antigen expression in the crude TISEKC as well as in 20 of the clonal lines. Importantly a single clone, TISEKC-5, was found to lack expression of SV40 T antigen in any of the cells, although there was no difference in cell proliferation compared to the 20 other clones. PCR analysis was performed to examine the presence of SV40 T antigen gene in total DNA from TISEKC-5 and TISEKC-6 at passages 5 and 20 as well as crude TISEKC and SEKC using the primers described in “Material and Methods.” As shown in Fig. 3b, the 1 kb PCR product was successfully amplified at both times of passages with DNAs among TISEKC-6 (lanes 1 and 2), TISECK-5 (lanes 3 and 4), and crude TISEKC (lane 5). No PCR product was amplified with the DNA of SEKC and negative control. This latter corresponded to PCR mixture without DNA and the last lane corresponds to a positive control using classical actin primers generating a 0.5 kb PCR product. Both the clones and crude cells were found to be positive for cytokeratin and negative for vimentin markers (Fig. 4). All TISEKC clonal lines retained phenotypical epithelial characteristics similar to that of the crude and the parental cells. Comparison of crude TISEKC and the TISEKC-5 clone showed that the epithelial phenotype was well conserved, both in size and form of the cells (Fig. 5).

Detection of large T SV40 antigen expression and viral DNA. (a) Immunocytochemistry was performed for all TISEKC (1–21) clones. The TISEKC-5 clone (5) showed no detectable expression of a large T SV40 antigen. Positive control (22) corresponds to TIGEF cells (Da Silva Teixeira et al., 1997) showed positive signal; and negative control (21) corresponds to TIGEF treated with PBS in replacement of the primary Ab showed no signal. (b) PCR and analysis to confirm the presence of SV40 T antigen gene in total DNA from TISEKC, TISEKC-5, and TISEKC-6. (1) TISEKC clone 6, passage 5; (2) TISEKC clone 6, passage 20; (3) TISEKC-5, passage 5; (4) TISEKC-5, passage 20; (5) crude TISEKC, passage 5; (C−) negative control lacking DNA; (C+) positive control with conventional actin primers yielding a 500 bp PCR product.

Immunodetection of cytokeratin and vimentin protein expression. Immunocytochemistry was performed for crude TISEKC cells, TISEKC-5 and 6 lines, and the non-immortalized parental SEKC epithelial cells. Negative control corresponds to cells treated with PBS and showed no positive staining. MAbs and secondary polyclonal antibodies are described in the Material and Method section. In the left panels are cells observed with the light microscopy and in the right panels are cells observed with fluorescent microscopy.

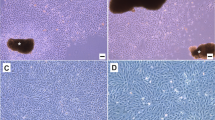
Morphological aspects of TISEKC cells. Crude TISEKC (a) and TISEKC-5 (b) cell culture stained by May-Grünwald Giemsa observed with phase contrast microscopy at passage 20. A similar morphological shape of epithelial cells is observed (scale bars, 40 μm).
Karyotype examinations
The chromosome number in a standard sheep cell is (2n) = 54 comprising 52 autosomes and two sex chromosomes, XY or XX. There are three pairs of sub-metacentric and 24 pairs of acrocentric chromosomes, including X and Y chromosomes. Cytogenetic analysis was carried out on chromosomal preparation of the non-immortalized SEKC at passage 3, TISEKC clonal lines, and of the parental crude TISEKC at early P5 passage post-transfection (Fig. 6). Data of this analysis revealed a tetraploid stage with aneuploid cells (2n = 108 + 6) in 75 to 80% of cells in all immortalized cell preparations at passage 5 post-transfection. The remaining cells (20–25%) showed a usual diploid number of chromosomes (2n = 54). In contrast, non-immortalized SEKC at passage 3 showed a usual diploid number of chromosomes (2n = 54) in all examined cells, like in Fig. 7b and d. After serial passages of crude TISEKC and TISEKC clones, a chromosomal re-examination of cells at passage 20 revealed a reversion of the chromosome number to normal diploid (2n = 54) in cells of all examined TISEKC crude and clones like in Fig. 7a and c.

Cytogenetic examination of karyotypes of TISEKC cells. Illustrations of G-band staining of TISEKC-5 (a) and crude TISEKC (b) metaphase chromosomes, as well as R-band stains TISEKC-5 (c) and crude TISEKC (d) mitoses stained with May-Grünwald Giemsa (magnification × 100).

Transfection efficiency of TISEKC-5 cells. This is revealed by GFP-fluorescence (green) observed by fluorescence microscopy after transfections performed using either phosphate calcium or TransIT LTI methods (scale bars, 20 μm).
These results demonstrate that abnormal chromosome numbers seen in the early passages post-transfection of SEKC were only transient, and established cell lines became all stably diploid. Of note, no structural chromosomal rearrangements were found within the limit of the karyotype resolution, which does not, however, exclude a potential small rearrangement or gene disorder.
Test of the susceptibility of cell lines to transfection
To verify that the immortalized TISEKC cells can be used to transfect plasmids expressing proteins of interest, we tested the GFP plasmid. Transfections were done using either calcium phosphate or TransIT-LT1 reagents. Fluorescent microscopy revealed that the TISEKC cells could effectively be transfected near 20% with TransIT-LT1 and 15% with calcium phosphate as indicated by the presence of expressed GFP (Fig. 7). It appears that the TransIT-LT1 transfection method is more efficient than calcium chloride, as the proportion of fluorescent cells observed with TransIT-L1 is higher Fig. 7.
Discussion
Sheep embryo kidney–derived cell lines, TISEKC, were successfully established following the transfection of primary SEKC with SV40 T antigen–expressing plasmid and serial passages. Unlike the parental SEKC that supports only a limited number of passages (P5–P6), TISEKC was passaged over 30 times with no sign of senescence or slow proliferation rate. The proliferation rate was found to be increased compared to that of the parental cells showing 17 h doubling time for cells in culture between the seeding time and 24 h later. This doubling time change to between 20 and 25 h during the exponential phase of growth. Interestingly, we noted the absence of adaptation time (0–24 h) for the cell line, unlike the parental cells (SEKC), which had a doubling time of 40 h between 0 and 24 h (adaptation time). All these results indicated that the new cell lines were stable and proliferated rather rapidly, and their characterization revealed some improved proprieties compared to the primary cells.
Several approaches exist for immortalizing mammalian cells in cell culture systems using viral genes. These include human papillomavirus (HPV) E6 and E7 (type16) (Cascio 2001), Epstein-Barr virus (EBV), Simian virus 40 (SV40) T antigen. This latter induces immortalization by inactivating the tumor suppressor genes (P53; PRb) (Schafer 1998). This gene has been widely used in basic biological research and also for the generation of transgenic mouse models of human cancers (Colvin 2014). It has been demonstrated that expression of large T antigen alone is sufficient for the immortalization of mammalian cells, including human cells. This antigen expression initiates S-phase and pushes cells to proliferate (Banks-Schlegel and Howlev 1983; Jat and Sharp 1986) but not to undergo inducing neoplasia (Bloomfield and Duesberg 2015). In the present experiments, primary cells were immortalized using a plasmid that encodes the SV40 large T antigen. Obtained crude TISEKC and clones displayed a normal morphology, as evaluated by contrast microscopy, compared to the parental cells. Interestingly, their dependency on fetal bovine serum in culture indicated that the TISEKC were rather immortalized TISEKC than transformed. Indeed, they still require serum factors to proliferate in the cell culture system, indicating that the TISEKC has similar biological characteristics like the parental cells. Not all T antigen–transfected cells can escape the crisis and, only those that survive the crisis became immortal (Shay and Wright 1989) indicating that the large T antigen–induced immortalization is genetically based (Ray and Kraemer 1993). The mechanism by which the large T antigen induces immortalization is well established. It regulates some key cellular pleiotropic switches that coordinately confer different modified properties on cells. The SV40 large T antigen is a multifunctional phosphoprotein that targets multiple cellular pathways (Ahuja et al. 2005; An et al. 2012). The importance of the large T antigen interaction with Rb-proteins and with p53 is also well established. Genetic studies suggest that inactivation of Rbp and p53 may not account for the full transformation potential of T antigen (Sachsenmeier and Pipas 2001; Wei et al. 2003; Lock et al. 2004; Martini et al. 2007; An et al. 2012). Several other T antigen–binding proteins have been identified that have also the potential to contribute to immortalization, such as (i) transcriptional adapter proteins p300/CBP or p185; (ii) tumor suppressor Cul7 (Culine 7) (Ali et al. 2004); (iii) transcription factor TEF-1; (iv) Nbs1, a component of the MRN complex DNA repair (Wu et al. 2004); (v) Fbw7, a component of the ubiquitination machinery (Welcker and Clurman 2005); and (vi) Bub1 protein, a mitotic spindle checkpoint protein. The interaction of large T antigen with Bub1 results in perturbation of the spindle checkpoint (Cotsiki et al. 2004; Hein et al. 2009; Hu et al. 2013) and maybe one explanation for aneuploidy and genetic instability induced by T antigen. Indeed, the ability of T antigen to cause such karyotypic instability in human cells has been found to correlate with its ability to deregulate normal mitotic checkpoints (Chang et al. 1997). This may explain several previous reports that demonstrated the ability of T antigen to induce aneuploidy and genetic instability, giving rise to both structural and numerical chromosome aberrations (Ray et al. 1990; Stewart and Bacchetti 1991; Levine et al. 1991; Woods et al. 1994). The karyotype was examined both at early and late stages during the TISEKC lifespans. Several clones exhibited abnormal high chromosomes numbers (tetraploidy and aneuploidy), and later, the TISEKC clones restored their diploid genotype. Moreover, the karyotype had stabilized, because at the latest passage chromosomes number remained normal (2n = 54).
These exciting results showed that cell lines remain proliferative despite some genomic instability during early stages post-transfection. However, genotype instability is quickly lost in time, and cell lines regain genetic stability (diploid). The reversion of TISEKC-5 line to a stable diploid genotype observed at passage 20 is not unique. Previous studies have reported that original tumorigenic cells with genomic instability can revert to the non-tumorigenic karyotype following serial in vitro passages (Schiller et al. 1998).
Conclusion
We concluded that TISEKC clones would be useful tools for basic molecular research including proteomics and genomics. However, the loss of expression of the oncogenic large T antigen in the TISEKC-5 cell line while retaining the immortal characteristics makes it an excellent tool for veterinary vaccine production. Indeed, this cell line offers many advantages such as fast cell division for large-scale biotechnological production, reproducibility since the same stock of cells will be used, availability at any time as stocks can be frozen in liquid nitrogen at − 196°C and rapidly restarted in culture.
References
Ahuja D, Sáenz-Robles MT, Pipas JM (2005) SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 24:7729–7745
Ali SH, Kasper JS, Arai T, De Caprio JA (2004) Cul7/p185/p193 binding to simian virus 40 large T antigen has a role in cellular transformation. J Virol 78:2749–2757
An P, Sáenz Robles MT, Pipas JM (2012) Large T antigens of polyomaviruses: amazing molecular machines. Annu Rev Microbiol 66:213–236
Babiuk S, Parkyn G, Copps J, Larence JE, Sabara MI, Bowden TR, Boyle DB, Kitching RP (2007) Evaluation of an ovine testis cell line (OA3.Ts) for propagation of capripoxvirus isolates and development of an immunostaining technique for viral plaque visualization. J Vet Diagn Investig 19:486–491. https://doi.org/10.1177/104063870701900505
Banks-Schlegel SP, Howlev PM (1983) Differentiation of human epidermal cells transformed by SV40. J Cell Biol 96:330–337
Bloomfield M, Duesberg P (2015) Karyotype alteration generates the neoplastic phenotypes of SV40-infected human and rodent cells. Mol Cytogenet 8:79
Cascio SM (2001) Novel strategies for immortalization of human hepatocytes. Artif Organs 25:529–538
Chang TH, Ray FA, Thompson DA, Schlegel R (1997) Disregulation of mitotic checkpoints and regulatory proteins following acute expression of SV40 large T antigen in diploid human cells. Oncogene 14:2383–2393
Colvin EK, Weir C, Ikin RJ, Hudson AL (2014) SV40 TAg mouse models of cancer. Semin Cell Dev Biol 27:61–73
Cotsiki M, Lock RL, Cheng Y, Williams GL, Zhao J, Perera D, Freire R, Entwistle A, Golemis EA, Roberts TM, Jat PS, Gjoerup OV (2004) Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc Natl Acad Sci U S A 101:947–952
Da Silva Teixeira MF, Lambert V, Mselli-Lakahl L, Chettab A, Chebloune Y, Morex JF (1997) Immortalization of caprine fibroblasts permissive for replication of small ruminant lentiviruses. AJVR 58(6):579–584
De Lange M (1959) The histology of the cytopathic changes produced in monolayer of epithelial cultures by viruses associated with lumpy skin disease. Onderstepoort J Vet Res 28:245–255
European Food Safety Authority (EU body or agency) (2018) Veterinary sector and animal health, slaughter animal, slaughter of animals. https://doi.org/10.2805/270833
Gaush CR, Hard WL, Smith TF (1966) Characterization of an established line of canine kidney cells (MDCK). Proc Soc Exp Biol Med 122:931–935
Gluzman Y, Otsuka H, Kit S (1980) Origin-defective mutants of SV40. Cold Spring Harb Symp Quant Biol 44:293–300
Gómez-Lechón MJ, Donato MT, Castell JV, Jover R (2003) Human hepatocytes as a tool for studying toxicity and drug metabolism. Curr Drug Metab 4(292):312–312. https://doi.org/10.2174/1389200033489424
Graham FL, Smiley J, Russell WC, Nairn R (1977) Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 36:59–74. https://doi.org/10.1099/0022-1317-36-1-59
Hein J, Boichuk S, Wu J, Cheng Y, Freire R, Jat PS, Roberts TM, Gjoerup OV (2009) Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J Virol 83(1):117–127
Hu L, Filippakis H, Huang H, Yen TJ, Gjoerup OV (2013) Replication stress and mitotic dysfunction in cells expressing simian virus 40 large T antigen. J Virol 87(24):13179–13192
Jat PS, Sharp PA (1986) Large T antigen of simian virus 40 and polyomavirus efficiently establish primary fibroblasts. J Virol 59:746–750
Levine DS, Sanchez CA, Rabinovitch PS, Reid BJ (1991) Formation of the tetraploid intermediate is associated with the development of cells with more than four centrioles in the elastase-simian virus 40 tumor antigen transgenic mouse model of pancreatic cancer. Proc Natl Acad Sci U S A 88:6427–6431
Lock RL, Benvenuti S, Jat PS (2004) Immortalization by Sv40 large T antigen. In: Stein GS, Pardee AB (eds) Cell Cycle and Growth Control: Biomolecular Regulation and Cancer second, pp 15–92
Madin SH, Darby NB Jr (1958) Established kidney cell lines of normal adult bovine and ovinorigin. Proc Soc Exp Biol Med 98:574–576
Martini F, Corallini A, Balatti V, Sabbioni S, Pancaldi C, Tognon M (2007) Simian virus 40 in humans. Infect Agent Cancer 2:13
Matsuda T, Cepko CL (2004) Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A 101(1):16–22
Ray FA, Kraemer PM (1993) Iterative chromosome mutation and selection as a mechanism of complete transformation of human diploid fibroblasts by SV4O large T antigen. Carcinogenesis 14:15111516
Ray FA, Peabody DS, Cooper JL, Cram LS, Kraemer PM (1990) SV40 T antigen alone drives karyotype instability that precedes neoplastic transformation of human diploid fibroblasts. J Cell Biochem 42:13–31
Sachsenmeier KF, Pipas JM (2001) Inhibition of Rb and p53 is insufficient for SV40 T-antigen transformation. Virology 283:40–48
Schafer KA (1998) The cell cycle: a review. Vet Pathol 35:461–478
Schiller JH, Bittner G, Wu SQ, Meisner L (1998) Karyotypic changes associated with spontaneous acquisition and loss of tumorigenicity in a human transformed bronchial epithelial cell line: evidence for in vivo selection of transformed clones. In Vitro Cell Dev Biol Anim 34:283–289
Shay JW, Wright WE (1989) Quantitation of the frequency of immortalization of normal human diploid fibroblasts by SV4O large T-antigen. Exp Cell Res 184:109–118
Simizu B et al (1967) Characterization of the Tacaribe group of arboviruses. I. Propagation and plaque assay of Tacaribe virus in a line of African green monkey kidney cells (Vero). Proc Soc Exp Biol Med 125:19–123
Stewart N, Bacchetti S (1991) Expression of SV40 large T antigen, but not small t antigen, is required for the induction of chromosomal aberrations in transformed human cells. Virology 180:49–57
Taylor WP, Abegunde A (1979) The isolation of peste des petits ruminants virus from Nigerian sheep and goats. Res Vet Sci 26(1):94–96
Wei W, Jobling WA, Chen W, Hahn WC, Sedivy JM (2003) Abolition of cyclin-dependent kinase inhibitor p16Ink4a and p21Cip1/Waf1 functions permits Ras-induced anchorage-independent growth in telomerase-immortalized human fibroblasts. Mol Cell Biol 23:2859–2870
Welcker M, Clurman BE (2005) The SV40 large T antigen contains a decoy phosphodegron that mediates its interactions with Fbw7/hCdc4. J Biol Chem 280:7654–7658
Woods C, LeFeuvre C, Stewart N, Bacchetti S (1994) Induction of genomic instability in SV40 transformed human cells: sufficiency of the N-terminal 147 amino acids of large T antigen and role of pRB and p53. Oncogene 9:2943–2950
Wu X, Avni D, Chiba T, Yan F, Zhao Q, Lin Y, Heng H, Livingston D (2004) SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes Dev 18:1305–1316
Acknowledgments
The authors wish to gratefully thank Myriam Khattabi and Marcelline Samadane for excellent technical help.
Funding
This work was supported by the Algerian Ministry of High Education and Scientific Research, the University of Sciences and Technology “Houari Boumediene” Algiers, the French INRA, and the University of Grenoble Alps.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: Tetsuji Okamoto
Rights and permissions
About this article

Cite this article
Seridi, N., Hamidouche, M., Belmessabih, N. et al. Immortalization of primary sheep embryo kidney cells. In Vitro Cell.Dev.Biol.-Animal 57, 76–85 (2021). https://doi.org/10.1007/s11626-020-00520-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-020-00520-y