
Abstract
This review examines the effects of cannabinoids on immune function, with a focus on effects on T-cells, as well as on resistance to infection. The paper considers the immune modulating capacity of marijuana, of ∆9-THC extracted from the marijuana plant, and synthetic cannabinoids. Of particular interest are synthetic compounds that are CB2 receptor (CB2R) selective agonists. As the CB2R is principally expressed on cells of the immune system, agonists that target this receptor, and not CB1 (which is mainly expressed on neurons), have the possibility of altering immune function without psychoactive effects. The overall conclusion of the studies discussed in this review is that cannabinoids that bind to the CB2 receptor, including ∆9-THC and CB2 selective agonists are immunosuppressive. The studies provide objective evidence for potentially beneficial effects of marijuana and ∆9-THC on the immune system in conditions where it is desirable to dampen immune responses. Evidence is also reviewed supporting the conclusion that these same compounds can sensitize to some infections through their immunosuppressive activities, but not to others. An emerging area of investigation that is reviewed is evidence to support the conclusion that CB2 selective agonists are a new class of immunosuppressive and anti-inflammatory compounds that may have exceptional beneficial effects in a variety of conditions, such as autoimmune diseases and graft rejection, where it is desirable to dampen the immune response without psychoactive effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
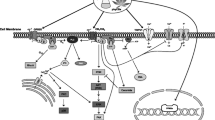
Introduction: Cannabinoids, Cannabinoid Receptors, and the Immune System
Shortly after cloning of the CB1 receptor (CB1R) from rat brain in 1990 (Matsuda et al. 1990), a second cannabinoid receptor (CB2R) was discovered in 1993 (Munro et al. 1993) using a human premonocytic cell line (HL60). The two receptors demonstrated only 44 % homology and showed anatomical separation, with the CB1 receptor primarily expressed on neurons in the brain, and the CB2 receptor primarily expressed on cells of the immune system (Galiegue et al. 1995; Munro et al. 1993). This observation was novel in terms of neuroimmune interactions in regard to drugs of abuse, as the opioid receptors (mu, kappa and delta) were shown to be expressed on neurons and also on cells of the immune system, but in the case of the opioid receptors, it was the same receptors in the diverse anatomical locations. From an evolutionary point of view, it is intriguing to ask why a divergence in cannabinoid receptors in the neural and immune systems would be beneficial. Of interest is the observation that the phytocannabinoid ligand, ∆9-tetrahydrocannbinol (∆9-THC), as well as the currently known endogenous cannabinoid agonists, anandamide (arachidonylethanolamide) and 2-arachidonyl glycerol, bind to both CB1R and CB2R (Devane et al. 1992; Felder et al. 1995; Mechoulam et al. 1995), in spite of the structural divergence of the receptors. However, synthetic CB2R selective agonists have been generated, although to date there is no CB1R selective agonist. Interestingly, CB2R was found to be most abundant, based on mRNA expression, in B-cells>natural killer cells> monocytes>neutrophils>T-cells in rat, mouse and human tissues and cells. (Bouaboula et al. 1993; Galiegue et al. 1995). In spite of T-cells being lowest on this list, cannabinoids have been shown to have powerful effects on their function. This seeming anomaly can be explained by the fact that both CB1 and CB2 receptor expression is modulated by immune cell activation (Börner et al. 2007; Daaka et al. 1996; Lee et al. 2001; Munro et al. 1993). It is also important to remember that CB1R is also expressed in immune cells although at a considerably lower level than CB2R (Bouaboula et al. 1993; Kaminski et al. 1992). Thus, it is necessary to determine which receptor is mediating functional effects of ligands that activate both CB1R and CB2R. This review will examine the effect of both ∆9-THC and CB2R selective agonists on T-cell function and resistance to infection. There are several published reviews on effects of cannabinoids on the immune system, but they were broader in scope (Croxford and Yamamura 2005; Friedman et al. 2003; Klein et al. 1998; Klein 2005). This review will concentrate on experimental paradigms where variables can be closely controlled, as opposed to studies in marijuana smokers. Constituents of Cannabis sativia, other than ∆9-THC, including cannabidiol, have also been reported to have immunomodulatory activity, but it has not been firmly established whether these effects are through CB1, CB2 or other receptors (Pertwee 2008). Further, most of the reported effects of cannabidiol on the immune system are on cell migration by microglia, macrophages, and neutrophils, not T-cells. Therefore, cannabidiol and related compounds will not be considered in this review.
Effects of ∆9-THC on T-cells
Studies begun in the 1990s began to catalog the effects of cannabinoids on immune function. Many of these studies used the major active ingredient in the marijuana plant, ∆9-THC, and tested its effect in vitro on a variety of responses of human peripheral blood T-cells and murine splenic T- cells. Most of the results found that ∆9-THC was immunosuppressive. Ability to respond to T-cell mitogens (PHA or Con A) was a test used by several laboratories, all of whom found that the cannabinoid suppressed mitogen-stimulated replication (Börner et al. 2009; Klein et al. 1985; Luo et al. 1992; Nakano et al. 1992). ∆9-THC given to rats in vivo suppressed splenic T-cell responses to Con A when cells were harvested 1 week after treatment, but not after 1 h (Massi et al. 1998). Other investigators found that ∆9-THC inhibited the proliferative response to anti-CD3 treatment when added in vitro to mouse spleen cells (Schatz et al. 1993) or to peripheral blood human T-cells (Yuan et al. 2002). A singular finding was the observation that low doses of ∆9-THC stimulated spleen cells of adult mice treated with ant-CD3 antibody (Nakano et al. 1992). ∆9-THC was also shown to depress proliferative responses induced by the Mixed Lymphocyte Reaction (MLR) of human peripheral blood T-cells exposed to allogeneic antigen presenting cells (Yuan et al. 2002) and murine spleen cells (Robinson et al. 2013). A considerable number of studies examined which T-cell subsets were affected by the cannabinoid, and the mechanisms by which T-cell function was altered. ∆9-THC was found to differentially suppress the number of CD8 T-cells and reduce their cytolytic activity (Klein et al. 1991; Pross et al. 1990). A major cytokine involved in T-helper 1 (Th1) cell activity is IL-2. IL-2 is an autocrine growth factor for Th1 cells, governing their expansion in response to stimulation. Treatment of mice with ∆9-THC resulted in persistent inhibition of IL-2 production even 7 days post cannabinoid treatment (Massi et al. 1998). In vitro treatment of murine spleen cells with ∆9-THC and exposure to T-cell mitogens also depressed IL-2 (Nakano et al. 1992). Further, it was observed that IL-2 secretion and IL-2 gene expression that were stimulated by PMA/Io were markedly inhibited by ∆9-THC, in an IL-2 secreting murine thymoma cell line (Condie et al. 1996). These effects were linked to the capacity of ∆9-THC to down-regulate the forskolin-stimulated increase in cAMP in these cells. Both CB1R and CB2R are Gi/o coupled receptors. A consequence of cAMP elevation in cells is release of PKA catalytic subunits, which have been shown to activate transcriptional regulators, including the CREB/ATF family. ∆9-THC was found to inhibit PKA in a dose dependent manner in the murine thymoma cells (Condie et al. 1996). CREB can dimerize with Fos and Jun, which then binds to AP-1 sites in the IL-2 promoter. A reduction in CREB by ∆9-THC would inhibit IL-2 production (Kaminski 1996). More recent studies have further elucidated the mechanisms by which ∆9-THC inhibits IL-2 production by T-cells. Börner (Börner et al. 2009) used primary peripheral blood human T-cells and the human Jurkat T-cell line to show that both CB1 and CB2 receptors mediate depression of IL-2 synthesis. Further, Jurkat cells transfected with reporter CAT constructs with binding sites for AP-1, NF-κB and NFAT were shown to have depressed responses in expression of these transcription factors when stimulated with anti-CD3/anti-CD28 antibodies in the presence of ∆9-THC. Additional experiments showed that in Jurkat T-cells, both CB1R and CB2R inhibited the anti-CD3/anti-CD28 induced dephosphorylation of LcK, a major transducer of T-cell activation upon antigen engagement of the T-cell receptor, which leads to IL-2 production. These investigators further probed the role of cAMP in regulation of these signal transduction cascades and found that ∆9-THC initially down-regulated cAMP for an hour, but subsequently resulted in its sustained elevation for 48 h (Börner et al. 2009). The cAMP activated PKA and PKC, and phosphorylated LcK, blocking T-cell receptor signaling. The discrepancy in the findings between Condie et al. (Condie et al. 1996) and Börner et al. (Börner et al. 2009) is not resolved.
A number of other assays have been used to test the functional effect of ∆9-THC on immune responses. Some of the models require more than one immune cell type to cooperate to produce the functional end-point read-out. One of these assays measures the capacity of mouse spleen cells to mount an antibody response to sheep red blood cells (SRBCs) used as antigen. Cooperation between antigen-presenting cells, T-cells and B-cell is required. A number of laboratories had shown that ∆9-THC inhibited this antibody forming cell (AFC) assay. (Baczynsky and Zimmerman 1983; Eisenstein et al. 2007; Lefkowitz and Klager 1978; Smith et al. 1978; Watson et al. 1983). An unanswered question was whether ∆9-THC directly inhibited capacity of the B-cells to synthesize and excrete antibody, or whether antigen-presenting cells or T-helper cell function were being depressed by the cannabinoid, resulting in a deficit of needed signals to activate the B-cells. Kaminski’s laboratory showed that ∆9-THC given per os for 7 days differentially suppressed antibody responses to a T-dependent antigen, SRBCs, but not to a T-independent antigen, DNP-Ficoll, supporting the conclusion that the effect is on T-cells or on antigen-presenting cells that are needed for T-dependent responses. In these preceding experiments the antigens were administered in vivo on the 8th day after the start of ∆9-THC administration, and spleen cell antibody responses were tested ex vivo (Schatz et al. 1993). The demonstration that in vitro addition of ∆9-THC to normal, untreated spleen cell cultures blocked the response to anti-CD3 treatment, showed that ∆9-THC could have a direct effect on T-cells (Schatz et al. 1993). It should be noted that conclusions from studies using in vivo administered drugs need to be interpreted cautiously, as there is always the possibility that the immunosuppression is indirect, through activation of the HPA axis or the sympathetic nervous system. In this case, the supporting evidence that similar suppression could be obtained by treating drug naïve spleen cells in vitro, abrogated that caveat. In further in vitro studies by this group, it was shown that ∆9-THC blocked induction of ICOS (Inducible T-cell Costimulator), a molecule expressed on T-cells activated by anti-CD3/CD28 or by PMA/Io, in mouse spleen cells and in a thymoma T-cell line (Lu et al. 2009), adding additional supporting evidence for a direct effect of the cannabinoid on T-cells.
Several laboratories have examined the effect of ∆9-THC on cytokine profiles of immune cells and found that ∆9-THC polarizes the immune response towards a Th2 phenotype. Friedman’s laboratory first showed, in the context of the effect of ∆9-THC on murine infection with Legionella pneumophila, that cannabinoid treatment in vivo down-regulated IL-2, IL-12, and IFN-γ, which are Th1 cytokines, as well as the IL-12 receptor (Klein et al. 2000; Newton et al. 1994). In a model using lung tumor bearing mice, it was found by the Roth laboratory that ∆9-THC treatment decreased IFN-γ production and increased IL-10 and TGF-γ levels, both anti-inflammatory and immunosuppressive cytokines. Administration of anti-IL-10 or anti-TGF-γ antibodies abrogated the tumor promoting effect of ∆9-THC. Further, T-cells from ∆9-THC-treated mice enhanced tumor growth in untreated mice, showing that immunosuppression was an active, not a passive process (Zhu et al. 2000). Cytokine polarization has also been observed using human peripheral blood T-cells. In the MLR assay, IFN-γ was depressed in culture supernatants and there was a trend towards elevation of IL-4 levels, thus decreasing the Th1/Th2 balance (Yuan et al. 2002). These results were further supported by intracellular staining for cytokine levels in human T-cells stimulated with anti-CD3/anti-CD28. ∆9-THC reduced the percentage of T-cells expressing IFN-γ and the average level of intracellular IFN-γ detected per cell in both CD4 and CD8 T-cell subsets (Yuan et al. 2002). ∆9-THC has also been shown to alter histone methylation and acetylation patterns in lymph node cells of mice stimulated with Staphylococcal enterotoxin B. ∆9-THC treatment resulted in histone modifications associated with up-regulation of Th2 cytokine genes and down-regulation of Th1 cytokine genes (Yang et al. 2014). Another important observation is the report that ∆9-THC can induce TGF-γ production in normal human peripheral blood lymphocytes stimulated with anti-CD3 (Gardner et al. 2002). As TGF-γ inhibits a spectrum of immune responses, confirmation of its up-regulation by ∆9-THC provides another mechanism by which the cannabinoid exerts suppressive effects. This group also showed that ∆9-THC modulated TGF-γ levels via the CB2 receptor, and that TGF-γ could down-regulate CB2 receptor mRNA, revealing a negative autocrine loop (Gardner et al. 2002).
Which Receptor Modulates Immune Effects of ∆9-THC?
An important question is which receptor(s) modulate effects of ∆9-THC on T-cells, CB1R or CB2R? In standard pharmacological assays, ∆9-THC has been shown to be a partial agonist at both receptors (reviewed in Pertwee 2008). Several approaches have been used to provide definitive answers: use of pharmacologically selective agonists and antagonists, and use of CB1R or CB2R knock-out (k/o) mice. In the paper describing the development of the CB2 k/o mouse, the Zimmer laboratory tested the effect on one parameter of immune function, the effect of ∆9-THC on the capacity of macrophages to present antigen to T-cells in vitro. When macrophages from wild-type (WT) mice were used, ∆9-THC inhibited the reaction, whereas macrophages from CB2 k/o mice were not inhibited, showing that ∆9-THC was acting through the CB2 receptor (Buckley et al. 2000). Ziring et al. carried out a study of the effect of genetic CB2 ablation on immune cell subsets and found that CB2 k/o mice had decreased numbers of splenic and peritoneal B-cell subsets, decreased numbers of CD4 and CD8 memory cells in the spleen, but increased numbers of intraepithelial CD4 T-cells in small intestine (Ziring et al. 2006). In many of the experimental paradigms, as will be detailed below, CB2 seems to predominate in modulating T-cell function. Gardner (Gardner et al. 2002) found that stimulation of human peripheral blood T-cells with anti-CD3 up-regulated CB2 receptors, and ∆9-THC induced the cytokine, TGF-β, in a CB2 dependent manner. Börner reported that primary human T-cells and the Jurkat T-cell line constitutively expressed CB2 (Börner et al. 2009). In addition, they showed that treatment with IL-4 induced the cells to express CB1. In the absence of induction, ∆9-THC was active through CB2R, and CB2 selective agonists, but not CB1 selective agonists, modulated immune function. As cited above, Zhu found that ∆9-THC exacerbated tumor growth in mice, and this effect was mediated by CB2 (Zhu et al. 2000). The Eisenstein group showed that in vitro inhibition of antibody formation by anandamide and ∆9-THC by mouse spleen cells was CB2 dependent, as the CB2 selective antagonist, but not the CB1 selective antagonist, abrogated the immunosuppressive effect (Eisenstein et al. 2007). Using a similar assay Kaminski’s laboratory found no diminution in the ex vivo response of murine spleen cells taken from WT or CB1−/CB2− k/o mice when ∆9-THC was administered in vivo which leads to the conclusion that the cannabinoid is acting via a mechanism independent of either cannabinoid receptor (Springs et al. 2008). In this type of assay, T-cells participate in helping the B-cells to make antibody, but neither study described above examined whether the effect of the cannabinoid was on T-cells, macrophages, or B-cells. For in vivo administration of ∆9-THC, it is also possible that the immunosuppression is not due to a direct effect of the drug on cells of the immune system, but is indirect via other suppressive mechanisms such as release of cortisone. Further exploration of the role of ∆9-THC and the cannabinoid receptors in modulating T-cell CD40L expression was carried out by the Kaminski laboratory (Ngaotepprutaram et al. 2012). CD40L is expressed by T-cells and binds to its cognate molecule, CD40, on antigen presenting cells and B-cells, resulting in T-cell activation. CD40L expression can be induced in vitro by treating T-cells with anti-CD3/CD28 or with PMA/Io. In this study, ∆9-THC was shown to inhibit CD40L expression by mouse splenic T-cells using anti-CD3/CD28, but not PMA/Io. The inhibition was still evident using spleen cells from CB1−/CB2− mice, again pointing to the conclusion that the immunosuppression by the cannabinoid was independent of these receptors. Additional evidence for action of ∆9-THC by mechanisms outside of the classical CB1 and CB2 receptors was presented in studies using ∆9-THC to inhibit cytotoxic T-lymphocyte function (CD8 cells). Spleen cells from wild-type or CB1−/CB2− mice were mixed in vitro with an MHC Class I incompatible cell line, P815 mastocytoma cells, which activates T-cells in the spleen to kill the P815 cells. ∆9-THC decreased the cytotoxicity of the spleen cells, which is attributed to cytotoxic CD8 cells (Karmaus et al. 2012). However, similar inhibition of T-cell function was observed in ∆9-THC spleen cells taken from the CB1−/CB2− mice, leading to the conclusion that the cannabinoid receptors were not involved. Evidence was presented implicating Ca++ activation in the ∆9-THC mediated suppression (Karmaus et al. 2012). In work from the Eisenstein laboratory, ∆9-THC was shown to inhibit the Mixed Lymphocyte Reaction (MLR), in which spleen cells from two MHC class I mis-matched strains are placed in culture. Cells of one strain are inhibited by treatment with mitomycin C, leaving the cells of the other strain to divide and take up 3H-thymidine in response to the foreign cells. Using this system, these investigators showed that ∆9-THC inhibited the MLR reaction, blocking responder cell division, and that the effect was mediated by the CB2 receptor, as a CB2 selective antagonist, but not a CB1 selective antagonist, blocked the cannabinoid induced immunosuppression (Robinson et al. 2013). No suppression was observed in spleen cells taken from CB2R k/o mice (Robinson et al. 2013). These data lead to the conclusion that the CB2 receptor is mediating these immune responses to ∆9-THC.
The CB2 Receptor and Immune Responses
As described above, in the case of ∆9-THC, which is a partial agonist at both CB1 and CB2 receptors, in many cases, although not all, its immunomodulatory activity was shown to be through the CB2 receptor, using receptor selective agonists and antagonists and CB2 receptor k/o mice. With the synthesis of CB2R selective agonists (Hanuš et al. 1999; Huffman et al. 1996; Huffman et al. 1999), the effect of cannabinoids on immune responses could be more precisely investigated. As might be predicted, these molecules, which have significantly higher affinity for CB2 receptors as compared to CB1 receptors, (sometimes orders of magnitude higher) would have robust effects on the immune system, since this system expresses most of the CB2 receptors in the body. Several reviews have examined the effect of CB2 on immune responses (Basu and Dittel 2011; Rieder et al. 2010). This review will concentrate on effects on T-cells, but it should be appreciated that there are also significant effects on B-cells, macrophages and dendritic cells. in vitro studies have found that the CB2 selective agonists, JWH-015 and JWH-133, inhibited chemotaxis of primary human peripheral blood T-cells (Coopman et al. 2007; Ghosh et al. 2006) and the human Jurkat T-cell line (Ghosh et al. 2006), to the chemokine CXCL12 (SDF-1). As noted in the preceding sections on effects of ∆9-THC, T-cell proliferation and IL-2 secretion are consequences of T-cell activation. The CB2 selective agonist, JWH-133, has been shown to inhibit antigen-specific mouse T-cell proliferation and mRNA for IL-2 (Maresz et al. 2007). JWH-015 has been shown to inhibit T-cell proliferation and IL-2 secretion in the Mixed Lymphocyte Reaction, described above, in a dose dependent manner (Robinson et al. 2013). CB2 selective agonists have also been shown to inhibit IL-10 production in the MLR (Robinson et al. 2013). The biological endpoints were shown to be dependent on CB2 engagement, as the CB2 selective antagonist, but not the CB1 selective antagonist, blocked the suppression (Robinson et al. 2013), and cells taken from CB2R k/o mice were not suppressed (Maresz et al. 2007; Robinson et al. 2013). In studies using primary human peripheral blood T-cells, anandamide was shown to suppress proliferation and release of IL-2, INF-γ and TNF-α from stimulated cells. This occurred via the CB2 receptor as shown using receptor selective antagonists. In the same study, JWH-015 was also shown to inhibit these immune end-points (Cencioni et al. 2010). Another aspect of effects of cannabinoids on cells of the immune system relates to expression of the CB2 receptor. Castaneda (Castaneda et al. 2013) reported, using human peripheral blood leukocytes and flow cytometric analysis with tagged antibodies, that only B-cells express CB2 on their surface (and also intracellularly). In T-cells and monocytes, CB2 is intracellular in resting cells (Castaneda et al. 2013). Further, activation of immune cells in the MLR increased mRNA for CB1 in human T-cells (Börner et al. 2007; Daaka et al. 1996) and CB2 in both murine T-cells and macrophages (Robinson et al. 2013). TGF-β, the immunosuppressive cytokine induced by ∆9-THC, also down-regulated receptor expression, providing a feed-back loop (Gardner et al. 2002).
The CB2 Receptor and Autoimmune Diseases
CB2 selective agonists and CB2 k/o mice have been used in several mouse models of inflammatory and autoimmune conditions where T-cells are known to play an important role. In many of the studies, effects on T-cells in vivo could be demonstrated, but whether the effects of endogenous or exogenous CB2 ligands are due to direct effects on the T-cells was not ascertained. In other studies, these models have provided systems that are amenable to in depth examination of the mechanisms by which CB2 agonists alter T-cell function. Experimental autoimmune encephalitis (EAE) is an accepted mouse model for multiple sclerosis, induced by injection of myelin basic proteins leading to a demyelinization mediated by T-cells recognizing myelin basic proteins. Maresz, in an elegant paper, showed that the disease was significantly more severe in CB2 k/o mice compared to WT mice. Using conditional CB1 k/o mice with either a deficiency of this receptor in neurons or T-cells, they found that CB1 on T-cells did not play a role in disease development (Maresz et al. 2007). In further experiments, these investigators definitely established that the CB2 receptor modulates the pathological potential of the T-cells. They first transferred T-cells from WT or CB2 k/o mice induced to have EAE and compared their encephalitic potential. Cells taken from the CB2 k/o mice resulted in much more severe disease in naïve recipients. Second, when cells from WT EAE mice were compared with similar cells from CB2 k/o mice by placing them in culture and re-stimulating them with antigen, the CB2 agonist, JWH-133, inhibited cell proliferation and elaboration of the Th1 cytokines, IFN-γ and IL-2 from WT but not CB2 k/o cells. Mice deficient in CB2R had higher numbers of encephalitogenic T-cells in the CNS, and the cells had greater rates of proliferation and mRNA levels for IFN-γ and IL-2 cytokine production in the spinal cord, but not the spleen, than the WT controls. Encephalitogenic T-cells purified from EAE animals showed higher levels of mRNA for these cytokines if harvested from the CB2 k/o mice, showing that the differences in cytokine mRNA were not due to just infiltration by more cells, but an actual increase in these cytokines on a per cell basis. These results led to the conclusion that the CB2 receptor on T-cells homeostatically inhibits aggressive T-cell responses via endogenous cannabinoid ligands (Maresz et al. 2007). This work is supported by another study that showed that therapeutic administration of the CB2 agonist, O-1966, resolved the primary exacerbation in EAE and dampened relapses (Zhang et al. 2009b). In studies in two different mouse models of systemic sclerosis, a connective tissue disorder characterized by collagen deposition and fibrosis of skin and organs, the CB2 agonist, JWH-133, ameliorated disease end-points including reduction in infiltrating lymphocytes. An interesting ancillary observation by two other groups using the systemic sclerosis paradigms is that CB2 k/o mice have heightened immune responses, reinforcing the conclusion that the CB2 receptor mediates immunosuppression (Akhmetshina et al. 2009; Servettaz et al. 2010). In another model of autoimmune disease, it was found that therapy with the CB2 agonists, JWH-133 or AM1241, reduced symptoms of chemically induced murine colitis, and a CB2 receptor antagonist, AM630 blocked their efficacy (Storr et al. 2009). The role of T-cells was not defined in these studies. However, using two different mouse models of inflammatory bowel disease, one in IL-10 k/o mice and the other chemically induced (Storr et al. 2009), JWH-133 was shown to have a therapeutic effect on clinical scores that could be blocked by a CB2 selective antagonist. Effects on multiple cells of the immune system in the gastrointestinal tract were observed, among them, a decline in the percentage of activated T-cells, which was attributed to apoptosis (Singh et al. 2012). In a mouse model of autoimmune uveoretinitis the effect of a CB2 selective agonist on T-cells was better defined. JWH-133 was shown to inhibit T-cell infiltrates into the eye by down-regulating their adhesion molecules CD162 (P-selectin glycoprotein ligand 1) and CD11a (LFA-1) (Xu et al. 2007). It was also shown that T-cells harvested from JWH-133 treated mice with uveoretinitis, when tested ex vivo, had reduced cell division upon stimulation with retinal peptide or the T-cell mitogen, Con A. In another example of cannabinoid inhibition of an autoimmune response, ∆9-THC was reported to ameliorate streptozotocin induced murine diabetes, but the mechanism, in terms of immune cells, was not investigated (Li et al. 2001). The effects of cannabinoids on models of autoimmunity and graft rejection are summarized in Table 1.
Other Mechanisms of Effects of ∆9-THC and CB2 Selective Cannabinoids on T-cells: Apoptosis and Treg Cells
One of the mechanisms that has been proposed for the effect of cannabinoids on T-cells is induction of apoptosis. In 1994 Schwarz (Schwarz et al. 1994) observed apoptosis in human peripheral blood T- cells treated with anti-CD3 with addition of anandamide, an endogenous cannabinoid ligand, or with ∆9-THC. A year later, the Klein/Friedman group reported that mouse spleen cells activated with Con A showed evidence of DNA fragmentation that was increased with concomitant treatment with ∆9-THC (Zhu et al. 1998). In addition, peritoneal macrophages stimulated with bacterial LPS showed DNA fragmentation only when ∆9-THC was added to the cultures (Zhu et al. 1998). The mechanisms were shown to be through a ∆9-THC-induced decrease in Bcl-2 and via caspase activation (Zhu et al. 1998). The Nagarkattis’ laboratory reported that ∆9-THC given in vivo caused thymic atrophy that was due to apoptosis (McKallip et al. 2002). Administration of ∆9-THC to pregnant mice induced thymic atrophy in the fetus that correlated with T-cell apoptosis (Lombard et al. 2011). This group also showed that a CB2 selective agonist could induce apoptosis in thymocytes in culture, and inhibit in vitro responses of T and B cells to mitogens by apoptosis. Similarly to what was observed for ∆9-THC, in vivo administration of a CB2 agonist induced thymic atrophy and inhibited T-cell ex vivo responses to mitogens. Caspases 3, 8, and 9 were shown to be involved (Lombard et al. 2007). It has been suggested that the beneficial immunosuppressive effect of cannabinoids in murine models of colitis and hepatitis may be partially due to induction of apoptosis (Rieder et al. 2010; Singh et al. 2012). In contrast, Robinson et al. used two CB2 selective agonists added to mouse spleen cells in vitro and observed suppression of T-cells in the Mixed Lymphocyte Reaction (MLR) (see end of Section III). However, Live/Dead staining and TUNEL analysis showed that the cannabinoids, even at relatively high doses, were not toxic, and did not induce apoptosis greater than that observed in control cultures without cannabinoid (Robinson et al. 2013). Also, cells taken from spleens of CB2 k/o mice gave normal proliferative responses in the MLR assay in the presence of the higher doses of CB2 agonists, so these drugs are not nonspecifically toxic or apoptotic (Robinson et al. 2013). Supporting evidence for immunosuppression without apoptosis was provided in a separate study by Schwarz (Schwarz et al. 1994), who found that the CB2 agonist, CP55940, was ten times more immunosuppressive than ∆9-THC or anandamide, but did not induce apoptosis in human peripheral blood lymphocytes, as was observed for the less active cannabinoids.
Another mechanism that has been proposed for the immunosuppressive activity of cannabinoids is up-regulation of Foxp3+ Tregs (T-regulatory cells), a relatively new class of T-cells which down-regulate immune responses. ∆9-THC was found to increase numbers of Tregs in a mouse model of chemically induced hepatitis (Hegde et al. 2008). In the Mixed Lymphocyte Reaction CB2 selective agonists were found to increase the percentage of T-regs in cultures (Robinson and Eisenstein unpublished observations).
Summary of Immunosuppressive Mechanisms of Cannabinoids
In summary, there appear to be many mechanisms by which cannabinoids can inhibit T-cell responses. As noted in detail at the end of section II, ∆9-THC has been shown to induce immunosuppressive cytokines including IL-10 and TGF-γ, and to inhibit IL-2, a cytokine that stimulates T-cell division and expansion. Thus, cannabinoids can exert immunosuppressive effects by increasing immunosuppressive cytokines and decreasing T-cell activating cytokines. Finally, cannabinoids have been shown to decrease adhesion capacity of leukocytes to extravasate into sites of inflammation (Ramirez et al. 2012; Xu et al. 2007; Zhang et al. 2009a). Apoptosis and induction of Treg cells may potentially add to the pathways of cannabinoid-mediated immunosuppression. The effects of ∆9-THC, anandamide and CB2 selective agonists on immune responses are summarized in Table 2.
Cannabinoids and Infection
Early experimental studies examining the effect of cannabinoids on resistance to infection showed that ∆9-THC treatment sensitized to several microbial infections (Friedman et al. 2003). Among the first to be documented was herpes simplex virus, type 2. Initial work was carried out using a guinea pig model of vaginal infection, which showed that ∆9-THC resulted in a more rapid onset and greater severity of disease, with greater virus shedding and more frequent recrudescence of the virus (Cabral et al. 1986). These findings were confirmed using a mouse model of the disease. Mice treated intraperitoneally with the cannabinoid for 4 days had enhanced viral replication and shedding from the vagina, more rapid progression of symptoms, increased mortality, greater systemic dissemination of organisms, and a higher frequency of disease recurrence when the challenge was 1 day after the start of the cannabinoid (Mishkin and Cabral 1985; Morahan et al. 1979). ∆9-THC was also shown to sensitize to infection in murine models of Friend Leukemia virus and Listeria monocytogenes (Morahan et al. 1979; Specter et al. 1991). Intensive studies on the mechanisms by which ∆9-THC suppressed host defenses to infection were undertaken by the Friedman/Klein group, using a murine model of Legionella pneumophila. It was discovered that ∆9-THC given 18 h prior to a primary sub-lethal infection sensitized to a secondary challenge with Legionella, indicating that the drug interfered with the adaptive immune response to the organism (Newton et al. 1994). This effect was shown to be due to depressed Th1 responses, including decreased IFN-γ, IL-12, and IL-12β2 receptor mRNA, and increased IL-4 and the IL-4 transcription factor, GATA-3, indicating a shift in cytokine profile from Th1 to Th2 (Klein et al. 2000; Newton et al. 2009). The profile of the antibody response to the organism also was shifted by ∆9-THC from IgG2a (type 1) to IgG1 (type 2) (Newton et al. 1994). It was shown that CB1R mediates the decrease in the type 1 cytokines, and the increase in IL-4 is dependent on CB2R (Newton et al. 2009). Cannabinoids have also been shown to sensitize to viral infections in murine models. Kaminski’s laboratory reported that orally administered ∆9-THC increased viral replication in lungs of mice given influenza virus intranasally, which correlated with reduced influx of T-cells and macrophages into the lung (Buchweitz et al. 2007). This laboratory also investigated the effect of genetic deficiency of both CB1 and CB2 receptors on this model of murine influenza and found that lack of the cannabinoid receptors, with or without ∆9-THC, led to higher levels of immune responses as measured by CD4 T-cell numbers and IFN-γ levels in bronchoalveolar lavage fluid (Buchweitz et al. 2008). More intense analysis using this model showed that CB1−/CB2− mice infected with influenza had increased inflammatory responses compared to WT mice as determined by microarray analysis (Karmaus et al. 2011). Bone marrow derived antigen-presenting cells (BMDC) of the deficient animals were able to activate ovalbumin specific T-cells without LPS stimulation, but WT BMDCs could not (Karmaus et al. 2011). These studies led to the conclusion that endogenous cannabinoids suppress immune responses. In studies from another laboratory using a model of vaccinia virus infection (the organism used in the smallpox vaccine which causes cowpox), it was shown that ∆9-THC increased the severity and duration of symptoms (Huemer et al. 2011). There is also a report that ∆9-THC sensitizes mice to intranasal infection with an amoeba, Acanthamoeba castellanii, which results in amoebic encephalitis. The mechanism of cannabinoid action was suppression of microglial anti-microbial functions (Cabral and Marciano-Cabral 2004).
Of particular interest are reports that ∆9-THC alters immune responses to SIV and HIV. The Roth/Baldwin group carried out a ground-breaking study using humanized severe combined immunodeficient mice (huPBL-SCID) treated with ∆9-THC and infected with an HIV reporter virus construct that infects mice (Roth et al. 2005). They found that ∆9-THC treated animals had a 50-fold increase in viral load. ∆9-THC alone decreased the number of IFN-γ secreting cells, which is compatible with the evidence presented earlier in this review showing that ∆9-THC decreases Th1 cytokines. Using a slightly different mouse model in which HIV infected human monocyte-derived macrophages were injected intra-cranially into huPBL-SCID mice, another group found that a CB2 selective agonist, Gp1a, administered orally, protected animals against HIV-induced encephalitis (HIVE) (Gorantla et al. 2010). In this hu-PBL/HIVE model, the cannabinoid reduced infiltration of human cells from the periphery into the mouse brain, providing a protective effect.
Dr. Molina’s laboratory has carried out a series of studies on the effect of ∆9-THC on SIV infection using rhesus macaques. Her group found that chronic administration of ∆9-THC, starting 28 days prior to SIV inoculation, decreased early mortality from the virus and viral load in CSF and plasma, but did not alter total lymphocyte counts, the CD4/CD8 ratio, or increase a marker of apoptosis in drug-treated, SIV infected animals (Molina et al. 2011). Examination of a panel of 10 cytokines and chemokines in three different brain regions showed a significant reduction only in MCP-1 (CCL2) (Winsauer et al. 2011). Another study from this group examined the effect of chronic ∆9-THC in macaques that were not SIV infected. Again, no effect of the drug was observed on lymphocytes subsets, naive versus memory cell subsets, or markers of proliferation or apoptosis of T-cells. The one difference that was found was an increase in expression of CXCR4 on CD4 and CD8 T-cells (LeCapitaine et al. 2011). These animals were later infected with SIV after 17 months of chronic THC administration. (Molina et al. 2014). The cannabinoid treated animals trended towards a decrease in plasma and peripheral blood viral load that was not statistically significant. Examination of mucosal immune responses using duodenal biopsies obtained 5 months after infection showed that there was an increase in Th2 cytokine expression (IL-4, IL-5) and a trend towards higher levels of IL-17, as well as decreased apoptosis of duodenal cells. This group of studies is significant because the results show that ∆9-THC did not increase viral load, and showed a trend towards its decrease, a result that is perhaps contrary to what would have been predicted. The lack of statistical significance in some parameters can be attributed to the small number of animals in the vehicle versus the ∆9-THC groups (n = 4).
There is a small literature on the effect of CB2 selective agonists on HIV infection that was recently reviewed by Purohit (Purohit et al. 2014). In regard to cannabinoids and T-cells, an in vitro study tested the effect of several CB2 selective agonists (JWH-133, JWH-150), and showed that they inhibited productive HIV infection in primary human T-cells, and a CB2 antagonist blocked the effect (Costantino et al. 2012). The mechanism of cannabinoid action was shown to be by interfering with the signal transduction of CXCR4 that ultimately decreased F-actin accumulation. Investigators hypothesized that the F-actin defect prevented movement of viral pre-integration complexes to the nucleus. Another mechanism that could be important in viral infectivity is the observation that the CB2 agonists inhibited T-cell activation induced by anti-CD3/anti-CD28, as viral infection is potentiated in activated cells (Costantino et al. 2012).
If one surveys the small literature on the effects of cannabinoids on infection, there is a dichotomy in the outcomes. For the majority of the experimental infections tested in mice, ∆9-THC sensitized to the microbes. In one murine model using huPBL-SCID mice, HIV replication was exacerbated by ∆9-THC, and in another model a CB2 selective agonist was protective. The models differed in that one used a viral construct and the other used transfer of human mononuclear cells infected with HIV. Whether the differences in outcomes are due to the difference in the cannabinoid or the virus cannot be determined at this time. The finding that ∆9-THC affords protection against SIV in macaques is also interesting, and provides a second example of a model where a cannabinoid rather than sensitizing to infection is protective. Perhaps the difference between this model and the other models of viral infection is that activated T-cells potentiate SIV infection, and cannabinoids suppress T-cell activation, whereas in infection with Herpes simplex or influenza, suppression of T-cells may lead to reduced immune responses necessary to control the infection. The observation that cannabinoids can block chemokine signaling, and thus prevent the necessary second signal for HIV replication, is also another potential mechanism by which these drugs interfere with retroviral replication, a mechanism that may be extraneous for other microbes.
Table 3 summarizes the effects of cannabinoids on infections. One interesting observation is that there are no papers in the literature on the effect of a CB2 agonist on a microbial infection, other than HIV.
Summary
The literature cited in this review provides incontrovertible evidence that cannabinoids are immunosuppressive. The discovery of a cannabinoid receptor, CB2, primarily expressed on cells of the immune system and not neurons, with strong immunosuppressive effects on T-cells, opens the possibility of developing drugs that target this receptor in conditions where the immune system is over-active, as in autoimmune diseases and in graft rejection. Such drugs are expected to be without psychoactive effects. These studies also give credence to medicinal uses of ∆9-THC that relate to dampening immune function. This review has focused on T-cells, but ∆9-THC as well as the CB2 selective agonists, all have suppressive effects on B-cells, monocyte/macrophages, and dendritic cells. T-cells play a major role as effectors in a variety of autoimmune conditions, so demonstration of their down-regulation by cannanbinoids is of special importance in terms of possible therapeutic value of this class of compounds. Further, the observations by several laboratories that the endogenous cannabinoid, anandamide, which binds to both CB1 and CB2 receptors, is also immunosuppressive, and that genetic ablation of the CB2 receptor activates the immune system, strongly indicates that the cannabinoid system is important in immune homeostatis.
Abbreviations
- BMDC:
-
bone marrow-derived dendritic cells
- ConA:
-
concanavalin A
- ∆9-THC:
-
∆9-tetrahydrocannabinol
- HPA:
-
Hypothalamic-Pituitary-Adrenal Axis
- Io:
-
Ionomycin
- K/O:
-
Knock-out
- Lck:
-
Lymphocyte-specific protein tyrosine kinase
- MLR:
-
Mixed Lymphcyte Reaction
- PHA:
-
Phytohemaggluttinin
- PKA:
-
Protein Kinase A
- PMA:
-
Phorbol Myristic Acid
- SCID:
-
Severe Combined Immunodeficiency
- SRBC:
-
Sheep Red Blood Cells
- WT:
-
Wild Type
References
Akhmetshina A, Dees C, Busch N, Beer J, Sarter K, Zwerina J, Zimmer A, Distler O, Schett G, Distler JH (2009) The cannabinoid receptor CB2 exerts antifibrotic effects in experimental dermal fibrosis. Arthritis Rheum 60:1129–1136
Baczynsky WO, Zimmerman AM (1983) Effects of Δ9-tetrahydrocannabinol, cannabinol and cannabidiol on the immune system of mice. I. in vivo investigation of the primary and secondary immune response. Pharmacology 26:1–11
Basu S, Dittel BN (2011) Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol Res 51:26–38
Börner C, Höllt V, Kraus J (2007) Activation of human T cells induces upregulation of cannabinoid receptor type 1 transcription. Neuroimmunemodulation 14:281–286
Börner C, Smida M, Höllt V, Schraven B, Kraus J (2009) Cannabinoid receptor type 1- and 2-mediated increase in cyclic AMP inhibits T cell receptor-triggered signaling. J Biol Chem 284:35450–35460
Bouaboula M, Rinaldi M, Carayon P, Carillon C, Delpech B, Shire D, Le Fur G, Casellas P (1993) Cannabinoid-receptor expression in human leukocytes. Eur J Biochem 214:173–180
Buchweitz JP, Karmaus PWF, Harkema JR, Williams KJ, Kaminski N (2007) Modulation of airway responses to influenza A/PR/8/34 by Δ9-tetrahydrocannabinol in C57BL/6 mice. J Pharmacol Exp Ther 323:675–683
Buchweitz JP, Karmaus PW, Williams KJ, Harkema JR, Kaminski NE (2008) Targeted deletion of cannabinoid receptors CB1 and CB2 produced enhanced inflammatory responses to influenza A/PR/8/34 in the absence and presence of Δ9-tetrahydrocannabinol. J Leukoc Biol 83:785–796
Buckley NE, McCoy KL, Mezey E, Bonner T, Zimmer A, Felder CC, Glass M (2000) Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. Eur J Pharmacol 396:141–149
Cabral GA, Marciano-Cabral F (2004) Cannabinoid-mediated exacerbation of brain infection by opportunistic amebae. J Neuroimmunol 147:127–130
Cabral GA, Mishkin EM, Marciano-Cabral F, Coleman P, Harris L, Munson AE (1986) Effect of delta-9-tetrahydrocannabinol on herpes simplex virus type 2 vaginal infections in the guinea pig. Proc Soc Exp Biol Med 182:181–186
Castaneda JT, Harui A, Kiertscher SM, Roth JD, Roth MD (2013) Differential expression of intracellular and extracellular CB2 cannabinoid receptor protein by human peripheral blood leukocytes. J Neuroimm Pharmacol 8:323–332
Cencioni MT, Chiurchiu V, Catanzaro G, Borsellino G, Bernardi G, Battistini L, Maccarrone M (2010) Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB2 receptors. PLoS ONE 5:e8688 [Electronic Resource]
Condie R, Herring A, Koh WS, Lee M, Kaminski NE (1996) Cannabinoid inhibition of adenlate cyclase-mediated signal transduction and IL-2 expression in the murine T-cell line, EL4.IL-2. J Biol Chem 271:13175–13183
Coopman K, Smith LD, Wright KL, Ward SG (2007) Temporal variation in CB2R levels following T lymphocyte activation: evidence that cannabinoids modulate CXCL12-induced chemotaxis. Int Immunopharmacol 7:360–371
Costantino CM, Gupta A, Yewdall AW, Dale BM, Devi LA, Chen BK (2012) Cannabinoid receptor 2-mediated attenuation of CXCR4-tropic HIV infection in primary CD4+ T cells. PLoS ONE 7:e33961 [Electronic Resource]
Croxford JL, Yamamura T (2005) Cannabinoids and the immune system: potential for the treatment of inflammatory diseases? J Neuroimmunol 166:3–18
Daaka Y, Friedman H, Klein TW (1996) Cannabinoid receptor proteins are increased in jurkat, human T-cell line after mitogen activation. J Pharmacol Exp Ther 276:776–783
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949
Eisenstein TK, Meissler JJ, Wilson Q, Gaughan JP, Adler MW (2007) Anandamide and Δ9-tetrahydrocannabinol directly inhibit cells of the immune system via CB2 receptors. J Neuroimmunol 189:17–22
Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, Mitchell RL (1995) Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol 48:443–450
Friedman H, Newton C, Klein TW (2003) Microbial infections, immunomodulation, and drugs of abuse. Clin Micro Rev 16:209–219
Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P (1995) Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem 232:54–61
Gardner B, Zu LX, Sharma S, Liu Q, Makriyannis A, Tashkin DP, Dubinett SM (2002) Autocrine and paracrine regulation of lymphcyte CB2 receptor expression by TGF-β. Biochem Biophys Res Comm 290:91–96
Ghosh S, Preet A, Groopman JE, Ganju RK (2006) Cannabinoid receptor CB2 modulates the CXCL12/CXCR4-mediated chemotaxis of T lymphocytes. Mol Immunol 43:2169–2179
Gorantla S, Makarov E, Roy D, Finke-Dwyer J, Murrin LC, Gendelman HE, Poluektova L (2010) Immunoregulation of a CB2 receptor agonist in a murine model of neuroAIDS. J Neuroimmune Pharmacol 5:456–468
Hanuš L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M, Pertwee RG, Ross RA, Mechoulam R, Fride E (1999) HU-308: a specific agonist for CB2, a peripheral cannabinoid receptor. Proc Natl Acad Sci 96:14228–14233. doi:10.1073/pnas.96.25.14228:
Hegde VL, Hegde S, Cravatt BF, Hofseth LJ, Nagarkatti M, Nagarkatti PS (2008) Attenuation of experimental autoimmune hepatitis by exogenous and endogenous cannabinoids: Involvement of regulatory T cells. Mol Pharmacol 74:20–33
Huemer HP, Lassnig C, Bernhard D, Sturm S, Nowotny N, Kitchen M, Pavlic M (2011) Cannabinoids lead to enhanced virulence of the smallpox vaccine (vaccinia) virus. Immunobiology 216:670–677
Huffman JW, Yu S, Showalter V, Abood ME, Wiley JL, Compton DR, Martin BR, Bramblett RD, Reggio PH (1996) Synthesis and pharmacology of a very potent cannabinoid lacking a phenolic hydroxyl with high affinity for the CB2 receptor. J Med Chem 39:3875–3877
Huffman JW, Liddle J, Yu S, Aung MM, Abood ME, Wiley JL, Martin BR (1999) 3-(1′,1′-dimethylbutyl)-1-deoxy-delta8-THC and related compounds: synthesis of selective ligands for the CB2 receptor. Bioorg Med Chem 7:2905–2914
Kaminski NE (1996) Immune regulation by cannabinoid compounds through the inhibition of the cyclic AMP signaling cascade and altered gene expression. Biochem Pharmacol 52:1133–1140
Kaminski NE, Abood ME, Kessler FK, Martin BR, Schatz AR (1992) Identification of a functionally relevant cannabinoid receptor on mouse spleen cells that is involved in cannabinoid-mediated immune modulation. Mol Pharmacol 42:736–742
Karmaus PWF, Chen W, Crawford RB, Harkema JR, Kaplan BLF, Kaminski NE (2011) Deletion of cannabinoid receptors 1 and 2 exacerbates APC function to increase inflammation and cellular immunity during influenza infection. J Leukoc Biol 90:983–995
Karmaus PW, Chen W, Kaplan BL, Kaminski NE (2012) Δ9-tetrahydrocannabinol suppresses cytotoxic T lymphocyte function independent of CB1 and CB 2, disrupting early activation events. J Neuroimmune Pharmacol 7:843–855
Klein TW (2005) Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol 5:400–411
Klein TW, Newton CA, Widen R, Friedman H (1985) The effect of delta-9-tetrahydrocannabinol and 11-hydroxy-delta-9-tetrahydrocannabinol on T-lymphocyte and B-lymphocyte mitogen responses. J Immunopharmacol 7:451–466
Klein TW, Kawakami Y, Newton C, Friedman H (1991) Marijuana components suppress inducion and cytolytic function of murine cytotoxic T-cells in vitro and in vivo. J Toxicol Environ Health 32:465–477
Klein TW, Friedman H, Specter S (1998) Marijuana, immunity and infection. J Neuroimmunol 83:102–115
Klein TW, Newton CA, Nakachi N, Friedman H (2000) Δ9-tetrahydrocannabinol treatment suppresses immunity and early IFN-γ, IL-12 and IL-12 receptor β2 responses to legionella pneumophila infection. J Immunol 164:6461–6466
LeCapitaine NJ, Zhang P, Winsauer P, Walker E, Stouwe CV, Porretta C, Molina PE (2011) Chronic Δ9-tetrahydrocannabinol administration increases lymphocyte CXCR4 expression in rhesus macaques. J Neuroimmune Pharmacol 6:540–545
Lee SF, Newton C, Widen R, Friedman H, Klein TW (2001) Differential expression of cannabinoid CB2 receptor mRNA in mouse immune cells subpopulations and following B cell stimulation. Eur J Pharmacol 423:235–241
Lefkowitz S, Klager K (1978) Effect of Δ9-tetrahydrocannabinol on in vitro sensitization of mouse splenic lymphocytes. Immunol Commun 7:557–566
Li X, Kaminsky NE, Fischer LJ (2001) Examination of the immunosuppressive effect of Δ9-tetrahydrocannabinol in streptozotocin-induced autoimmune diabetes. Int Immunopharmacol 1:699–712
Lombard C, Nagarkatti M, Nagarkatti P (2007) CB2 cannabinoid receptor agonist, JWH-015, triggers apoptosis in immune cells: Potential role for CB2-selective ligands as immunosuppressive agents. Clin Immunol 122:259–270
Lombard C, Hegde VL, Nagarkatti M, Nagarkatti PS (2011) Perinatal exposure to Δ9-tetrahydorcannabinol triggers profound defects in T cell differentiation and function in fetal and postnatal stages of life, including decreased responsiveness to HIV antigens. J Pharmacol Exp Ther 339:607–317
Lu H, Kaplan BL, Ngaotepprutaram T, Kaminski NE (2009) Suppression of T cell costimulator ICOS by Delta9-tetrahydrocannabinol. J Leukoc Biol 85:322–329
Luo YD, Patel MK, Wiederhold MD, Ou DW (1992) Effects of cannabinoids and cocaine on the mitogen-induced transformations of lymphocytes of human and mouse origins. Int J Immunopharmacol 14:49–56
Maresz K, Pryce G, Ponomarev ED, Marsicano G, Croxford JL, Shriver LP, Ledent C, Cheng X, Carrier EJ, Mann MK, Giovannoni G, Pertwee RG, Yamamura T, Buckley NE, Hillard CJ, Lutz B, Baker D, Dittel BN (2007) Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat Med 13:492–497
Massi P, Sacerdote P, Ponti W, Fuzio D, Manfredi B, Viganó D, Rubino T, Bardotti M, Parolaro D (1998) Immune function alterations in mice tolerant to Δ9-tetrahydrocannabinol: functional and biochemical parameters. J Neuroimmunol 92:60–66
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564
McKallip RJ, Lombard C, Martin BR, Nagarkatti M, Nagarkatti PS (2002) Δ9-tetrahydrocannabinol-induced apoptosis in the thymus and spleen as a mechanism of immunosuppression in vitro and in vivo. J Pharmacol Exp Ther 302:451–465
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50:83–90
Mishkin EM, Cabral GA (1985) Delta-9-tetrahydrocannabinol decreases host resistance to herpes simplex virus type 2 vaginal infection in the B6C3F1 mouse. J Gen Virol 66:2539–2549
Molina PE, Amdee A, LeCapitaine NJ, Zabaleta J, Mohan M, Winsauer P, Stouwe CV (2011) Cannabinoid neuroimmune modulation of SIV disease. J Neuroimmune Pharmacol 6:516–527
Molina PE, Amedee AM, LeCapitaine NJ, Zabaleta J, Mohan M, Winsauer PJ, Stouwe CV, McGoey RR, Auten MW, LaMotte L, Chandra LC, Birke LL (2014) Modulation of gut-specific mechanisms by chronic D9-tetrahydrocannabinol administration in male rhesus macaques infected with simian immunodeficiency virus: a systems biology analysis. AIDS Res Hum Retrovir 30:567–578
Morahan PS, Klykken PC, Smith SH, Harris LS, Munson AE (1979) Effects of cannabinoids of host resistance to Listeria monocytogenes and herpes simples virus. Infect Immun 23:670–674
Munro S, Thomas KK, Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65
Nakano Y, Pross S, Friedman H (1992) Contrasting effect of delta-9-tetrahydrocannabinol on IL-2 activity in spleen and lymph node cells of mice of different ages. Life Sci 52:41–51
Newton CA, Klein TW, Friedman H (1994) Secondary immunity to Legionella pneumophila and Th1 activity are suppressed by delta-9-tetrahydrocannabinol injection. Infect Immun 62:4015–4020
Newton CA, Chou P, Perkins I, Klein TW (2009) CB1 and CB2 cannabinoid receptors mediate different aspects of delta-9-tetrahydrocannabinol (THC)-induced T helper cell shift following immune activation by Legionella pneumophila infection. J Neuroimmune Pharmacol 4:92–102
Ngaotepprutaram T, Kaplan BL, Crawford RB, Kaminski NE (2012) Differential modulation by Δ9-tetrahydrocannabinol (Δ9-THC) of CD40 ligand (CD40L) expression in activated mouse splenic CD4+ T cells. J Neuroimmune Pharmacol 7:969–980
Pertwee RG (2008) The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol, and Δ9-tetrahydrocannabivarin. Br J Pharmacol 153:199–215
Pross SH, Klein TW, Newton CA, Smith J, Widen R, Friedman H (1990) Differential suppression of T-cell subpopulations by THC (delta-9-tetrahydrocannabinol). Int J Immunopharmacol 12:539–544
Purohit V, Rapaka RS, Rutter J (2014) Cannabinoid receptor-2 and HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol 9:447–453
Ramirez SH, Hasko J, Skuba A, Fan S, Dykstra H, McCormick R, Reichenbach N, Krizbai I, Mahedevan A, Zhang M, Tuma R, Son Y, Persidsky Y (2012) Activation of cannabinoid receptor 2 attenuates leukocyte-endothelial cell interactions and blood–brain barrier dysfunction under inflammatory conditions. J Neurosci 32:4004–4016
Rieder SA, Chauhan A, Singh U, Nagarkatti M, Nagarkatti P (2010) Cannabinoid-induced apoptosis in immune cells as a pathway to immunosuppression. Immunobiology 215:598–605
Robinson RH,Meisser JJ, Breslow-Deckman JM,Gaughan J,AdlerMW, Eisenstein TK (2013) Cannabinoids inhibit T-cells via cannabinoid receptor 2 in an in vitro assay for graft rejection, the mixed lymphocyte reaction. J Neuroimmune Pharmacol 8:1239–1250
Roth MD, Tashkin DP, Whittaker KM, Choi R, Baldwin GC (2005) Tetrahydrocannabinol suppresses immune function and enhances HIV replication in the huPBL-SCID mouse. Life Sci 77:1711–1722
Schatz AR, Koh WS, Kaminski NE (1993) Δ9-tetrahydrocannabinol selectively inhibits T-cell dependent humoral immune responses through direct inhibition of accessory T-cell function. Immunopharmacology 26:129–137
Schwarz H, Blanco FJ, Lotz M (1994) Anandamide, an endogenous cannabinoid receptor agonist inhibits lymphocyte proliferation and induces apoptosis. J Neuroimmunol 55:107–115
Servettaz A, Kavian N, Nicco C, Deveaux V, Chereau C, Wang A, Zimmer A, Lotersztajn S, Weill B, Batteux F (2010) Targeting the cannabinoid pathway limits the development of fibrosis and autoimmunity in a mouse model of systemic sclerosis. Am J Pathol 177:187–196
Singh UP, Singh NP, Singh B, Price RL, Nagarkatti M, Nagarkatti PS (2012) Cannabinoid receptor-2 (CB2) agonist ameliorates colitis in IL-10(−/−) mice by attenuating the activation of T cells and promoting their apoptosis. Toxicol Appl Pharmacol 258:256–267
Smith SH, Harris LS, Uwayday IM, Munson AE (1978) Structure-activity relationships of natural and synthetic cannabinoids in suppression of humoral and cell-mediated immunity. J Pharmacol Exp Ther 207:165–170
Specter S, Lancz G, Westrich G, Friedman H (1991) Delta-9-tetrahydrocannabinol augments murine retroviral induced immunosuppression and infection. Int J Immunopharmacol 13:411–417
Springs AEB, Karmaus PWF, Crawford RB, Kaplan BLF, Kaminski NE (2008) Effects of targeted deletion of cannabinoid receptors CB1 and CB2 on immune competence and sensitivity to immune modulation by Δ9-tetrahydrocannabinol. J Leukoc Biol 84:1574–1584
Storr MA, Keenan CM, Zhang H, Patel KD, Makriyannis A, Sharkey KA (2009) Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflamm Bowel Dis 15:1678–1685
Watson ES, Murphy JC, El Sohly HN, El Sohly MA, Turner CE (1983) Effects of the administration of coca alkaloids on the primary immune responses of mice: Interaction with Δ9-tetrahydrocannabinol and ethanol. Toxicol Appl Pharmacol 71:1–13
Winsauer PJ, Molina PE, Amedee AM, Filipeanu CM, McGoey RR, Troxclair DA, Walker EM, Birke LL, Stouwe CV, Howard JM, Leonard ST, Moerschbaecher JM, Lewis PB (2011) Tolerance to chronic delta-9-tetrahydrocannabinol (Δ9-THC) in rhesus macaques infected with simian immunodeficiency virus. Exp Clin Psychopharmacol 19:154–172
Xu H, Cheng CL, Chen M, Manivannan A, Cabay L, Pertwee RG, Coutts A, Forrester JV (2007) Anti-inflammatory property of the cannabinoid receptor-2-selective agonist JWH-133 in a rodent model of autoimmune uveoretinitis. J Leukoc Biol 82:532–541
Yang X, Hegde VL, Rao R, Zhang J, Nagarkatti PS, Nagarkatti M (2014) Histone modifications are associated with Δ9-tetrahydrocannabinol-mediated alterations in antigen-specific T cell responses. J Biol Chem 289:18707–18718
Yuan M, Kiertscher SM, Cheng Q, Zoumalan R, Tashkin DP, Roth MD (2002) Δ9-tetrahydrocannabinol regulates Th1/Th2 cytokine balance in activated human T cells. J Neuroimmunol 133:124–131
Zhang M, Adler MW, Abood ME, Ganea D, Jallo J, Tuma RF (2009a) CB2 receptor activation attenuates microcirculatory dysfunction during cerebral ischemic/reperfusion injury. Microvasc Res 78:86–94
Zhang M, Martin BR, Adler MW, Razdan RJ, Kong W, Ganea D, Tuma RF (2009b) Modulation of cannabinoid receptor activation as a neuroprotective strategy for EAE and stroke. J Neuroimmune Pharmacol 4:249–259
Zhu W, Friedman H, Klein TW (1998) Δ9-tetrahydrocannabinol induces apoptosis in macrophages and lymphocytes: involvement of bcl-2 and capsase-1. J Pharmacol Exp Ther 286:1103–1109
Zhu LX, Sharma S, Stolina M, Gardner B, Roth MD, Tashkin DP, Dubinett SM (2000) Δ9-tetrahydrocannabinol inhibits antitumor immunity by a CB2 receptor-mediated, cytokine-dependent pathway. J Immunol 165:373–380
Ziring D, Wei B, Velazquez P, Schrage M, Buckley NE, Braun J (2006) Formation of B and T cell subsets require the cannabinoid receptor CB2. Immunogenetics 58:714–725
Acknowledgments
This work was supported by grant DA013429 from the National Institute on Drug Abuse and a grant from the Pennsylvania Department of Health as part of the Tobacco Settlement funds to Temple University.
Conflict of Interest
The author declares that she has no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Eisenstein, T.K., Meissler, J.J. Effects of Cannabinoids on T-cell Function and Resistance to Infection. J Neuroimmune Pharmacol 10, 204–216 (2015). https://doi.org/10.1007/s11481-015-9603-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-015-9603-3