
Abstract
Combining a rational design strategy with a simple synthetic method to produce an eco-friendly material presents a unique challenge in photocatalysis technology. This work reports the successful synthesis of a heteroanionic titanium oxycarbide photocatalyst—TiO0.25C0.75 (TiOC) via a solid-state reaction and employed for the decomposition of Congo red (CR) and H2O2 generation from water under visible light radiation. Initially, TiOC demonstrated a moderate CR degradation efficiency of 54% (1 g/L) in 120 min and evolved a minimal amount of H2O2 after 90 min (2 mmol) and 120 min (10 mmol) of light exposure. This clearly indicates that the localization of hybridization of O-2p and C-2p orbitals and a more positive conduction band edge (CB) of TiOC lead to reduced carrier mobility and, consequently, high recombination of photogenerated charge carriers. Therefore, sacrificial reagents such as H2O2 and isopropyl alcohol (ISA) were employed to improve CR removal and H2O2 evolution rate, resulting in 70% and 21.5 mmol (120 min), respectively.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the past few decades, research interest has risen in developing materials that can show the capability of both generating greener energy and degrading organic pollutants using solar radiation [1,2,3,4,5]. Mainly, the discharge of organic dyes and medical wastes into water bodies from the industries is causing growing concern about water pollution [6]. More specifically, Congo red (CR) dye is a highly hazardous and carcinogenic anionic dye. It is used in the textile, and rubber industries as a colorant. The consumption of CR mixed water causes cancer and mutations in plants, animals, and humans [7, 8]. Therefore, it is of the utmost need to create unique materials and approaches to combat this severe problem to alleviate the pure drinking water shortage. Moreover, the demand for clean and green energy sources in recent years has created many challenges, including the need to find alternatives to conventional energy sources, which cause significant damage to the ecosystem. Based on this demand, hydrogen peroxide (H2O2) was found to be a promising eco-friendly compound that generates greener energy during combustion without producing other toxic compounds. Besides, it possesses good calorific value and oxidation capability and evolves greener products (H2O and O2) during combustion [9, 10]. These characteristics made H2O2 to play a significant role in the petrochemical industry, making pulp and papers, sewage treatment for disinfection, etc., [11, 12]. The anthraquinone (AQ) process is the most widely employed conventional method for generating a large amount of hydrogen peroxide, which possesses few drawbacks, such as the complex process yields toxic by-products and is costlier [13, 14]. While considering these bottlenecks, it is necessary to devise a cost-effective and reliable approach for removing CR from drinking water resources and generating hydrogen peroxide from water using solar radiation.
Therefore, photocatalysis is found to be a viable technique for effective CR removal and hydrogen peroxide production due to its mild reaction conditions, lower energy consumption, and absence of secondary pollutants [15, 16]. It is an emerging approach and has gained much popularity in recent years since it uses safe, cheap materials that harvest a large amount of solar radiation and are easy to fabricate. The materials used in this approach have an excellent capability to promote the redox reactions in the reaction chamber, which plays a promising role in the decomposition of organic pollutants and solar fuel generation [17,18,19]. Using these advantages, researchers designed various types of photocatalyst materials for organic pollutant degradation and solar fuel generation. Initially, conventional photocatalysts such as ZnO, TiO2, and Bi2S3 were utilized for the decomposition of organic dye and solar fuel generation [20,21,22,23]. These photocatalysts were valued for their stability, affordability, and non-toxicity. However, they encountered significant issues such as a wider band gap, low visible light absorption, photo-corrosion, and rapid recombination rate [24,25,26]. The effectiveness of traditional photocatalysts in meeting the increasing demand for energy and degrading emerging stable pollutants is being questioned due to their limitations. As a result, researchers have turned to heteroanionic materials as a potential alternative for future energy generation and water purification. These materials are attractive due to their superior light absorption capacity, narrower band gap, and enhanced photo-corrosion resistance [27, 28]. Heteroanionic photocatalysts are primarily composed of metal cations that are linked with oxygen and other anion atoms. These photocatalysts have more than one anion in their structure, which provides an opportunity for valence band engineering by carefully manipulating the anions [29, 30]. In these materials, the valence band (VB) is occupied by a combination of anion-p and oxygen-2p orbitals, while the conduction band is composed of d0 or d10 orbitals of metal ions. This arrangement results in a narrower band gap and a more negative valence band compared to conventional photocatalysts [31, 32]. Heteroanionic photocatalysts can be classified based on the type of anion present in their systems, such as metal oxynitrides, oxysulfides, oxyhalides, and oxycarbides [27, 33].
More specifically, titanium oxycarbide, a heteroanionic photocatalyst, has demonstrated great potential in photocatalysis applications [34, 35]. This material contains a metal ion that is linked to oxygen and carbon atoms, and its electrical and thermal properties can be easily adjusted. In addition, its oxidation resistance, chemical inertness, and photon absorption capabilities are exceptional. The valence band of TiOC is occupied by hybridized C-2p and O-2p orbitals, while its conduction band is composed of empty d orbitals of metal ions [36]. Although there are very few reports on the applications of TiOC in photocatalysis, it has been shown to exhibit good photocatalytic activity towards organic pollutants [37, 38]. This is mainly due to its wider surface area, good light absorption ability, and narrower band gap energy. It is believed to have combined characteristics of its oxide and carbide compounds. Moreover, Ti-based photocatalysts generally show promising photocatalytic activity towards organic pollutants because of their wider surface area and good stability. These catalysts can also be used for solar-fuel generation using sunlight [39,40,41].
Therefore, we chose titanium oxycarbide as a photocatalyst which was synthesized through a simple and cost-effective approach and employed it for the decomposition of CR and H2O2 generation from water using visible light radiation. We also examined how the photocatalytic efficiency of TiOC on CR degradation and H2O2 generation was influenced by its process parameters such as the amount of catalyst and the presence of sacrificial agents. From the literature survey, it was found that the adoption of TiOC photocatalysts for the decomposition of CR and H2O2 generation from the water was not investigated elsewhere.
Experimental Procedure
Synthesis of titanium oxycarbide
All the chemicals were of analytical grade and were utilized as purchased without any additional purification. Titanium oxycarbide was synthesized by blending titanium (II) oxide (TiO) and titanium carbide (TiC) in a mortar with acetone for 4 h. The mixed powders were then dried and compressed into pellets using a two-ton uniaxial press. The resultant pellets were heated in two phases at 1000°C and 1500°C for 6 h, with a heating rate of 5°C/min in a vacuum atmosphere, as shown in Fig. 1. The chemical reactions involved in the formation of TiOC are represented as follows:

Preparation of TiO0.25C0.75 using solid-state reaction
Material characterization
The crystalline structure and morphology feature of synthesized photocatalysts was studied using X-ray diffraction (PANalytical X’accelerator, λ = 0.15406 nm) and scanning electron microscope (JEOL JSM-5600) and a transmission electron microscope (JEOL-2010). Elemental mapping of the synthesized samples was observed using energy-dispersive X-ray spectroscopy (EDX) attached to the SEM instrument. The optical characteristics were calculated using the ultraviolet−visible diffuse absorbance spectrum (UV-Vis-DRS) on a JASCO V-670 UV-Vis spectrophotometer.
Photodegradation test of CR
The photocatalytic performance of TiOC was evaluated for the decomposition of CR in an aqueous medium under visible light irradiation. A stock solution of Congo red (20 mg/L) was prepared by dissolving 20 mg of CR dye in one liter of deionized water. Then, different concentrations (0.25, 0.50, 0.75, and 1 g/L) of TiOC photocatalysts were added to 100 ml of the CR solution. The TiOC-suspended CR solution was kept in the dark for 1 h in order to achieve the adsorption–desorption equilibrium. After reaching equilibrium, the dark-treated solution was exposed to a 300 W Xenon light source in the photocatalytic chamber (CAE-HXF 300). The relative concentration of congo red in the light-treated solution was evaluated using a Shimadzu UV-Vis spectrophotometer at every interval of 30 min.
Photocatalytic H2O2 production test
For the hydrogen peroxide generation test, 50 mg of TiOC photocatalysts were added to 100 mL of deionized water. Then, the prepared solution was kept under a 300 W Xenon light source. The generation of H2O2 was calculated based on redox titration with KMnO4, as per the previously reported method [42,43,44]. During the photocatalytic reaction, 1 mL of the solution was withdrawn from the reaction chamber at every 30-min interval and purified using a 0.45-μm filter. Then, 1 mL of purified solution, 2 mL of KMnO4 solution (40 mM), and 1 mL of H2SO4 solution (1 M) were mixed together and held for 3 min to reduce KMnO4 into MnSO4 by H2O2. To determine how much KMnO4 was used in the reaction, the intensity of the light absorbed by the solution at 523 nm was measured at various reaction times. Based on the following equation and formula, the concentration of the generated H2O2 can be determined.
Results and discussion
Powder XRD analysis on titanium oxycarbide
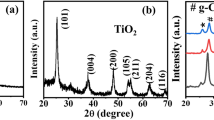
X-ray diffraction patterns of TiOC were measured in the 2Ɵ range from 30° to 90° at a scan rate of 1°/min, as shown in Fig. 2. The synthesized TiOC depicts the moderately intense peak, confirming the formation of well-crystalline pure phase. It clearly revealed that the XRD signals of TiOC are well-matched with the JCPDS files of TiO and TiC as shown in Fig. 2 [45]. It provides a confirmation that synthesized TiOC has a cubic structure with space group Fm-3m. Mainly, all the peaks of TiO0.25C0.75 have shifted and broadened a little towards higher 2θ values, which provides an additional clue for the addition of the carbon atom in the TiO structure. A similar kind of observation was reported in earlier research works on the TiOC [46, 47]. In addition, no other satellite peaks were found in the XRD pattern confirming the purity of the synthesized TiOC photocatalysts.

XRD patterns of TiO0.25C0.75 matching with standard JCPDS files of TiO (red) and TiC (blue)
With the help of Williamson-Hall (W-H) formulas [48], the crystalline size and strain of the prepared TiO0.25C0.75 photocatalysts have been calculated, as shown in Fig. 3. It has been found that TiOC possesses a large crystalline size and with less strain in it (Table 1). This may be due to the decrease in crystalline defects and higher synthesis temperature applied for TiO0.25C0.75.

Williamson-Hall (W-H) analysis for the TiO0.25C0.75
SEM and TEM analysis on titanium oxycarbide
It is believed that the surface morphology feature of the prepared TiOC plays a dominant role in improving its photocatalytic character. Therefore, the surface morphology of synthesized TiOC was captured using SEM, as shown in Fig. 4(a).

(a) SEM image and (b) elemental mapping of prepared TiO0.25C0.75 photocatalysts
It has been found that the TiOC surface was made up of uniformly distributed porous and spherical-shaped particles without much agglomeration. Moreover, the higher temperature supplied during the synthesis process help to create a wider surface area with a large crystalline size in TiOC, which induces facial migration of excited electrons and holes to participate in succeeding photocatalytic reactions. This also creates a strong interface between different anions in TiOC, acting as the charge carrier transfer channel. For instance, Negi et al. observed similar observations when carbon was doped in the titanium dioxide structure, which supports the reduction of its agglomeration behavior and provides a wider surface area [49]. This significantly increased the decomposition efficiency of C-TiO2 over the methylene blue (MB) dye. Moreover, the SEM image of the C-TiO2 closely matches the surface morphology of TiO0.25C0.75 found in our observations. The elemental mapping using EDX also confirmed the fact that elements such as Ti, O, and C are uniformly distributed throughout the sample, as shown in Fig. 4(b). The TEM image and selected area electron diffraction (SAED) pattern of the synthesized TiO0.25C0.75 photocatalyst are displayed in Fig. 5(a) and (b). These observations again confirmed the phase purity of the TiO0.25C0.75 photocatalysts, as discussed in the XRD analysis.

(a) TEM image of the synthesized TiO0.25C0.75 photocatalysts, along with its (b) SAED pattern
UV-Vis-NIR analysis on titanium oxycarbide
The optical and energy band gap characteristics of the synthesized TiOC were analyzed using UV-Vis absorption spectra and its tauc plot. As shown in Fig. 6(a), it was found that TiOC exhibited an absorption edge around ~500 nm and possessed good light absorption characteristics. The band gap energy (Eg) of TiOC was computed using the Kubelka-Munk (KM) function [50, 51], and its tauc plot is presented in Fig. 6(b). The tauc plot indicates that TiOC has a narrower band gap energy (Eg) value of 1.0 eV. This result was primarily obtained by an upward shift of the VB maximum, not by moving the CB maximum downward, and also by strong hybridization between the C-2p and O-2p orbitals. Furthermore, the VB of TiOC was occupied by hybridized orbitals of carbon and oxygen, while its CB was composed of d orbitals of Ti-ions. A similar kind of band gap reduction was also found by Negi et al. [49] in the C-TiO2 structure. This kind of electronic arrangement exhibits suitable band edge positions in TiOC, which are crucial in promoting the reduction and oxidation reactions during the decomposition of CR and H2O2 generation.

(a) Represents the UV-Vis absorption spectra of synthesized TiOC photocatalysts and (b) its tauc plot
Photocatalytic degradation activity of titanium oxycarbide
The photocatalytic behavior of TiOC was assessed by observing the degradation of the CR in an aqueous solution under visible light illumination. When no photocatalyst was present, the CR was found to be more stable under visible light illumination. Then,TiOC photocatalysts of different concentrations were suspended in the prepared CR solution and kept in the dark environment to obtain adsorption–desorption equilibrium. Subsequently, the dark-treated solution was exposed to visible light illumination, and the absorption spectra of the light-treated solution were recorded at 30-min intervals using a UV-Vis spectrophotometer. Most importantly, the impact of various process parameters on the CR degradation activity, such as the various catalyst amount and the presence of sacrificial agents, was examined. The CR degradation efficiencies and its pseudo-first-order kinetics (Langmuir–Hinshelwood model) of TiOC can be determined using the following formulas [52, 53]:
where, C0 and Ct are the concentrations of CR at times 0 and t, respectively, under visible light exposure; “k” is the pseudo-first-order rate constant of photodegradation (min−1) of CR degradation.
Initially, the effect of catalyst loading on the photocatalytic degradation of Congo red was studied by varying the amount of TiOC from 0.25 to 1 g/L. The photocatalytic degradation of CR over time using different amount of TiOC is shown in Fig. 7(a). From the figure, it can be observed that the concentration of CR gradually decreased with an increase in the amount of TiOC catalyst. The maximum CR degradation efficiency of TiOC was observed at 1 g/L, which took only 120 min to degrade 54% of CR. Similarly, at 0.25 g/L, 0.50 g/L, and 0.75 g/L of TiOC, it degraded 23%, 46%, and 51% of CR at 120 min, respectively. The CR degradation efficiency for various TiOC photocatalyst amounts has been calculated using eq. (4) and is plotted in Fig. 7(b). It is evident that at higher doses of catalyst, more active sites were created on the surface of TiOC due to the massive absorption of light, leading to the creation of an excessive amount of hydroxyl radicals during the degradation of CR. Similarly, the first-order reaction kinetics of CR degradation has been computed for various TiOC amounts. The linear fitting is plotted in Fig. 7(c), and the R2 values are tabulated in Table 2.

(a) Comparison of the photocatalytic CR degradation rates using different amounts of TiOC photocatalyst and in the presence of sacrificial reagents; (b) its degradation efficiency; (c) and (d) pseudo-first-order kinetics of the photocatalytic CR degradation rates using different amounts of TiOC photocatalyst and in the presence of sacrificial reagents, and (e) reusability nature of TiOC photocatalysts
Based on the above results, it has been found that TiOC showed average CR degradation efficiency even at a higher amount of catalysts compared to commercial photocatalysts. This may be due to the quick recombination of photogenerated electron-hole pairs. Therefore, H2O2 was introduced as an electron sacrificial agent to prevent the quick recombination of charge carriers and enhance the Congo red degradation efficiency of TiOC. We dispersed 1 ml of H2O2 (1 mmol) and 0.50 g/L of TiOC into 100 ml of the prepared CR solution. Surprisingly, after the addition of H2O2, it was observed that the CR decomposition increased to 70%, performing even better than the CR solution with 1 g/L of TiOC, and its degradation characteristics can be seen in Fig. 7(a), (b), and (d). This is because, when H2O2 was added to the CR solution, the photogenerated electrons on the TiOC surface were captured by electron sacrificial reagents, reducing the charge carrier recombination rate and providing sufficient time for the creation of more hydroxyl radicals (•OH) using photogenerated charge carriers. In addition to this, the strong CR adsorption on the TiOC photocatalysts could also play a promising role in enhancing the photodegradation efficiency.
Similarly, we also examined the role of the photogenerated holes in the degradation of CR. To analyze this fact, we mixed 1 ml of ethanol into the prepared CR solution along with 0.50 g/L of TiOC. The CR degradation characteristics of TiOC using ethanol were plotted in Fig. 7(a), (b), and (d). It was found that the CR removal rate of TiOC decreased after the inclusion of ethanol. This clearly indicates that the photogenerated holes of TiOC were entirely consumed by ethanol for its self-degradation to generate CH3CHO and 2H+ ions. Overall, the photogenerated holes of TiOC played a vital role during the decomposition of CR. A similar kind of experimental observation was observed by Xinjian Xie et al. [54] during the decomposition of organic pollutant MB using CuWO4. They used various sacrificial reagents to suppress the effect of the quick recombination rate of charge carriers.
Furthermore, the stability of TiOC was studied towards CR with a continuous run of three cycles, each lasting 2 h, for a total of 6 h. It was found that the stability of TiOC (1 g/L) remained almost constant until the completion of the third run. After each cycle, the titanium oxycarbide powders were recovered via a centrifugation process and dried for 2 h in the oven. Then, the dried recovered samples were reused for the photodegradation of CR under light illumination, which could run for another 2 h. The absorbance values remained almost the same for all runs. The graph representing the reusability of TiOC is shown in Fig. 7(e).
Furthermore, a comparative analysis of the water pollutant removal activity of a few heteroanionic photocatalysts is shown in Table 3. It can be found from the table that the efficiency of the prepared titanium oxycarbide is far better than that of other reported different types of photocatalysts.
Photocatalytic H2O2 generation using titanium oxycarbide
Photocatalytic H2O2 generation rates on TiOC photocatalysts were measured every 30 min with/without the addition of the sacrificial reagent such as isopropyl alcohol (ISA) under visible light illumination. In order to make the calibration curves, the absorption curve was evaluated for different volumes of KMnO4 (0.2 to 2 ml) which was mixed with one ml of H2SO4 and 2 ml of H2O and held for 3 min. Then, a linear fitting plot was made based on the absorbance value observed at 523 nm, as shown in Fig. 8(a) and (b). Initially, the H2O2 evolution rate for TiOC was evaluated without adding ISA under visible light radiation. It produced a minimal amount of H2O2, such as 2 and 10 mmol, after 90 and 120 min of visible light irradiation, respectively, as shown in Fig. 8(c). This indicates that pure TiOC is minimally active under visible light illumination. This was reasonably anticipated because the photogenerated electrons in the CB of TiOC do not possess enough reduction potential to drive the proton reduction reaction and also faces a quick recombination rate of charge carriers. These results confirmed once again that TiOC was capable of showing better photocatalytic character only after 120 min of light exposure, which was also observed during the photodegradation of CR using different concentrations of the TiOC photocatalyst.

(a) UV-Vis absorption spectra of 40 mM KMnO4 in aqueous solution with different solution volumes and (b) its linear fitting; (c) and (d) comparison of photocatalytic H2O2 evolution rate of TiOC with/without the presence of ISA
Interestingly, the H2O2 evolution rate of TiOC was significantly improved after adding 2ml of ISA as a sacrificial agent. ISA-loaded TiOC produced a good amount of H2O2 of 7.5 mmol at 30 min of light irradiation. Upon the completion of 120 min, the H2O2 production rate increased to 21.5 mmol, which found to be double the amount of H2O2 evlolved during the reaction without ISA and is shown in Fig. 8(d). Therefore, the H2O2 evolution rate of TiOC can be increased further only after adding sacrificial reagents, which react irreversibly with the photogenerated holes to enhance electron/hole separation efficiency.
Possible mechanism of the photocatalytic CR degradation and H2O2 generation using TiOC
The photocatalytic behavior of any compound can be proposed based on its CB and VB edge arrangements. The CB and VB edge positions of TiOC can be determined using the Mulliken electronegativity method in order to present the photocatalytic mechanism that occurred during the degradation of CR and H2O2 generation. This method employs two equations to calculate the valence (EVB) and conduction band (ECB) edge positions of TiOC [6, 53].
where X (~5.37 eV) and Eg (~1.0 eV) is the electronegativity and band gap energy value of the TiOC; Ee is the energy of a free electron on a hydrogen scale (4.5 eV).
Based on this technique, the CB and VB edges of TiOC were determined to be 0.4 and 1.4 eV, respectively. The acceptable photocatalytic character exhibited by TiOC in the presence of the sacrificial reagents can be attributed to the following factors:
During the CR degradation process, the TiOC absorb light radiation larger/equal to its band gap energy and encourages the creation of the electron-hole charge carriers and migrates their electrons from the VB of TiOC to its CB, which acts as the trapping site of electrons. In general, the electrons in the conduction band generate superoxide radical (•O2−) by reacting with O2 molecules in the reaction chamber. Then these superoxide radicals (•O2−) react with H2O to create •OH. At the same time, photogenerated holes in the VB oxidize OH− and H2O to produce •OH radicals.
When the H2O2 is introduced as a sacrificial reagent in the reaction chamber, it helps to create more hydroxyl radicals either by interaction with light radiation and superoxide anion or by reacting the electrons from the CB of TiOC as shown in eqs. (13–15). Hydroxyl radical (•OH) and superoxide radical (•O2−) are the main species that degrade CR into CO2 and H2O under light irradiation. Moreover, this reaction reduces the recombination rate of the charge carriers by providing sufficient charge separation. This induces a strong internal electric field between the different anions in TiOC, and increased the photocatalytic CR degradation efficiency.
The chemical reactions involved in the degradation of CR using TiOC in the presence of H2O2 are as follows;
(or)
(or)
(or)
In addition, we have proposed a possible mechanism for the generation of H2O2 using TiOC, as shown in Fig. 9. When TiOC is exposed to visible light radiation, it generates electron-hole charge carriers. The electrons in the CB of TiOC react with molecular oxygen in the reaction chamber to create superoxide radicals. The holes at the VB of TiOC oxidize water molecules to produce hydroxyl radicals and protons. These hydroxyl radicals combine with each other to form H2O2. Then, the generated protons and superoxide radicals react with another electron to generate H2O2. However, due to the quick recombination of charge carriers in TiOC, it produces a minimal amount of H2O2 even after 120 min of light exposure. Therefore, ISA has been introduced into the reaction chamber to consume the excessive holes from the valence band to generate protons, greatly enhancing the rate of hydrogen peroxide production along with the conventional process. The reaction involved during the H2O2 evolution reaction using TiOC can be described as follows [62,63,64,65].

The possible mechanism of photocatalytic congo red degradation and H2O2 production from TiOC photocatalyst
When the ISA sacrificial reagent is introduced,
Therefore, supplying and separating sufficient electrons and holes in TiOC will automatically trigger redox reactions, leading to enhanced photocatalytic activity.
During the production of H2O2, some decomposition reactions of H2O2 occur simultaneously, which are unavoidable. This decomposition is caused by the cumulative electrons in TiOC, which decompose the generated H2O2 into H2O (water) and •OH (hydroxyl radicals). This process drastically reduces the H2O2 production efficiency of TiOC [66, 67]:
However, after the inclusion of the ISA sacrificial reagent, the decomposition rate of H2O2 may be reduced because the cumulative electrons involved in the decomposition of H2O2 may be utilized for the production reaction.
Conclusions
In summary, titanium oxycarbide photocatalysts were successfully prepared by supplying two calcination temperatures such as 1000°C and 1500°C, for 6 h. The XRD, SEM, TEM, and elemental mapping confirmed the purity of the TiOC. It was found that the synthesized TiOC photocatalysts possess maximum absorption edge at ~500 nm and exhibited a narrower band gap energy of 1.0 eV due to the formation of hybridized C-2p and O-2p orbitals at the valence band. During photocatalytic studies, TiOC initially demonstrated a moderate CR degradation efficiency of 54% using 1 g/L (TiOC) in 120 min and evolved a minimal amount of H2O2 after 90 min and 120 min of light exposure. The results of CR degradation and H2O2 generation without any sacrificial agent reveal that due to more positive CB position than the standard H+/H2O reduction potential and localization of hybridization of O 2p-orbitals and C 2p-orbitals lead to the reduced carrier mobility and increased quick recombination of photogenerated charge carriers. However, in the presence of sacrificial reagents such as H2O2 and ISA, the CR removal rate and H2O2 evolution rate of TiOC are significantly improved to 70% and 21.5 mmol in 120 min, respectively. This enhancement in the photocatalytic activity is due to the suppression of the recombination of photogenerated charge carriers. It provided sufficient time for the creation of a large number of hydroxyl radicals (•OH) and H+ ions after the addition of sacrificial reagents. In future work, novel titanium oxycarbide photocatalysts with good light absorption character and less recombination rate of the charge carriers by adding dopant/formation of heterostructures with other visible light semiconductors. To the best of our knowledge, this is the first investigation of the photocatalytic activity of TiOC through the degradation of congo red and H2O2 generation experiment under visible light radiation.
References
Tahir MB, Asiri AM, Nawaz T (2020) A perspective on the fabrication of heterogeneous photocatalysts for enhanced hydrogen production. Int J Hydrogen Energy 45(46):24544–24557. https://doi.org/10.1016/j.ijhydene.2020.06.301
Chen D et al (2020) Photocatalytic degradation of organic pollutants using TiO2-based photocatalysts: a review. J Clean Prod 268:121725. https://doi.org/10.1016/j.jclepro.2020.121725
Dhanasekaran A et al (2022) Computational fluid dynamics for protonic ceramic fuel cell stack modeling: a brief review. Energies (Basel) 16(1):208. https://doi.org/10.3390/en16010208
Omeiza LA et al (2023) Nanostructured electrocatalysts for advanced applications in fuel cells. Energies (Basel) 16(4):1876. https://doi.org/10.3390/en16041876
Subramanian Y et al (2021) Artificial intelligence technique based performance estimation of solid oxide fuel cells. Mater Today Proc. https://doi.org/10.1016/j.matpr.2021.06.412
Subramanian Y et al (2022) Efficient degradation of endocrine-disrupting compounds by heterostructured perovskite photocatalysts and its correlation with their ferroelectricity. New J Chem 46(24):11851–11861. https://doi.org/10.1039/D2NJ00785A
Hernández-Zamora M, Martínez-Jerónimo F, Cristiani-Urbina E, Cañizares-Villanueva RO (2016) Congo red dye affects survival and reproduction in the cladoceran Ceriodaphnia dubia. Effects Direct Diet Ex, Ecotoxicol 25(10):1832–1840. https://doi.org/10.1007/s10646-016-1731-x
Oladoye PO, Bamigboye MO, Ogunbiyi OD, Akano MT (2022) Toxicity and decontamination strategies of Congo red dye. Groundw Sustain Dev 19:100844. https://doi.org/10.1016/j.gsd.2022.100844
Chang A-L, Nguyen V-H, Lin K-YA, Hu C (2020) Selective synthesis of ZIFs from zinc and nickel nitrate solution for photocatalytic H2O2 production. Arab J Chem 13(11):8301–8308. https://doi.org/10.1016/j.arabjc.2020.04.027
Jiang Z, Zhang Y, Zhang L, Cheng B, Wang L (2022) Effect of calcination temperatures on photocatalytic H2O2-production activity of ZnO nanorods. Chi J Catal 43(2):226–233. https://doi.org/10.1016/S1872-2067(21)63832-9
Samanta C (2008) Direct synthesis of hydrogen peroxide from hydrogen and oxygen: an overview of recent developments in the process. Appl Catal A Gen 350(2):133–149. https://doi.org/10.1016/j.apcata.2008.07.043
Pi L et al (2020) Generation of H2O2 by on-site activation of molecular dioxygen for environmental remediation applications: a review. Chem Eng J 389:123420. https://doi.org/10.1016/j.cej.2019.123420
Kim H, Choi Y, Hu S, Choi W, Kim J-H (2018) Photocatalytic hydrogen peroxide production by anthraquinone-augmented polymeric carbon nitride. Appl Catal B 229:121–129. https://doi.org/10.1016/j.apcatb.2018.01.060
Gao G, Tian Y, Gong X, Pan Z, Yang K, Zong B (2020) Advances in the production technology of hydrogen peroxide, Chi J Catal 41(7):1039–1047. https://doi.org/10.1016/S1872-2067(20)63562-8
Byrne C, Subramanian G, Pillai SC (2018) Recent advances in photocatalysis for environmental applications. J Environ Chem Eng 6(3):3531–3555. https://doi.org/10.1016/j.jece.2017.07.080
Khan S et al (2021) Degradation of Congo red dye using ternary metal selenide-chitosan microspheres as robust and reusable catalysts. Environ Technol Innov 22:101402. https://doi.org/10.1016/j.eti.2021.101402
Nakata K, Fujishima A (2012) TiO2 photocatalysis: design and applications. J Photochem Photobiol C: Photochem Rev 13(3):169–189. https://doi.org/10.1016/j.jphotochemrev.2012.06.001
Thomas M, Naikoo GA, Sheikh MUD, Bano M, Khan F (2016) Effective photocatalytic degradation of Congo red dye using alginate/carboxymethyl cellulose/TiO2 nanocomposite hydrogel under direct sunlight irradiation. J Photochem Photobiol A Chem 327:33–43. https://doi.org/10.1016/j.jphotochem.2016.05.005
Subramanian Y et al (2023) Structural, photoabsorption and photocatalytic characteristics of BiFeO3-WO3 nanocomposites: an attempt to validate the experimental data through SVM-based artificial intelligence (AI). J Electron Mater 52(4):2421–2431. https://doi.org/10.1007/s11664-022-10188-7
Ljubas D, Smoljanić G, Juretić H (2015) Degradation of Methyl orange and Congo red dyes by using TiO2 nanoparticles activated by the solar and the solar-like radiation. J Environ Manage 161:83–91. https://doi.org/10.1016/j.jenvman.2015.06.042
Sambathkumar C, Manirathinam V, Manikandan A, Krishna Kumar M, Sudhahar S, Devendran P (2021) Solvothermal synthesis of Bi2S3 nanoparticles for active photocatalytic and energy storage device applications. J Mater Sci: Mater Elec 32(15):20827–20843. https://doi.org/10.1007/s10854-021-06596-w
Chaudhary YS et al (2012) Visible light-driven CO2 reduction by enzyme coupled CdS nanocrystals. Chem Commun 48(1):58–60. https://doi.org/10.1039/C1CC16107E
Reza MS, Ahmad NBH, Afroze S, Taweekun J, Sharifpur M, Azad AK Hydrogen production from water splitting through photocatalytic activity of carbon-based materials. Chem Eng Technol. https://doi.org/10.1002/ceat.202100513
Dong H et al (2015) An overview on limitations of TiO2-based particles for photocatalytic degradation of organic pollutants and the corresponding countermeasures. Water Res 79:128–146. https://doi.org/10.1016/j.watres.2015.04.038
Xu X, Liu G, Azad AK (2015) Visible light photocatalysis by in situ growth of plasmonic Ag nanoparticles upon AgTaO3. Int J Hydrogen Energy 40(9):3672–3678. https://doi.org/10.1016/j.ijhydene.2015.01.046
Xu X, Azad AK, Irvine JTS (2013) Photocatalytic H2 generation from spinels ZnFe2O4, ZnFeGaO4 and ZnGa2O4. Catal Today 199(1):22–26. https://doi.org/10.1016/j.cattod.2012.03.013
Chatterjee K, Skrabalak SE (2021) Durable metal heteroanionic photocatalysts. ACS Appl Mater Interfaces 13(31):36670–36678. https://doi.org/10.1021/acsami.1c09774
Yaghoubi-berijani M, Bahramian B (2020) Synthesis, and new design into enhanced photocatalytic activity of porphyrin immobilization on the surface of bismuth oxyhalides modified with polyaniline. J Inorg Organomet Polym Mater 30(11):4637–4654. https://doi.org/10.1007/s10904-020-01652-0
Moriya Y, Takata T, Domen K (2013) Recent progress in the development of (oxy)nitride photocatalysts for water splitting under visible-light irradiation, Coord Chem Rev 257(13–14):1957–1969. https://doi.org/10.1016/j.ccr.2013.01.021
Maeda K, Takata T, Domen K (2011) (Oxy)nitrides and oxysulfides as visible-light-driven photocatalysts for overall water splitting. 487–529. https://doi.org/10.1007/978-0-85729-638-2_14
Takata T, Pan C, Domen K (2015) Recent progress in oxynitride photocatalysts for visible-light-driven water splitting. Sci Technol Adv Mater 16(3):033506. https://doi.org/10.1088/1468-6996/16/3/033506
Wang Q et al (2019) Oxysulfide photocatalyst for visible-light-driven overall water splitting. Nat Mater 18(8):827–832. https://doi.org/10.1038/s41563-019-0399-z
Subramanian Y, Dhanasekaran A, Omeiza LA, Somalu MR, Azad AK (2023) A review on heteroanionic-based materials for photocatalysis applications. Catalysts 13(1):173. https://doi.org/10.3390/catal13010173
Huang K, Li Y, Xing Y (2013) Carbothermal synthesis of titanium oxycarbide as electrocatalyst support with high oxygen evolution reaction activity. J Mater Res 28(3):454–460. https://doi.org/10.1557/jmr.2012.353
Lopes D et al (2020) Design of multifunctional titania-based photocatalysts by controlled redox reactions. Materials 13(3):758. https://doi.org/10.3390/ma13030758
Cuan J et al (2019) Multiple anionic transition-metal oxycarbide for better lithium storage and facilitated multielectron reactions. ACS Nano 13(10):11665–11675. https://doi.org/10.1021/acsnano.9b05580
Huang K, Li Y, Xing Y (2013) Carbothermal synthesis of titanium oxycarbide as electrocatalyst support with high oxygen evolution reaction activity. J Mater Res 28(3):454–460. https://doi.org/10.1557/jmr.2012.353
Guan S, Hao L, Yoshida H, Pan F, Asanuma H, Lu Y (2016) Enhanced photocatalytic activity of photocatalyst coatings by heat treatment in carbon atmosphere. Mater Lett 167:43–46. https://doi.org/10.1016/j.matlet.2015.12.074
Qin Y et al (2021) Au nanorods decorated TiO2 nanobelts with enhanced full solar spectrum photocatalytic antibacterial activity and the sterilization file cabinet application. Chi Chem ers 32(4):1523–1526. https://doi.org/10.1016/j.cclet.2020.10.020
Qian X et al (2022) Heterostructuring 2D TiO2 nanosheets in situ grown on Ti3C2T MXene to improve the electrocatalytic nitrogen reduction. Chin J Catal 43(7):1937–1944. https://doi.org/10.1016/S1872-2067(21)64020-2
Fang B, Xing Z, Sun D, Li Z, Zhou W (2022) Hollow semiconductor photocatalysts for solar energy conversion. Adv Powder Mat 1(2):100021. https://doi.org/10.1016/j.apmate.2021.11.008
Chong B, Li H, Xu B, Yang G (2022) Hollow double-shell stacked CdS@ZnIn2S4 photocatalyst incorporating spatially separated dual cocatalysts for the enhanced photocatalytic hydrogen evolution and hydrogen peroxide production. Catal Today 405–406:227–234. https://doi.org/10.1016/j.cattod.2022.05.020
Wei Y, Zhang J, Zheng Q, Miao J, Alvarez PJ, Long M (2021) Chemosphere 279:130556. https://doi.org/10.1016/j.chemosphere.2021.130556
Zhang E et al (2021) Visually resolving the direct Z-scheme heterojunction in CdS@ZnIn2S4 hollow cubes for photocatalytic evolution of H2 and H2O2 from pure water. Appl Catal B 293:120213. https://doi.org/10.1016/j.apcatb.2021.120213
Zhang B, Xiao J, Jiao S, Zhu H (2021) A novel titanium oxycarbide phase with metal-vacancy (Ti1-C O1-): structural and thermodynamic basis. Ceram Int 47(11):16324–16332. https://doi.org/10.1016/j.ceramint.2021.02.212
Jiang B et al (2013) Structural studies of TiC1−xOx solid solution by Rietveld refinement and first-principles calculations. J Solid State Chem 204:1–8. https://doi.org/10.1016/j.jssc.2013.05.009
Miller DN et al (2016) Studies on the crystal structure, magnetic and conductivity properties of titanium oxycarbide solid solution (TiO 1−x C x ). J Mater Chem A Mater 4(15):5730–5736. https://doi.org/10.1039/C6TA00042H
Subramanian Y et al (2019) Investigations on the enhanced dye degradation activity of heterogeneous BiFeO3–GdFeO3 nanocomposite photocatalyst. Heliyon 5(6):e01831. https://doi.org/10.1016/j.heliyon.2019.e01831
Negi C et al (2021) Carbon-doped titanium dioxide nanoparticles for visible light driven photocatalytic activity. Appl Surf Sci 554:149553. https://doi.org/10.1016/j.apsusc.2021.149553
Kumar P et al (2022) Photocatalytic activity of a hydrothermally synthesized γ-Fe2O3@Au/MoS2 heterostructure for organic dye degradation under green light. J Photochem Photobiol A Chem 433:114186. https://doi.org/10.1016/j.jphotochem.2022.114186
Apopei P, Catrinescu C, Teodosiu C, Royer S (2014) Mixed-phase TiO2 photocatalysts: crystalline phase isolation and reconstruction, characterization and photocatalytic activity in the oxidation of 4-chlorophenol from aqueous effluents. Appl Catal B 160–161:374–382. https://doi.org/10.1016/j.apcatb.2014.05.030
Subramanian Y, Mishra B, Mandal S, Gubendiran R, Chaudhary YS (2021) Design of heterostructured perovskites for enhanced photocatalytic activity: insight into their charge carrier dynamics. Mater Today Proc 35:179–185. https://doi.org/10.1016/j.matpr.2020.04.215
Subramanian Y, Ramasamy V, Gubendiran RK, Srinivasan GR, Arulmozhi D (2018) Structural, optical, thermal and photocatalytic dye degradation properties of BiFeO3–WO3 nanocomposites. J Electron Mater 47(12):7212–7223. https://doi.org/10.1007/s11664-018-6654-2
Xie X et al (2016) Efficient photo-degradation of dyes using CuWO4 nanoparticles with electron sacrificial agents: a combination of experimental and theoretical exploration. RSC Adv 6(2):953–959. https://doi.org/10.1039/C5RA18788E
Fatima S, Ali SI, Iqbal MZ, Rizwan S (2020) Congo red dye degradation by graphene nanoplatelets/doped bismuth ferrite nanoparticle hybrid catalysts under dark and light conditions. Catalysts 10(4):367. https://doi.org/10.3390/catal10040367
Karuppasamy K et al (2021) Ternary Zn1-xNixSe nanostructures as efficient photocatalysts for detoxification of hazardous Congo red, methyl orange, and chrome yellow dyes in wastewater sources. Environ Res 201:111587. https://doi.org/10.1016/j.envres.2021.111587
Sadek Kadari A et al (2022) Growth, properties and photocatalytic degradation of congo red using Gd:ZnO thin films under visible light. Inorg Chem Commun 142:109626. https://doi.org/10.1016/j.inoche.2022.109626
Akika FZ et al (2018) Structural and optical properties of Cu-substitution of NiAl2O4 and their photocatalytic activity towards Congo red under solar light irradiation. J Photochem Photobiol A Chem 364:542–550. https://doi.org/10.1016/j.jphotochem.2018.06.049
García CR, Oliva J, Arroyo A, Garcia-Lobato MA, Gomez-Solis C, Torres LAD (2018) Photocatalytic activity of bismuth doped SrAl2O4 ceramic powders. J Photochem Photobiol A Chem 351:245–252. https://doi.org/10.1016/j.jphotochem.2017.10.039
Nawaz A, Rani A, Zarrin H, Saravanan P (2022) Construction of highly efficient separable p-n junction based light driven composite (NiFe2O4/MnWO4) for improved solar light utilisation. Colloids Surf A Physicochem Eng Asp 642:128716. https://doi.org/10.1016/j.colsurfa.2022.128716
Jiang R et al (2016) Fabrication of novel magnetically separable BiOBr/CoFe2O4 microspheres and its application in the efficient removal of dye from aqueous phase by an environment-friendly and economical approach. Appl Surf Sci 364:604–612. https://doi.org/10.1016/j.apsusc.2015.12.200
Chen Y, Gu W, Tan L, Ao Z, An T, Wang S (2021) Photocatalytic H2O2 production using Ti3C2 MXene as a non-noble metal cocatalyst. Appl Catal A Gen 618:118127. https://doi.org/10.1016/j.apcata.2021.118127
Meng X et al (2020) Au-nanoparticle-supported ZnO as highly efficient photocatalyst for H2O2 production. Catal Commun 134:105860. https://doi.org/10.1016/j.catcom.2019.105860
Liu Y et al (2021) Template-free preparation of non-metal (B, P, S) doped g-C3N4 tubes with enhanced photocatalytic H2O2 generation. J Mater Sci Technol 95:127–135. https://doi.org/10.1016/j.jmst.2021.03.025
Wang J, Yang L, Zhang L (2021) Constructed 3D hierarchical micro-flowers CoWO4@Bi2WO6 Z-scheme heterojunction catalyzer: two-channel photocatalytic H2O2 production and antibiotics degradation. Chem Eng J 420:127639. https://doi.org/10.1016/j.cej.2020.127639
Zhou L et al (2020) Ultrathin g-C3N4 nanosheet with hierarchical pores and desirable energy band for highly efficient H2O2 production. Appl Catal B 267:118396. https://doi.org/10.1016/j.apcatb.2019.118396
Tsukamoto D et al (2012) Photocatalytic H2O2 production from ethanol/O2 system using TiO2 loaded with Au–Ag bimetallic alloy nanoparticles. ACS Catal 2(4):599–603. https://doi.org/10.1021/cs2006873
Acknowledgments
The authors, Yathavan Subramanian, Anitha Dhanasekaran, and Lukman Ahmed Omeiza would like to thank the Universiti of Brunei Darussalam for the UGS scholarship for their PhD research.
CRediT authorship contribution statement
Yathavan Subramanian: Investigation, methodology, performed experiments, writing—original draft; Anitha Dhanasekaran: investigation, writing—review and editing; Lukman Ahmed Omeiza: contributed reagents, materials, analysis tools or data; Juliana Haji Zaini: visualization, writing—review and editing, conceptualization, resources, supervision; .John T.S. Irvine: conceptualization, resources, supervision, visualization, review and editing: Abul Kalam Azad: visualization, writing—review and editing, conceptualization, resources, supervision.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Subramanian, Y., Dhanasekaran, A., Omeiza, L.A. et al. Influence of various sacrificial reagents on congo red degradation and H2O2 production based on heteroanionic titanium oxycarbide photocatalyst and its mechanism. Ionics 29, 2435–2447 (2023). https://doi.org/10.1007/s11581-023-05006-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-023-05006-x