
Abstract
HIV-1 transactivator of transcription (Tat) protein disrupts the dopamine (DA) neurotransmission by inhibiting DA transporter (DAT) function, leading to increased neurocognitive impairment in HIV-1 infected individuals. Through integrated computational modeling and pharmacological studies, we have demonstrated that mutation of tyrosine470 (Y470H) of human DAT (hDAT) attenuates Tat-induced inhibition of DA uptake by changing the transporter conformational transitions. The present study examined the functional influences of other substitutions at tyrosine470 (Y470F and Y470A) and tyrosine88 (Y88F) and lysine92 (K92M), two other relevant residues for Tat binding to hDAT, in Tat-induced inhibitory effects on DA transport. Y88F, K92M and Y470A attenuated Tat-induced inhibition of DA transport, implicating the functional relevance of these residues for Tat binding to hDAT. Compared to wild type hDAT, Y470A and K92M but not Y88F reduced the maximal velocity of [3H]DA uptake without changes in the Km. Y88F and K92M enhanced IC50 values for DA inhibition of [3H]DA uptake and [3H]WIN35,428 binding but decreased IC50 for cocaine and GBR12909 inhibition of [3H]DA uptake, suggesting that these residues are critical for substrate and these inhibitors. Y470F, Y470A, Y88F and K92M attenuated zinc-induced increase of [3H]WIN35,428 binding. Moreover, only Y470A and K92M enhanced DA efflux relative to wild type hDAT, suggesting mutations of these residues differentially modulate transporter conformational transitions. These results demonstrate Tyr88 and Lys92 along with Tyr470 as functional recognition residues in hDAT for Tat-induced inhibition of DA transport and provide mechanistic insights into identifying target residues on the DAT for Tat binding.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
HIV-1-associated neurocognitive disorder (HAND) that encompasses neurological and psychiatric complications remains highly prevalent within people living with HIV-1 even in the era of highly active antiretroviral therapy (Heaton et al. 2010; Simioni et al. 2010; Mothobi and Brew 2012). It is commonly accepted that HIV-1 infection and replication within CNS system play a central role in the development of HAND (Gaskill et al. 2009). Substance abuse-HIV-1 comorbidity such as cocaine has been shown to increase the incidence and exacerbate the severity of HAND by enhancing viral replication (Nath et al. 2001; Ferris et al. 2008; Buch et al. 2011; Nair and Samikkannu 2012). HAND is highly correlated with neurotoxic effects of HIV-1 viral proteins that are exuded from infected microglial cells (Bansal et al. 2000; Kaul et al. 2001; Mattson et al. 2005; Kaul and Lipton 2006). HIV-1 transactivator of transcription (Tat), a non-structural viral protein required for productive infection of virus, is one of the major neurotoxins responsible for neurotoxicity and oxidative stress in central nervous system (Pocernich et al. 2005; Wallace et al. 2006). While HIV-1 virus enters the brain and produces proviral DNA in the early stage of HIV-1 infection (Nath and Clements 2011), antiretroviral agents cannot prevent the production of HIV-1 viral proteins, such as Tat protein, in infected brain cells (McArthur et al. 2010; Nath and Clements 2011). Tat has been detected in brains of patients with HIV-1 infection (Del Valle et al. 2000; Hudson et al. 2000; Lamers et al. 2010), and implicated in the pathophysiology of HAND (Li et al. 2009). Tat interacting with cocaine exacerbates the progression of the neurocognitive impairment (Gannon et al. 2011). Currently, there are no promising therapeutic approaches for such HIV-1-associated neurocognitive impairments.
Dysregulation of dopamine (DA) neurotransmission is associated with abnormal neurocognitive function and pathophysiology observed in HAND (Berger and Arendt 2000; Purohit et al. 2011). DA levels are decreased in DA-rich brain areas (Sardar et al. 1996; Kumar et al. 2009), but increased in the CSF (Scheller et al. 2010) of HAND patients. The psychostimulant action of cocaine is mediated by increased extracellular DA in the brain.
The presynaptic DA transporter (DAT), which is critical for neurocognitive function (Chudasama and Robbins 2006), is a major molecular target for both Tat and cocaine to impact the DA system (Zhu et al. 2009). DAT activity is strikingly reduced in HIV-1-infected patients with a history of cocaine use (Wang et al. 2004; Chang et al. 2008). Our previous studies have shown that Tat protein and cocaine synergistically impair DAT function as demonstrated both in vivo (Harrod et al. 2008) and in vitro (Ferris et al. 2009; Zhu et al. 2011). Further we also demonstrated that Tat inhibits DA transport and reduces DAT cell surface expression in rat striatal synaptosomes (Zhu et al. 2009; Midde et al. 2012), and directly binds to the DAT in an allosteric modulation manner (Zhu et al. 2009; Zhu et al. 2011). Cocaine acts as a competitive DAT inhibitor and blocks DA transport activity (Beuming et al. 2008). Importantly, the elevated DA induced by Tat and cocaine stimulates adjacent microglia, leading to increase of viral replication and Tat release (Gaskill et al. 2009), which has been implicated in the pathophysiology of HAND (Li et al. 2009). Considering oxidative stress-induced damage to dopaminergic neurons, long lasting exposure to viral proteins and elevated DA eventually lead to DAT(DA) deficit that potentiates severity and accelerates the progression of HAND (Purohit et al. 2011). Thus, understanding of the interplay of Tat with cocaine in disrupting DAT-mediated DA neurotransmission may provide therapeutic insights into HAND in concurrent cocaine abusers.
Through computational modeling and simulations, we have begun to define how Tat through its recognition binding sites on human DAT (hDAT) potentiates cocaine-induced inhibition on DAT function, resulting in dysfunction of the DA system. For example, we have demonstrated that mutating tyrosine470 to histidine of hDAT attenuates Tat-mediated inhibition of DA uptake and leads to alteration of transporter conformational transitions (Midde et al. 2013). In order to identify the binding pocket for Tat protein in DAT, it is necessary to identify the residues forming the crevice and understand the contribution of these recognition residues in substrate translocation, cocaine binding and conformational rearrangements in the transporter. In the current study, we investigated the role of additional substitutions at Tyr470 and other predicted potential Tat binding residues, Tyr88 and Lys92 of hDAT in Tat-induced decrease of DA translocation by generating point mutations Tyr470F (Y470F-hDAT), Tyr470A-hDAT (Y470A-hDAT), Tyr88Phe (Y88F-hDAT), Lys92Met (K92M-hDAT) and assessing their variability in function, surface expression, interaction with ligands and underlying mechanism for these alterations.
Materials and Methods
Predicting the Site for hDAT Binding with Tat
The binding structure of hDAT with HIV-1 clade B type Tat was modeled and simulated based on the nuclear magnetic resonance (NMR) structures of Tat (Peloponese et al. 2000) and the constructed structure of hDAT-DA complex. According to D-Y470 (D- refers to DAT, and hereafter) site-directed mutation experimental data as reported previously (Midde et al. 2013), Y470 of hDAT is a functional recognition residue for Tat-induced inhibition of DAT transport cycle. Therefore, Y470 of hDAT is expected to closely interact with Tat. The protein docking program ZDOCK (Pierce et al. 2011) was used to determine the initial binding structure of the hDAT-Tat complex. A total of 220,000 potential conformations were generated based on 11 NMR structures of Tat, then all of these conformations were evaluated and ranked by ZRANK (Pierce and Weng 2007). Top-3000 conformations selected from the protein-protein docking process were submitted to energy minimizations, and the docked structures were ranked according to the binding affinity estimated by using the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) method (Miller et al. 2012). Then top-256 conformations were selected for further evaluation by performing molecular dynamics (MD) simulations. Based on the MD simulations, the most favourable hDAT-Tat binding mode (with the best geometric matching quality and reasonable interaction between hDAT and Tat) was identified, and the final hDAT-Tat binding structure was energy-minimized for analysis.
Construction of Plasmids
All point mutations of Tyr88, Lys92, and Tyr470 in hDAT were selected based on the predictions of the three-dimensional computational modeling and simulations. Mutations in hDAT at Tyr88 and Tyr470 (tyrosine to phenylalanine, Y88F-hDAT and Y470F-hDAT) are expected to destroy the hydrogen bond only. Since methionine is nearly isosteric with lysine, substitution at Lys92 (lysine to methionine, K92M-hDAT) should abolish hydrogen bond with minimal perturbations to the native structure of the transporter. Substitution of tyrosine at 470 with alanine or tryptophan (Y470A-hDAT and Y470W-hDAT) is expected to eliminate both hydrogen bond and cation-π interactions that is similar to Y470H-hDAT, but with different spatial effects on structural organization of the transporter. All mutations in hDAT were generated based on wild type human DAT (WT hDAT) sequence (NCBI, cDNA clone MGC: 164608 IMAGE: 40146999) by site-directed mutagenesis. Synthetic cDNA encoding hDAT subcloned into pcDNA3.1+ (provided by Dr. Haley E Melikian, University of Massachusetts) was used as a template to generate mutants using QuikChange™ site-directed mutagenesis Kit (Agilent Tech, Santa Clara CA). The sequence of the mutant construct was confirmed by DNA sequencing at University of South Carolina EnGenCore facility. Plasmids DNA were propagated and purified using plasmid isolation kit (Qiagen, Valencia, CA, USA).
Cell Culture and DNA Transfection
Chinese hamster ovary (CHO, ATCC #CCL-61) cells were maintained in F12 medium supplemented with 10 % fetal bovine serum (FBS) and antibiotics (100 U/ml penicillin and 100 μg/mL streptomycin). Pheochromocytoma (PC12, ATCC #CRL-1721) cells were maintained in Dulbecco’s modified eagle medium supplemented with 15 % horse serum, 2.5 % bovine calf serum, 2 mM glutamine and antibiotics (100 U/ml penicillin and 100 μg/mL streptomycin). Both cells were cultured at 37 °C in a 5 % CO2 incubator. For hDAT transfection, cells were seeded into 24 well plates at a density of 1 × 105 cells/well. After 24 h, cells were transfected with WT or mutant DAT plasmids using Lipofectamine 2000 (Life Tech, Carlsbad, CA). Cells were used for the experiments after 24 h of transfection.
[3H]DA Uptake Assay
Twenty four hours after transfection, [3H]DA uptake in PC12 cells transfected with WT hDAT and mutants was performed as reported previously (Midde et al. 2013). To determine whether mutated hDAT alters the maximal velocity (Vmax) or Michaelis-Menten constant (Km) of [3H]DA uptake, kinetic analyses were conducted in WT hDAT and mutants. To generate saturation isotherms, [3H]DA uptake was measured in Krebs-Ringer-HEPES (KRH) buffer (final concentration in mM: 125 NaCl, 5 KCl, 1.5 MgSO4, 1.25 CaCl2, 1.5 KH2PO4, 10 D-glucose, 25 HEPES, 0.1 EDTA, 0.1 pargyline, and 0.1 L-ascorbic acid; pH 7.4) containing one of 6 concentrations of unlabeled DA (final DA concentrations, 1.0 nM–5 μM) and a fixed concentration of [3H]DA (500,000 dpm/well, specific activity, 21.2 Ci/mmol; PerkinElmer Life and Analytical Sciences, Boston, MA). In parallel, nonspecific uptake of each concentration of [3H]DA (in the presence of 10 μM nomifensine, final concentration) was subtracted from total uptake to calculate DAT-mediated uptake. The reaction was conducted at room temperature for 8 min and terminated by washing twice with ice cold uptake buffer. Cells were lysed in 500 μl of 1 % SDS for an hour and radioactivity was measured using a liquid scintillation counter (model Tri-Carb 2900TR; PerkinElmer Life and Analytical Sciences, Waltham, MA). Kinetic parameters (Vmax and Km) were determined using Prism 5.0 (GraphPad Software Inc., San Diego, CA). To determine the inhibitory effects of Tat on [3H]DA uptake, cells transfected with WT hDAT or mutants were preincubated with Tat1-86 or Tat Cys22 (500 nM, final concentration) for 20 min. Tat Cys22 was used as a negative control due to its null effect on DA uptake (Zhu et al. 2009).
The competitive inhibition DA uptake experiments were performed in duplicate in a final volume of 500 μl. Cells in each well were incubated in 450 μl KRH buffer containing 50 μl one of final concentrations of unlabeled DA (1 nM–1 mM), GBR12909 (1 nM–10 μM), cocaine (1 nM–1 mM), WIN 35,428 (1 nM–1 mM) or ZnCI2 (10 μM) at room temperature for 10 min and [3H]DA uptake was determined by addition of 50 μl of [3H]DA (0.1 μM, final concentration) for an additional 8 min.
[3H]WIN 35,428 Binding Assay
Binding assays were conducted to determine whether mutated hDAT alters the kinetic parameters (Bmax or Kd) of [3H]WIN 35,428 binding in PC12 cells transfected with WT hDAT and mutants. Twenty four hours after transfection, PC12 cells were dissociated with trypsin/EDTA (0.25 %/0.1 %, 1 mL for 100 mm dish) and resuspended in growth medium. After 10 min incubation at room temperature, the dissociated cells were harvested by centrifugation at 3000 rpm for 3 min and washed once with phosphate-buffered saline (PBS). The resulting cell pellets were resuspended in sucrose-phosphate buffer (final concentration in mM: 2.1 NaH2PO4, 7.3 Na2HPO47H2O, and 320 sucrose, pH 7.4) for binding assay.
To generate saturation isotherms, aliquots of cell suspensions (100 μl) were incubated with one of 8 concentrations of [3H]WIN 35,428 (0.5–30 nM final concentrations, 84 Ci/mmol, PerkinElmer Life and Analytical Sciences) in a final volume of 250 μl on ice for 2 h. In parallel, nonspecific binding at each concentration of [3H]WIN 35,428 (in the presence of 30 μM cocaine, final concentration) was subtracted from total binding to calculate the specific binding. For the competitive inhibition experiment, assays were performed in duplicate in a final volume of 500 μl. Aliquots of the cell suspensions (50 μl) were added to the assay tubes containing 50 μl of [3H]WIN 35,428 (final concentration, 5 nM) and one of 7 concentrations of unlabeled substrate DA (1 nM–100 μM), inhibitors cocaine (1 nM–100 μM) or GBR12909 (0.01 nM–1 μM) and incubated on ice for 2 h. Assays were terminated by rapid filtration onto Whatman GF/B glass fiber filters, presoaked for 2 h with assay buffer containing 0.5 % polyethylenimine, using a Brandel cell harvester. Filters were rinsed 3 times with 3 ml of ice-cold assay buffer. Radioactivity remaining on the filters was determined by liquid scintillation spectrometry.
Cell Surface Biotinylation
To determine whether DAT mutations alter DAT surface expression, biotinylation assays were performed as described previously (Zhu et al. 2005). CHO cells transfected with hDAT and mutants were plated on 6 well plates at a density of 105 cells/well. Cells were incubated with 1 ml of 1.5 mg/ml sulfo-NHS-SS biotin (Pierce, Rockford, IL) in PBS/Ca/Mg buffer (In mM: 138 NaCl, 2.7 KCl, 1.5 KH2PO4, 9.6 Na2HPO4, 1 MgCl2, 0.1 CaCl2, pH 7.3). After incubation, cells were washed 3 times with 1 ml of ice-cold 100 mM glycine in PBS/Ca/Mg buffer and incubated for 30 min at 4 °C in 100 mM glycine in PBS/Ca/Mg buffer. Cells were then washed 3 times with 1 ml of ice-cold PBS/Ca/Mg buffer and then lysed by addition of 500 ml of Triton X-100, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μM pepstatin, 250 μM phenylmethysulfonyl fluoride), followed by incubation and continual shaking for 20 min at 4 °C. Cells were transferred to 1.5 ml tubes and centrifuged at 20,000 g for 20 min. The resulting pellets were discarded, and 100 μl of the supernatants was stored at −20 °C for determining immunoreactivity of total DAT. Remaining supernatants were incubated with continuous shaking in the presence of monomeric avidin beads in Triton X-100 buffer (100 μl/tube) for 1 h at room temperature. Samples were centrifuged subsequently at 17,000 g for 4 min at 4 °C, and supernatants (containing the nonbiotinylated, intracellular protein fraction) were stored at −20 °C. Resulting pellets containing the avidin-absorbed biotinylated proteins (cell-surface fraction) were resuspended in 1 ml of 1.0 % Triton X-100 buffer and centrifuged at 17,000 g for 4 min at 4 °C, and pellets were resuspended and centrifuged twice. Final pellets consisted of the biotinylated proteins adsorbed to monomeric avidin beads. Biotinylated proteins were eluted by incubating with 75 μl of Laemmli sample buffer for 20 min at room temperature. If further assay was not immediately conducted, samples were stored at −20 °C until used.
Basal Efflux Assay
DAT-mediated basal substrate efflux was carried out, as described previously (Guptaroy et al. 2009). We have reported that Y470H-hDAT significantly increased DA efflux compared to WT hDAT (Midde et al. 2013). In this study, we compared the effects of 2 types of substrates, DA or MPP+ on basal efflux in WT hDAT and mutants. The MPP+ was chosen because MPP+ has less diffusive properties than DA in heterologous expression systems (Scholze et al. 2001). CHO cells were seeded into 24 well plates and transfected with WT hDAT and mutants. Twenty four hours after transfection cells at a density of 105 cells/well were washed 3 times with KRH buffer and preloaded with [3H]DA (50 nM, final concentration) or [3H]1-methyl-4-phenylpyridinium ([3H]MPP+, 5 nM, final concentration, specific activity, 83.9 Ci/mmol; PerkinElmer Life and Analytical Sciences) at room temperature for 20 or 30 min, respectively. After incubation, cells were washed 3 times with KRH buffer. To obtain an estimate of the total amount of [3H]DA or [3H]MPP+ in the cells at the zero time point, cells from a set of wells (4 wells/sample) were lysed rapidly in 1 % SDS. To determine the time course of the fractional basal efflux, fresh buffer (500 μl) was added into separate set of cell wells (4 wells/sample) and transferred to scintillation vials after 1 min as initial fractional efflux at 1 min, and another 500 μl buffer was added to the same wells (where the buffer was just removed for the 1 min point) and collected into vials after 10 min. Additional fractional efflux at 20, 30, 40 and 50 min, respectively, was repeated with the same procedure. After the last time point (50 min), cells were lysed in 1 % SDS and counted as total amount of [3H]DA remaining in the cells from each well.
Data Analyses
Descriptive statistics and graphical analyses were used as appropriate. Results are presented as mean ± SEM, and n represents the number of independent experiments for each experiment group. Kinetic parameters (Vmax, Km, Bmax, and Kd ) were determined from saturation curves by nonlinear regression analysis using a one-site model with variable slope. IC50 values for substrate and inhibitors inhibiting [3H]DA uptake or [3H]WIN 35,428 were determined from inhibition curves by nonlinear regression analysis using a one-site model with variable slope. For experiments involving comparisons between unpaired samples, unpaired Student’s t test was used to assess any difference in the kinetic parameters (Vmax, Km, Bmax, Kd or IC50) between WT and mutant; log-transformed values of IC50, Km or Kd were used for the statistical comparisons. Significant differences between samples were analyzed with separate ANOVAs followed by appropriate post-hoc tests. All statistical analyses were performed using IBM SPSS Statistics version 20, and differences were considered significant at p < 0.05.
Results
Structural Indications for the new Experimental Results
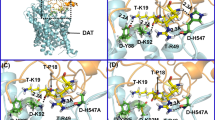
As shown in the typical binding structure of the MD-simulated hDAT-Tat complex (Fig. 1), key intermolecular interactions between hDAT and Tat include: (1) aromatic side chain of D-Y470 interacting with the positively charged N-terminus of T-M1 (T- refers to Tat, and hereafter) through cation-π interaction, (2) hydroxyl group of D-Y88 side chain forming a hydrogen bond with T-K19 side chain, and (3) D-K92 side chain forming a hydrogen bond with the carbonyl of the main chain of T-P18. Based on these interactions in the hDAT-Tat binding mode, eliminating the cation-π interaction between D-Y470 and T-M1 by mutating Y470 into a non-aromatic residue such as histidine or alanine would significantly weaken the binding between hDAT and Tat. On the contrary, the Y470F mutation on hDAT would not affect the cation-π interaction and, thus, should not significantly influent the binding affinity between hDAT and Tat. In addition, in light of the binding structure, mutating D-Y88 and D-K92 into residues without hydrogen-bonding capacity would also weaken the binding between DAT and Tat. As a result, the Y88F or K92M mutation on hDAT would be expected to decrease the binding between hDAT and Tat. These computational predictions are confirmed by the obtained experimental data (see below).

Modeled hDAT-Tat binding structure. a HIV-1 Tat protein is shown as ribbon and colored in golden, and hDAT is represented as semi-transparent surface and colored in cyan. Dopamine (DA) is shown as blue spheres. b Atomic interactions between HIV-1 Tat and hDAT as observed from the binding structure. Key residues for the intermolecular interactions between HIV-1 Tat and hDAT are shown in stick style. Residues T-M1, T-P18, and T-K19 of HIV-1 Tat are colored in purple, whereas residues D-Y470, D-K92, and D-Y88 are colored in green. Two dashed lines on the left side represent intermolecular hydrogen bonds with key distances labeled. The orange ball indicates the center of aromatic ring, and the dashed line pointing to the orange ball represents the cation-π interaction with the key distance labeled
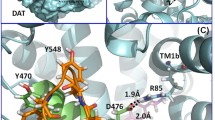
Furthermore, residues D-Y470, D-K92, and D-Y88 are also involved in the function of hDAT regarding the DA uptake. Based on the MD-simulated hDAT-Tat binding structure, we noted that residue Y470 of hDAT is a key component of a hydrophobic region, which is critical for stabilizing the compact structure of hDAT (Fig. 2). As a result, any significant change in the hydrophobic property or shape of Y470 (through mutation on Y470) would disturb the function of hDAT. Thus, the Y470H and Y470A mutation could decrease the Vmax of hDAT transporting function, whereas the Y470F mutation would not influence it. In addition, residues Y88, K92, and D313 also help to stabilize transmembrane helix 1b (TM1b) and transmembrane helix 6a (TM6a) through hydrophobic or electrostatic intramolecular interactions, which makes hDAT kept compact and ready for the conformational conversion associated with the DA transporting by hDAT. Specifically, the positively charged side chain of D-K92 forms a strong hydrogen bond with the negatively charged side chain of D-D313. As observed in our MD simulation, TM1b and TM6a move together during the conformational conversion associated with the DA transporting. Therefore, the hydrogen bond between D-K92 and D-D313 helps to synchronize the motion of TM1b and TM6a and, consequently, decrease the energy barrier of the conformational conversion. Hence, the K92M mutation would negatively impact the transporting kinetics of DA, i.e., decreasing the Vmax. Besides, the aromatic ring of the Y88 side chain is sandwiched between TM1b and extracellular loop 4 (EL4) in a hydrophobic region. As a result, the Y88F mutation would not expect to influence the function of hDAT, i.e., no significant change on Vmax. These computational insights are also consistent with the experimental data obtained for the K92M and Y88F mutants (see below).

Structural details of residues Y470, K92, and Y88 in the MD-simulated hDAT-Tat binding structure. Protein hDAT is represented as cyan ribbon, and its residues Y470, K92, Y88, and D313 are shown in sticks colored in green. The hydrogen bond between K92 and D313 is represented as a dashed line with the key distance given; the hydrogen bond favorably connects M1b with TM6a in hDAT
Mutations of Tyr470, Tyr88 and Lys92 Differentially Alter DA Transport
We have demonstrated that mutation of tyrosine (Y470H) in hDAT displayed a decrease in the Vmax values of [3H]DA uptake under a control condition (Midde et al. 2013). To further determine whether other substitutions at tyrosine 470 residue show differential effects on the basal DA transport, two mutations at this residue (tyrosine to phenylalanine, Y470F and tyrosine to alanine, Y470A) were generated by site-directed mutagenesis. We determined the pharmacological profiles of [3H]DA uptake in PC12 cells transfected with WT hDAT or these mutants. As shown in Fig. 3a, compared to WT hDAT (15.7 ± 0.9 pmol/min/105 cells), the Y470A-hDAT displayed a decrease in the Vmax values [1.2 ± 0.2 pmol/min/105 cells, t (6) = 23, p < 0.001, unpaired Student’s t test], whereas the Y470F-hDAT did not alter the Vmax and the Km values.

[3H]DA uptake and DAT surface expression in WT hDAT and mutants. a Kinetic analysis of [3H]DA uptake in WT hDAT, Y470F-hDAT and Y470A-hDAT. PC12 cells transfected with WT hDAT, Y470F-hDAT or Y470A-hDAT were incubated with one of 6 mixed concentrations of the [3H]DA as total rate of DA uptake. In parallel, nonspecific uptake of each concentration of [3H]DA (in the presence of 10 μM nomifensine, final concentration) was subtracted from total uptake to calculate DAT-mediated uptake. * p < 0.05 compared to control value (unpaired Student’s t test) (n = 5). b Cell surface expression of WT hDAT, Y470F-hDAT and Y470A-hDAT was analyzed by biotinylation assay. Top panel: representative immunoblots in CHO cells expressing WT hDAT (WT), Y470F-hDAT (Y470F) or Y470A-hDAT (Y470A). Bottom panel: DAT immunoreactivity is expressed as mean ± S.E.M. densitometry units from three independent experiments (n = 3)
As predicted by our computational modeling and simulations, Tyr88, Lys92 and Tyr470 in hDAT are associated to play a critical role in intermolecular interaction between Tat and DAT. Two mutations in hDAT (tyrosine to phenylalanine, Y88F-hDAT and lysine to methionine, K92M-hDAT) were generated by site-directed mutagenesis. As shown in Table 1 and Fig. 4a, the Vmax values were decreased in K92M-hDAT (4.5 ± 1.7 pmol/min/105 cells, t (5) = 3.3, p < 0.05, unpaired Student’s t test) but not altered in Y88F-hDAT (14.8 ± 3.7 pmol/min/105 cells) compared to WT hDAT (15.7 ± 0.9 pmol/min/105 cells). No difference in the Km values was observed. WIN 35,428 binding site shares pharmacological identity with the DA uptake carrier and is part of the cocaine binding domain (Pristupa et al. 1994). We also determined the effects of these mutants on the possible relationship between the binding site of Tat on DAT and the WIN 35,428 binding site. As shown in Table 2, the Bmax values of [3H]WIN 35,428 binding were decreased in Y88F-hDAT (5.8 ± 0.9 pmol/105 cells, t(6) = 2.1, p < 0.05, unpaired Student’s t test) and K92M-hDAT (2.4 ± 0.4 pmol/105 cells, t(6) = 6.3, p < 0.001, unpaired Student’s t test) compared with WT hDAT (9.7 ± 1.6 pmol/105 cells). The mutant-induced reduction of the Bmax values was accompanied by an increase in the Kd values in Y88F-hDAT (4.0 ± 0.6 nM, t(6) = 3.4, p < 0.05, unpaired Student’s t test) and K92M-hDAT (3.5 ± 1.1 nM, t(6) = 2.8, p < 0.05, unpaired Student’s t test) compared with WT hDAT 8.6 ± 1.1 nM).

[3H]DA uptake and DAT surface expression in WT hDAT and mutants. a Kinetic analysis of [3H]DA uptake in WT hDAT, Y88F-hDAT and K92M-hDAT. CHO cells transfected with WT hDAT, Y88F-hDAT or K92M-hDAT were incubated with one of 6 mixed concentrations of the [3H]DA as total rate of DA uptake. In parallel, nonspecific uptake of each concentration of [3H]DA (in the presence of 10 μM nomifensine, final concentration) was subtracted from total uptake to calculate DAT-mediated uptake. * p < 0.05 compared to control value (unpaired Student’s t test) (n = 5). b Cell surface expression of WT hDAT, Y88F-hDAT and K92M-hDAT was analyzed by biotinylation assay. Top panel: representative immunoblots in CHO cells expressing WT hDAT (WT), Y88F-hDAT (Y88F) or K92M-hDAT (K92M). Bottom panel: DAT immunoreactivity is expressed as mean ± S.E.M. densitometry units from three independent experiments (n = 3). * p < 0.05 compared to WT hDAT (unpaired Student’s t test)
To determine whether the mutations of Tyr88 and Lys92 influence selective binding sites on hDAT for DA, cocaine, GBR 12909 and WIN 35,428, we tested the ability of substrate and DAT inhibitors to inhibit [3H]DA uptake (Table 1) and [3H]WIN 35,428 binding (Table 2) in WT hDAT and its mutants. As shown in Table 1, the apparent affinity (IC50) for DA was significantly decreased Y88F-hDAT [2010 ± 60 nM, t(6) = 3.8, p < 0.01, unpaired Student’s t test] and K92M-hDAT (2868 ± 48 nM, t(6) = 3.8, p < 0.05, unpaired Student’s t test), respectively, compared to the WT hDAT (1730 ± 82 nM). However, the potencies of cocaine and GBR12909 for inhibiting [3H]DA uptake were ~4.0-fold greater in Y88F-hDAT and K92M-hDAT as compared with WT hDAT (unpaired Student’s t test). In addition, the potencies of WIN 35,428 for inhibiting [3H]DA uptake was not significantly altered in Y88F-hDAT (20 ± 2 nM) but more potent in K92M-hDAT [10 ± 1.0 nM, t(6) = 5.0, p < 0.01, unpaired Student’s t test] compared to WT hDAT (39 ± 10 nM). Regarding to the potencies of DA, cocaine and GBR12909 for inhibiting [3H]WIN 35,428 binding (Table 2), the apparent affinity (IC50) for DA was decreased in Y88F-hDAT (2071 ± 340 nM, t(8) = 3.5, p < 0.01) and K92M-hDAT (4211 ± 118 nM, t(8) = 2.7, p < 0.05), respectively, compared to than the WT hDAT (827 ± 120 nM). However, the potencies of cocaine were increased in Y88F-hDAT (85 ± 60 nM, t(8) = 5.8, p < 0.001) but not in K92M-hDAT compared to WT hDAT (150 ± 8.6 nM). No difference in IC50 of GBR 12909 for inhibiting [3H]WIN 35,428 binding was found among Y88F-hDAT, K92M-hDAT and WT hDAT.
To determine whether these mutations of hDAT alter DAT surface expression, the immunoreactivity of total DAT and surface DAT in CHO cells transfected with WT or Y470F-hDAT, Y470A-hDAT, Y88F-hDAT or K92M-hDAT was examined using cell surface biotinylation followed by Western blotting. As shown in Fig. 3b, no difference in the ratio of surface DAT (biotinylated DAT) to total DAT between WT hDAT and Y470F-hDAT (biotinylated/total: WT, 0.93 ± 0.09; and Y470F, 0.96 ± 0.1; p > 0.05, one-way ANOVA) was found. The immunoreactivity of total and surface DAT in Y470A-hDAT was undetectable (data not shown). Similarly, as shown in Fig. 4b, despite no differences in the ratio of surface DAT to total DAT between WT (1.0 ± 0.05), Y88F-hDAT (1.13 ± 0.13; p > 0.05, one-way ANOVA) and K92M-hDAT (1.09 ± 0.09; p > 0.05, one-way ANOVA), the absolute total DAT in K92M-hDAT was significantly decreased compared to WT hDAT (t(8) = 4.6, p < 0.01, unpaired Student’s t test). Thus, the decreased DA uptake in K92M-hDAT is not due to alteration of the available DAT on the cell surface.
Mutations of Tyr88, Lys92 and Tyr470 Differentially Influence Tat-Induced Inhibitory Effects on DA Transport
We have reported that mutation of either Tyr470 in hDAT or Cys22 in Tat attenuated Tat-induced decrease in DA uptake (Zhu et al. 2009; Midde et al. 2013). To determine whether Y470F-hDAT and Y470A-hDAT show differential effects on Tat-induced decrease in DA uptake, we examined the specific [3H]DA uptake in WT hDAT and the Tyr470 mutants in the presence or absence of recombinant Tat1-86 or recombinant Tat Cys22. As shown in Fig. 5a, two-way ANOVA on the specific [3H]DA uptake in WT hDAT and Tyr470 mutants revealed a significant main effect of mutation (F(2, 36) = 88.1; p < 0.001) and Tat treatment (F(2, 36) = 15.5; p < 0.001), as well as a significant mutation × Tat interaction (F(4, 36) = 5.2; p < 0.01). Tat (250 nM, final concentration) decreased the Vmax of [3H]DA uptake by 32 % in WT hDAT (F(1, 8) = 23.6; p < 0.01) and by 47 % in Y470F-hDAT (F(1, 8) = 15.4; p < 0.01), respectively; however, no effect of Tat was observed in Y470H-hDAT (p > 0.05) and Y470A-hDAT (p > 0.05), suggesting that the different substitutions at Tyr470 residue in hDAT differentially influence Tat-induced inhibition of DA uptake. In addition, Tat Cys22 (500 nM, final concentration) did not alter DA uptake in WT hDAT and three Tyr470 mutants compared to their respective controls, suggesting that Cys22 residue in Tat plays a critical role in Tat-induced regulation DAT function (Fig. 5a).

Effects of Tat or Tat Cys22 on kinetic analysis of [3H]DA uptake in WT hDAT and Tyr470 mutants. a PC12 cells transfected with WT hDAT (WT), Y470F-hDAT (Y470F), Y470H-hDAT (Y470H) or Y470A-hDAT (Y470A) were preincubated with or without recombinant Tat1-86 (rTat1-86) or Tat Cys22 (500 nM, final concentration) at room temperature for 20 min followed by the addition of 0.05 μM final concentration of the [3H]DA. Nonspecific uptake was determined in the presence of 10 μM final concentration of nomifensine. b [3H]DA uptake in cells transfected with WT hDAT (WT), Y88F-hDAT (Y88F) and K92M-hDAT (K92M) was determined in the presence or absence of Tat Cys22 or rTat1-86 (500 nM, final concentration). Data are expressed as means from seven independent experiments ± S.E.M. * p < 0.05 compared with the respective control values. # p < 0.05 compared to WT hDAT
We also determined the effects of Y88F-hDAT and K92M-hDAT on Tat-induced inhibition of DA uptake (Fig. 5b). A two-way ANOVA on the specific [3H]DA uptake revealed a significant main effect of mutation (F(2, 54) = 124; p < 0.001) and Tat treatment (F(2, 54) = 4.7; p < 0.05); however, mutation × Tat interaction (F(4, 54) = 1.9; p > 0.05) was not significant. Exposure to Tat decreased the Vmax of [3H]DA uptake by 32 % in WT hDAT (F(1, 12) = 8.0; p < 0.05; Fig. 5b); however, no effect of Tat was observed in Y88F-hDAT (F(1, 12) = 1.1; p > 0.05) and K92M-hDAT (F(1, 12) = 1.4; p > 0.05), suggesting that mutation of either Tyr88 or Lys92 in hDAT attenuates Tat-induced reduction of hDAT function. With regard to the effect of recombinant Tat Cys22 on DA uptake (Fig. 5b), exposure to Tat Cys22 did not alter DA uptake in Y88F-hDAT and K92M-hDAT compared to WT hDAT.
Effects of Tyr88, Lys92, and Tyr470 Mutants on Zinc Regulation of DAT Conformational Transitions and Basal DA Efflux
In general, occupancy of the endogenous Zn2+ binding site in WT hDAT stabilizes the transporter in an outward-facing conformation, which allows DA to bind but inhibits its translocation, thereby increasing [3H]WIN 35,428 binding (Norregaard et al. 1998; Moritz et al. 2013), but decreasing [3H]DA uptake (Loland et al. 2003). Addition of Zn2+ is able to partially reverse an inward-facing state to an outward-facing state (Norregaard et al. 1998; Loland et al. 2003). On the basis of this principle, we recently reported that Y470H-hDAT exhibit an attenuation of Zn2+-mediated decreased [3H]DA uptake and increased [3H]WIN35,428 binding observed in WT hDAT, which demonstrates a preference for the inward-facing conformation of the transporter (Midde et al. 2013). To investigate the role of additional substitutions at Tyr470 residue in DAT conformational transitions, we examined the effects of the respective mutations on Zn2+ modulation of [3H]DA uptake and [3H]WIN35,428 binding. As described in Fig. 6a, two-way ANOVA on the specific [3H]DA uptake in WT and Y470F-hDAT and Y470A-hDAT revealed a significant main effect of mutation (F(2, 18) = 99; p < 0.001), zinc (F(1, 18) = 40; p < 0.001) and a significant mutation × zinc interaction (F(2, 18) = 8.9; p < 0.01). The addition of Zn2+ significantly decreased [3H]DA uptake in WT hDAT, Y470F-hDAT and Y470A-hDAT by 47, 49 and 72 %, respectively (Fig. 6a, ps < 0.01 relative to control, unpaired Student’s t test), suggesting these mutants do not affect Zn2+-mediated regulation of DA transport. In contrast, as shown in Fig. 6b, a two-way ANOVA on the specific [3H]WIN35,428 binding in WT and Y470F-hDAT and Y470A-hDAT revealed a significant main effect of mutation (F(2, 18) = 73; p < 0.001), zinc (F(1, 18) = 10.1; p < 0.01); however, no significant mutation × zinc interaction (F(2, 18) = 1.8; p > 0.05) was observed. The addition of Zn2+ significantly increased [3H]WIN 35,428 binding in WT hDAT by 48 % (p < 0.05 relative to control, unpaired Student’s t test) but not in Y470F-hDAT and Y470A-hDAT, indicating that Y470F and Y470A mutants attenuate Zn2+-induced increase in WIN 35,428 binding sites.

Effects of Y470F, Y470A and Y470H mutants on transporter conformational transitions. Mutations of Tyr470 affect zinc regulation of [3H]DA uptake (a) and [3H]WIN 35,428 binding (b). CHO cells transfected with WT hDAT (WT), Y470F-hDAT (Y470F) and Y470A-hDAT (Y470A) were incubated with KRH buffer alone (control) or ZnCl2 (10 μM, final concentration) followed by [3H]DA uptake or [3H]WIN 35,428 binding (n = 6). The histogram shows [3H]DA uptake and [3H]WIN 35,428 binding expressed as mean ± S.E.M. of the respective controls set to 100 % for the mutant. * p < 0.05 compared to control. # p < 0.05 compared to WT hDAT with ZnCl2. (c) Functional DA efflux of DA properties of WT hDAT and mutants. CHO cells transfected with WT hDAT or mutants were preincubated with KRH buffer containing [3H]DA (0.05 μM, final concentration) at room temperature for 20 min. After incubation, cells were washed and incubated with fresh buffer at indicated time points. Subsequently, the buffer was removed from cells, and radioactivity in the buffer or remaining in the cells was counted. Each fractional efflux of [3H]DA in WT hDAT (WT), Y470F-hDAT (Y470F), Y470A-hDAT (Y470A) or Y470H-hDAT was expressed as percentage of total [3H]DA in the cells at the start of the experiment. Fractional [3H]DA efflux levels at 1, 10, 20, 30, 40 and 50 min are expressed as the percentage of total [3H]DA with preloading with 0.05 μM (WT hDAT: 26,837 ± 5089 dpm, Y470F-hDAT: 20,908 ± 4209 dpm, Y470A-hDAT: 1158 ± 123 dpm and Y470H-hDAT: 2488 ± 150 dpm) presented in the cells at the start of the experiment (n = 4). ×× p < 0.05 compared to WT hDAT (Bonferroni t-test). (d) Functional MPP+ efflux properties of WT hDAT and mutants. CHO cells transfected with WT hDAT or mutants were preincubated with KRH buffer containing [3H]MPP+ (5 nM, final concentration) at room temperature for 30 min. Fractional [3H]MPP+ efflux levels at each time point are expressed as the percentage of total [3H]MPP+ with preloading with 0.05 μM (WT hDAT: 12,120 ± 397 dpm, Y470F-hDAT: 7399 ± 359 dpm, Y470A-hDAT: 460 ± 46 dpm and Y470H-hDAT: 602 ± 28 dpm) presented in the cells at the start of the experiment (n = 4). ×× p < 0.05 compared to WT hDAT (Bonferroni t-test)
With regard to Y88F-hDAT and K92M-hDAT (Fig. 7a), a two-way ANOVA on the specific [3H]DA uptake revealed a significant main effect of mutation (F(1, 24) = 170; p < 0.001), zinc (F(1, 24) = 102; p < 0.001) and a significant mutation × zinc interaction (F(2, 24) = 8.3; p < 0.01). The addition of Zn2+ significantly decreased [3H]DA uptake in WT and Y88F-hDAT and K92M-hDAT by 48, 73 and 81 %, respectively (ps < 0.001 relative to control, unpaired Student’s t test). In addition, two-way ANOVA on the specific [3H]WIN35,428 binding in WT and Y88F-hDAT and K92M-hDAT revealed a significant main effect of mutation (F(1, 18) = 5.3; p < 0.05), zinc (F(1, 18) = 5.1; p < 0.05); however, significant mutation × zinc interaction (F(2, 18) = 0.7; p > 0.05) was not significant (Fig. 7b). Post-hoc analyses (unpaired Student’s t test) revealed that the addition of Zn2+ increased [3H]WIN 35,428 binding in WT hDAT by 54 % but did not significantly alter [3H]WIN 35,428 binding in Y88F-hDAT and K92M-hDAT, respectively, compared to their respective controls. These results suggest that Y88F and K92M mutants attenuate zinc modulation of [3H]WIN 35,428 binding sites but not [3H]DA uptake.

Effects of Y88F and K92M mutants on transporter conformational transitions. Mutations of Tyr88 and Lys92 affect zinc regulation of [3H]DA uptake (a) and [3H]WIN 35,428 binding (b). CHO cells transfected with WT hDAT (WT), Y88F-hDAT (Y88F) and K92M-hDAT (K92M) were incubated with KRH buffer alone (control) or ZnCl2 (10 μM, final concentration) followed by [3H]DA uptake or [3H]WIN 35,428 binding (n = 6). The histogram shows [3H]DA uptake and [3H]WIN 35,428 binding expressed as mean ± S.E.M. of the respective controls set to 100 % for the mutant. * p < 0.05 compared to control. # p < 0.05 compared to WT hDAT with ZnCl2. (c) Functional DA efflux of DA properties of WT hDAT and mutants. CHO cells transfected with WT hDAT or mutants were preincubated with KRH buffer containing [3H]DA (0.05 μM, final concentration) at room temperature for 20 min. After incubation, cells were washed and incubated with fresh buffer as indicated time points. Subsequently, the buffer was separated from cells, and radioactivity in the buffer and remaining in the cells was counted. Each fractional efflux of [3H]DA in WT hDAT (WT), Y88F-hDAT and K92M-hDAT was expressed as percentage of total [3H]DA in the cells at the start of the experiment. Fractional [3H]DA efflux levels at 1, 10, 20, 30, 40 and 50 min are expressed as the percentage of total [3H]DA with preloading with 0.05 μM (WT hDAT: 70,082 ± 8256 dpm, Y88F-hDAT: 41,805 ± 6887 dpm and K92M-hDAT: 9655 ± 2160 dpm) presented in the cells at the start of the experiment (n = 3). ×× p < 0.05 compared to WT hDAT (Bonferroni t-test). (d) Functional MPP+ efflux properties of WT hDAT and mutants. CHO cells transfected with WT hDAT or mutants were preincubated with KRH buffer containing [3H]MPP+ (5 nM, final concentration) at room temperature for 30 min. Fractional [3H]MPP+ efflux levels at each time point are expressed as the percentage of total [3H]MPP+ with preloading with 0.05 μM (WT hDAT: 15,516 ± 920 dpm, Y88F-hDAT: 5695 ± 450 dpm and K92M-hDAT: 967 ± 121 dpm) presented in the cells at the start of the experiment (n = 4)
To further determine the effects of the mutations of hDAT on transporter conformational transitions, we examined the fractional efflux levels of [3H]DA and [3H]MPP+ in WT hDAT and its mutants. As shown in Fig. 6c, after preloading with 0.05 μM [3H]DA for 20 min at room temperature, CHO cells transfected with WT hDAT, Y470H-hDAT, Y470F-hDAT and Y470A-hDAT were washed and fractional DA efflux samples were collected at the indicated times. A two-way ANOVA on the basal efflux of [3H]DA revealed significant main effects of mutation (F(3, 11) = 41.7; p < 0.001), time (F(5, 55) = 355.9; p < 0.001) and a significant mutation × time interaction (F(15,55) = 40.5; p < 0.001). Post-hoc analyses showed that compared to WT hDAT, DA efflux levels were elevated at 1, 10, 20 min in Y470H-hDAT and at 1 and 10 min in Y470A-hDAT (ps < 0.05, Bonferroni t-test). With regard to MPP+ efflux (Fig. 6d), a two-way ANOVA revealed significant main effects of mutation (F(3, 12) = 18.1; p < 0.001), time (F(5, 60) = 389.5; p < 0.001) and significant mutation × time interaction (F(15, 60) = 8.6; p < 0.001) for Y470F-hDAT, Y470H-hDAT and Y470A-hDAT compared to WT hDAT. Compared to WT hDAT, DA efflux levels were elevated in Y470H-hDAT and Y470A-hDAT at all-time points (ps < 0.05, Bonferroni t-test). As shown in Fig. 7c, a two-way ANOVA on the basal efflux of [3H]DA in Y88F-hDAT and K92M-hDAT revealed significant main effects of mutation (F(2, 6) = 28.1; p < 0.01), time (F(5, 30) = 156; p < 0.001) and significant mutation × time interaction (F(10, 30) = 511; p < 0.001). Post-hoc analysis revealed that compared to WT hDAT, DA efflux levels were elevated at 1 and 10 min in K92M-hDAT (p < 0.05, Bonferroni t-test) but not in Y88F-hDAT. Compared to WT hDAT, Y88F and K92M mutants did not alter the basal efflux of [3H]MPP+ at all-time points (Fig. 7d).
Discussion
We have recently demonstrated that mutation of Tyr470 of hDAT is critical for HIV-1 Tat-induced inhibition of DA transport and transporter conformational transitions (Midde et al. 2013). Through computational modeling and MD simulations, we identified that the residue Tyr470 is a key component of a hydrophobic region, which is important for stabilizing the compact structure of hDAT and maintaining DA transport. In addition, through hydrophobic or electrostatic intramolecular interactions, Tyr88 and Lys92, two relevant residues of hDAT, help to stabilize the compact structure of hDAT. Mutations of Tyr88, Lys92 or Tyr470 attenuated Tat-induced inhibition of DA transport, demonstrating the functional relevance of these residues for Tat binding to hDAT. Compared to WT hDAT, only Y470A and K92M decreased the Vmax of [3H]DA uptake without changing in the Km. Y88F and K92M decreased the potency for cocaine and GBR12909 but increased the affinity for DA in both [3H]DA uptake and [3H]WIN35,428 binding, suggesting that mutations of these residues in hDAT disrupt the binding sites for DA and inhibitors. Mutations of Tyr88, Lys92 or Tyr470 did not alter zinc-induced inhibition of DA uptake but reversed zinc-augmented [3H]WIN35,428 binding, implying a mechanistic context for the transporter conformational transitions by these mutants. Our results further provide a relatively comprehensive molecular insight into these important residues for intermolecular interactions between Tat and DAT.
The present results show that K92M but not Y88F decreased the Vmax with no changes in Km values; however, both mutants slightly increased IC50 values for DA inhibiting DA uptake compared to WT hDAT. Although K92M and Y88F exhibited a decrease in Bmax and Kd values of [3H]WIN35,428 binding, their surface DAT expression was not altered, suggesting that the decreased Vmax in these mutants is not due to DAT internalization. We have demonstrated that Y470H exhibits a reduction of the Vmax with no changes in Km (Midde et al. 2013); however, the current data show that Y470A but not Y470F decreased the Vmax without changing Km value because Y470F did not affect the hydrophobic property of Y470. These data support the computational prediction that Tyr 88, Lys 92 and Tyr 470, via interacting with TM1b and TM6a helices movement, stabilize DA transport function (Fig. 2). Mutations of these residues could disrupt the intermolecular interactions within DAT, thereby decreasing DA transport. On the other hand, mutations of these residues without changing Km for DA uptake suggest that these residues in DAT for Tat binding do not overlap with the binding site of substrate DA (Midde et al. 2013). In contrast, K92M had an increase in the DA uptake potency of cocaine, GBR12909 and WIN35,428, whereas Y88F only increased the potency of cocaine and GBR12909. With regard to the potency for [3H]WIN35,428 binding, Y88F and K92M increased IC50 value of DA, whereas only Y88F decreased IC50 of cocaine. These data are consistent with our previous finding showing an increase in the DA uptake potency of cocaine and GBR12909 in Y470H (Midde et al. 2013). GBR12909 labels the classic DA uptake site in rodent brain, binding to the piperazine acceptor site (Andersen et al. 1987), while cocaine competes with DA uptake and preferentially stabilizes the hDAT in the outward-facing conformational state, resulting in a reduction of DA uptake (Reith et al. 2001; Loland et al. 2002). We have demonstrated that Tat allosterically modulates DAT function (Zhu et al. 2011). Therefore, Tyr88, Lys92 and Tyr470 predicted by computational modeling represent the intermolecular binding sites in DAT for Tat and mutations of these residues may, via allosteric DAT modulation and conformational transitions, increase their potency of cocaine and GBR12909 inhibiting DA uptake and WIN binding. Recent studies have demonstrated that Tat and cocaine synergistically inhibit DAT function in vitro and in vivo (Ferris et al. 2008; Harrod et al. 2008; Paris et al. 2014). Thus, the current findings support our hypothesis that Tat, via allosteric modulation of DAT function, potentiates cocaine-mediated inhibitory effects on DA transport.
We have demonstrated that the Tat molecule is associated with DAT through intermolecular electrostatic attractions and complementary hydrophobic interactions (Midde et al. 2013). Further, the hDAT-Tat binding mode (Fig. 1) predicted that eliminating the cation-π interaction between Tyr470 in hDAT and T-M1 in Tat by mutating Y470 into a non-aromatic residue such as histidine or alanine would significantly weaken the binding between hDAT and Tat. Our previous study has demonstrated that mutating Tyr470 to histidine attenuates Tat-induced inhibitory effect on DA uptake (Midde et al. 2013). The present results show that Y470A but not Y470F also attenuated Tat-induced inhibition on DAT because mutating Tyr470 to phenylalanine (Y470F) does not affect the cation-π interaction. These findings indicate that the important role of Tyr470 in Tat-DAT interaction. In addition, in light of the Tat-DAT binding structure (Fig. 2), mutating Tyr88 or Lys92 to the residues without hydrogen-bonding capacity would diminish the binding affinity of Tat-DAT. The computational predictions were validated by experimental data showing that Y88F and K92M attenuate Tat-induced inhibition of DAT. In support of the Tat-DAT interaction model, the present results also demonstrate that mutation of Cys22 in Tat eliminates the inhibitory effect of wild type Tat on DA transport in WT hDAT and mutants, suggesting that the Cys22 residue is critical for Tat binding to DAT. Evidence shows that the cysteine rich domain (residues 22-37) in the first exon of Tat protein is critical for the biological function of Tat (Debaisieux et al. 2012; Bertrand et al. 2013). Our previous studies have demonstrated that mutation of Tat Cys22 had no effect on DAT function (Zhu et al. 2009; Midde et al. 2013). Understanding the functional relevance of additional residues in Tat on modulation of DAT will provide useful feedback for further refining the computationally predicted binding model of the DAT with Tat. These findings further support that the integrated computational-experimental studies is fairly reliable for identifying the recognition binding sites on hDAT for Tat and cocaine interaction.
Tat has been shown to allosterically modulate DAT function (Zhu et al. 2009; Zhu et al. 2011). The allosteric modulation of DAT is responsible for conformational transitions via substrate- and ligand-binding sites on DAT (Zhao et al. 2010; Shan et al. 2011). Mutation of Tyr470 (Y470H) leads to transporter conformational transitions by affecting zinc modulation of DA uptake and WIN binding as well as enhancing basal DA efflux (Midde et al. 2013). In the present study, Y88F, K92M, Y470F and Y470A did not affect Zn2+-induced inhibition of [3H]DA uptake but diminished Zn2+-augmented [3H]WIN 35,428 binding compared to WT hDAT. Evidence indicates that transporters that prefer inward facing state can reverse Zn2+ inhibitory effects on DA uptake and WIN35,428 binding (Norregaard et al. 1998). One possible explanation for the failure to reverse Zn2+ influence on DA uptake in these mutants could be due to less effect of these particular mutations on hydrogen bonds between Zn2+ and coordination residues, which is necessary for zinc-binding with outward-open state of DA transport (Alberts et al. 1998). However, our data showing an attenuation of Zn2+-enhanced [3H]WIN 35,428 binding by these mutants reveal one aspect of DAT conformational changes. The effect of mutations on conformational states of DAT was further validated by measuring basal substrate efflux. The present study confirmed Y470H-mediated enhancement of basal DA and MPP+ efflux, which is consistent with our previous report (Midde et al. 2013). Interestingly, mutant Y470A produces a similar increase in basal efflux levels for both DA and MPP+, whereas Y470F had no effect on both DA and MPP+ efflux. With regard to mutations of Tyr88 and Lys92, only K92M displayed an increase in basal efflux of DA but not MPP+. Nevertheless, the current results demonstrate the differential influences of DAT residues in the basal substrate efflux. We have demonstrated that Tat protein enhances DA efflux in WT hDAT (Midde et al. 2013). Thus, to fully understand the mechanisms by which Tat inhibits DAT function, future studies including amphetamine-stimulated efflux and MTSET will be necessary to further analyze the changes in the conformational transition attributed to the identified residues in DAT.
In summary, we have identified the residues Tyr470, Tyr88 and Lys92 as the specific recognition binding sites in DAT for Tat binding, as well as the molecular mechanism(s) that underlies how Tat, via these residues, allosterically modulates DA translocation. Considering that the dynamic and complex interactions between Tat and DAT involve multiple residues of DAT, testing the influence of multiple DAT mutations in Tat and DAT interaction in vitro and in vivo is an essential task in our future study. Our studies will provide a mechanistic basis to identify targets on the DAT for developing compounds that specifically block Tat binding site(s) in hDAT, thereby stabilizing physiological DA neurotransmission and improving neurocognitive function of HAND in concurrent cocaine abusers.
References
Alberts IL, Nadassy K, Wodak SJ (1998) Analysis of zinc binding sites in protein crystal structures. Protein Sci 7:1700–1716
Andersen PH, Jansen JA, Nielsen EB (1987) [3H]GBR 12935 binding in vivo in mouse brain: labelling of a piperazine acceptor site. Eur J Pharmacol 144:1–6
Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM (2000) Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res 879:42–49
Berger JR, Arendt G (2000) HIV dementia: the role of the basal ganglia and dopaminergic systems. J Psychopharmacol 14:214–221
Bertrand SJ, Aksenova MV, Mactutus CF, Booze RM (2013) HIV-1 Tat protein variants: critical role for the cysteine region in synaptodendritic injury. Exp Neurol 248:228–235
Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ (2008) The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci 11:780–789
Buch S, Yao H, Guo M, Mori T, Su TP, Wang J (2011) Cocaine and HIV-1 interplay: molecular mechanisms of action and addiction. J Neuroimmune Pharmacol 6:503–515
Chang L, Wang GJ, Volkow ND, Ernst T, Telang F, Logan J, Fowler JS (2008) Decreased brain dopamine transporters are related to cognitive deficits in HIV patients with or without cocaine abuse. Neuroimage 42:869–878
Chudasama Y, Robbins TW (2006) Functions of frontostriatal systems in cognition: comparative neuropsychopharmacological studies in rats, monkeys and humans. Biol Psychol 73:19–38
Debaisieux S, Rayne F, Yezid H, Beaumelle B (2012) The ins and outs of HIV-1 Tat. Traffic 13:355–363
Del Valle L, Croul S, Morgello S, Amini S, Rappaport J, Khalili K (2000) Detection of HIV-1 Tat and JCV capsid protein, VP1, in AIDS brain with progressive multifocal leukoencephalopathy. J Neurovirol 6:221–228
Ferris MJ, Mactutus CF, Booze RM (2008) Neurotoxic profiles of HIV, psychostimulant drugs of abuse, and their concerted effect on the brain: current status of dopamine system vulnerability in NeuroAIDS. Neurosci Biobehav Rev 32:883–909
Ferris MJ, Frederick-Duus D, Fadel J, Mactutus CF, Booze RM (2009) The human immunodeficiency virus-1-associated protein, Tat1-86, impairs dopamine transporters and interacts with cocaine to reduce nerve terminal function: a no-net-flux microdialysis study. Neuroscience 159:1292–1299
Gannon P, Khan MZ, Kolson DL (2011) Current understanding of HIV-associated neurocognitive disorders pathogenesis. Curr Opin Neurol 24:275–283
Gaskill PJ, Calderon TM, Luers AJ, Eugenin EA, Javitch JA, Berman JW (2009) Human immunodeficiency virus (HIV) infection of human macrophages is increased by dopamine: a bridge between HIV-associated neurologic disorders and drug abuse. Am J Pathol 175:1148–1159
Guptaroy B, Zhang M, Bowton E, Binda F, Shi L, Weinstein H, Galli A, Javitch JA, Neubig RR, Gnegy ME (2009) A juxtamembrane mutation in the N terminus of the dopamine transporter induces preference for an inward-facing conformation. Mol Pharmacol 75:514–524
Harrod SB, Mactutus CF, Fitting S, Hasselrot U, Booze RM (2008) Intra-accumbal Tat1-72 alters acute and sensitized responses to cocaine. Pharmacol Biochem Behav 90:723–729
Heaton RK et al (2010) HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER study. Neurology 75:2087–2096
Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, Male D, Everall I (2000) Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol 6:145–155
Kaul M, Lipton SA (2006) Mechanisms of neuroimmunity and neurodegeneration associated with HIV-1 infection and AIDS. J Neuroimmune Pharmacol: Off J Soc NeuroImmune Pharmacol 1:138–151
Kaul M, Garden GA, Lipton SA (2001) Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410:988–994
Kumar AM, Fernandez JB, Singer EJ, Commins D, Waldrop-Valverde D, Ownby RL, Kumar M (2009) Human immunodeficiency virus type 1 in the central nervous system leads to decreased dopamine in different regions of postmortem human brains. J Neurovirol 15:257–274
Lamers SL, Salemi M, Galligan DC, Morris A, Gray R, Fogel G, Zhao L, McGrath MS (2010) Human immunodeficiency virus-1 evolutionary patterns associated with pathogenic processes in the brain. J Neurovirol 16:230–241
Li W, Li G, Steiner J, Nath A (2009) Role of Tat protein in HIV neuropathogenesis. Neurotox Res 16:205–220
Loland CJ, Norregaard L, Litman T, Gether U (2002) Generation of an activating Zn(2+) switch in the dopamine transporter: mutation of an intracellular tyrosine constitutively alters the conformational equilibrium of the transport cycle. Proc Natl Acad Sci U S A 99:1683–1688
Loland CJ, Norgaard-Nielsen K, Gether U (2003) Probing dopamine transporter structure and function by Zn2 + -site engineering. Eur J Pharmacol 479:187–197
Mattson MP, Haughey NJ, Nath A (2005) Cell death in HIV dementia. Cell Death Differ 12(Suppl 1):893–904
McArthur JC, Steiner J, Sacktor N, Nath A (2010) Human immunodeficiency virus-associated neurocognitive disorders: mind the gap. Ann Neurol 67:699–714
Midde NM, Gomez AM, Zhu J (2012) HIV-1 Tat protein decreases dopamine transporter cell surface expression and vesicular monoamine transporter-2 function in rat striatal synaptosomes. J Neuroimmune Pharmacol 7:629–639
Midde NM, Huang X, Gomez AM, Booze RM, Zhan CG, Zhu J (2013) Mutation of tyrosine 470 of human dopamine transporter is critical for HIV-1 Tat-induced inhibition of dopamine transport and transporter conformational transitions. J Neuroimmune Pharmacol 8:975–987
Miller BR, McGee D, Swails JM, Homeyer N, Gohlke H, Roitberg AE (2012) MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput 8:3314–3321
Moritz AE, Foster JD, Gorentla BK, Mazei-Robison MS, Yang JW, Sitte HH, Blakely RD, Vaughan RA (2013) Phosphorylation of dopamine transporter serine 7 modulates cocaine analog binding. J Biol Chem 288:20–32
Mothobi NZ, Brew BJ (2012) Neurocognitive dysfunction in the highly active antiretroviral therapy era. Curr Opin Infect Dis 25:4–9
Nair MP, Samikkannu T (2012) Differential regulation of neurotoxin in HIV clades: role of cocaine and methamphetamine. Curr HIV Res 10:429–434
Nath A, Clements JE (2011) Eradication of HIV from the brain: reasons for pause. AIDS 25:577–580
Nath A, Maragos WF, Avison MJ, Schmitt FA, Berger JR (2001) Acceleration of HIV dementia with methamphetamine and cocaine. J Neurovirol 7:66–71
Norregaard L, Frederiksen D, Nielsen EO, Gether U (1998) Delineation of an endogenous zinc-binding site in the human dopamine transporter. EMBO J 17:4266–4273
Paris JJ, Carey AN, Shay CF, Gomes SM, He JJ, McLaughlin JP (2014) Effects of conditional central expression of HIV-1 tat protein to potentiate cocaine-mediated psychostimulation and reward among male mice. Neuropsychopharmacology 39:380–388
Peloponese JM Jr, Gregoire C, Opi S, Esquieu D, Sturgis J, Lebrun E, Meurs E, Collette Y, Olive D, Aubertin AM, Witvrow M, Pannecouque C, De Clercq E, Bailly C, Lebreton J, Loret EP (2000) 1H-13C nuclear magnetic resonance assignment and structural characterization of HIV-1 Tat protein. C R Acad Sci III 323:883–894
Pierce B, Weng Z (2007) ZRANK: reranking protein docking predictions with an optimized energy function. Proteins 67:1078–1086
Pierce BG, Hourai Y, Weng Z (2011) Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS One 6:e24657
Pocernich CB, Sultana R, Mohmmad-Abdul H, Nath A, Butterfield DA (2005) HIV-dementia, Tat-induced oxidative stress, and antioxidant therapeutic considerations. Brain Res Brain Res Rev 50:14–26
Pristupa ZB, Wilson JM, Hoffman BJ, Kish SJ, Niznik HB (1994) Pharmacological heterogeneity of the cloned and native human dopamine transporter: disassociation of [3H]WIN 35,428 and [3H]GBR 12,935 binding. Mol Pharmacol 45:125–135
Purohit V, Rapaka R, Shurtleff D (2011) Drugs of abuse, dopamine, and HIV-associated neurocognitive disorders/HIV-associated dementia. Mol Neurobiol 44:102–110
Reith ME, Berfield JL, Wang LC, Ferrer JV, Javitch JA (2001) The uptake inhibitors cocaine and benztropine differentially alter the conformation of the human dopamine transporter. J Biol Chem 276:29012–29018
Sardar AM, Czudek C, Reynolds GP (1996) Dopamine deficits in the brain: the neurochemical basis of parkinsonian symptoms in AIDS. Neuroreport 7:910–912
Scheller C, Arendt G, Nolting T, Antke C, Sopper S, Maschke M, Obermann M, Angerer A, Husstedt IW, Meisner F, Neuen-Jacob E, Muller HW, Carey P, Ter Meulen V, Riederer P, Koutsilieri E (2010) Increased dopaminergic neurotransmission in therapy-naive asymptomatic HIV patients is not associated with adaptive changes at the dopaminergic synapses. J Neural Transm 117:699–705
Scholze P, Sitte HH, Singer EA (2001) Substantial loss of substrate by diffusion during uptake in HEK-293 cells expressing neurotransmitter transporters. Neurosci Lett 309:173–176
Shan J, Javitch JA, Shi L, Weinstein H (2011) The substrate-driven transition to an inward-facing conformation in the functional mechanism of the dopamine transporter. PLoS One 6:e16350
Simioni S, Cavassini M, Annoni J-M, Rimbault Abraham A, Bourquin I, Schiffer V, Calmy A, Chave J-P, Giacobini E, Hirschel B, Du Pasquier RA (2010) Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS (London, England) 24:1243–1250
Wallace DR, Dodson S, Nath A, Booze RM (2006) Estrogen attenuates gp120- and tat1-72-induced oxidative stress and prevents loss of dopamine transporter function. Synapse (New York, NY) 59:51–60
Wang GJ, Chang L, Volkow ND, Telang F, Logan J, Ernst T, Fowler JS (2004) Decreased brain dopaminergic transporters in HIV-associated dementia patients. Brain 127:2452–2458
Zhao Y, Terry D, Shi L, Weinstein H, Blanchard SC, Javitch JA (2010) Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature 465:188–193
Zhu J, Apparsundaram S, Bardo MT, Dwoskin LP (2005) Environmental enrichment decreases cell surface expression of the dopamine transporter in rat medial prefrontal cortex. J Neurochem 93:1434–1443
Zhu J, Mactutus CF, Wallace DR, Booze RM (2009) HIV-1 Tat protein-induced rapid and reversible decrease in [3H]dopamine uptake: dissociation of [3H]dopamine uptake and [3H]2beta-carbomethoxy-3-beta-(4-fluorophenyl)tropane (WIN 35,428) binding in rat striatal synaptosomes. J Pharmacol Exp Ther 329:1071–1083
Zhu J, Ananthan S, Mactutus CF, Booze RM (2011) Recombinant human immunodeficiency virus-1 transactivator of transcription1-86 allosterically modulates dopamine transporter activity. Synapse 65:1251–1254
Acknowledgments
This research was supported by grant from the National Institute on Drug Abuse to Jun Zhu (DA035714).
Conflicts of Interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Midde, N.M., Yuan, Y., Quizon, P.M. et al. Mutations at Tyrosine 88, Lysine 92 and Tyrosine 470 of Human Dopamine Transporter Result in an Attenuation of HIV-1 Tat-Induced Inhibition of Dopamine Transport. J Neuroimmune Pharmacol 10, 122–135 (2015). https://doi.org/10.1007/s11481-015-9583-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-015-9583-3