
Abstract
The Tat protein of the human immunodeficiency virus (HIV) has been implicated in the pathophysiology of the neurocognitive deficits associated with HIV infection. This is the earliest protein to be produced by the proviral DNA in the infected cell. The protein not only drives the regulatory regions of the virus but may also be actively released from the cell and then interact with the cell surface receptors of other uninfected cells in the brain leading to cellular dysfunction. It may also be taken up by these cells and can then activate a number of host genes. The Tat protein is highly potent and has the unique ability to travel along neuronal pathways. Importantly, its production is not impacted by the use of antiretroviral drugs once the proviral DNA has been formed. This article reviews the pleomorphic actions of Tat protein and the evidence supporting its central role in the neuropathogenesis of the HIV infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Human immunodeficiency virus (HIV) infection frequently results in neurological complications such as decreased cognition, depression, and motor dysfunction. Several terms have been used to describe the syndrome including AIDS dementia complex and HIV-associated dementia. With the use of antiretroviral therapy the severe forms of neurocognitive impairment are seldom seen, and more commonly milder forms persist. This has been termed HIV-associated neurocognitive disorder or HAND (Antinori et al. 2007). The pathophysiology of this disorder in the presence of antiretroviral drugs is clearly different than that in the pre-antiretroviral era. This review has been focused on Tat protein of HIV for several reasons. Tat is the first protein produced during viral replication, it plays a critical role in driving viral replication by transactivation of the promoter region of the virus, there is no effective treatment that blocks Tat activity, and it is produced by infected cells once the proviral DNA is formed even in the presence of available antiretroviral drugs. In this review, we discuss the pleomorphic actions of the Tat protein on the various cell types within the brain, the molecular basis of these interactions and the pharmacological approaches to date developed to try and block its activity.
Tat Structure
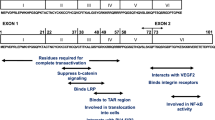
Tat is a transactivator of TAR (Tat associated region). It is a small protein of 101 amino acids but the length varies from strain to strain (Robert-Guroff et al. 1990). It is produced in the initial phase of viral transcription and localizes mainly to the nucleus and nucleolus (Stauber and Pavlakis 1998). It is a highly basic protein, and therefore, has the potential for a non specific binding with different RNAs (Ruben et al. 1989). The primary role of Tat is to regulate (productive and processive) transcription from the HIV promoter region termed the long terminal repeat (LTR) (Rosen et al. 1988). Residues 1–72 are encoded by the first exon and residues 73–101 or 104 are encoded by a second exon (Ruben et al. 1989). An 86 amino acid form of Tat, which exists in a few laboratory passaged virus strains (LAI, HXB2, and NL4-3), has been frequently used (Bilodeau et al. 1999). This version represents a truncated and not a naturally occurring full length protein. Tat can be divided into six functional regions (Fig. 1). Amino acids 1–48 represent a minimal activation domain of HIV-1 Tat required for LTR activation, the basic domain 49–72 contains a RKKRRQRRR motif, which confers TAR RNA binding and is important for nuclear localization signal (NLS) and uptake of Tat by cells (Jeang et al. 1999). Tat is believed to be functional as a monomer rather than dimer. Mutations in the region 1–21 amino acids are tolerant to changes without loss of biologic activity, in contrast, changes in amino acids 25–40 are generally deleterious for transactivation. Sequences from 22 to 37, a cysteine rich domain, binds with divalent cations like Zn2+ and Cd2+. It has been shown that the Zn binding property is important for dimerization of Tat and subsequently affects its biological functions (Frankel et al. 1988). Moreover, it has been demonstrated that substitutions at cysteine residues 22, 25, 27, and 37 alter the transactivation of HIV-LTR (Jeang et al. 1999). Acetylation of lysine at position 28 modulates the affinity and stability of Tat-CyclinT1-TAR complexes by enhancing an interaction with the CyclinT1 Tat-TAR recognition motif (D’Orso and Frankel 2009). The cysteine residue at position 31 is critical for binding to the NMDA receptor on neurons and mediating neurotoxicity (Li et al. 2008). Chemotactic properties have also been attributed to this residue (Ranga et al. 2004). The second exon of Tat, 73–101 or 104 amino acids, is less studied. Findings from HIV and SIV Tat are quite clear in demonstrating that this exon contributes toward optimal transactivation. There is also evidence that the second exon of HIV-1 Tat, in other assays is important for transactivation, transrepression of transcription factors such as AP-1 and NF-kB, and virus replication. The RGD motif in the second exon that binds to integrin receptors, is not found in HIV-2 and SIV Tat (Barillari et al. 1993). Moreover, the second exon has an ESKKKVE motif which is conserved in most HIV-1 Tat proteins and partially present in HIV-2 and SIV Tat (Kuppuswamy et al. 1989).

Structural and functional domains of HIV-Tat protein. The Tat protein is formed from two exons. The first exon is formed of 72 amino acids and the second exon varies in length and encodes amino acids 73–86 or 104. The protein is divided into six regions. The first five reside in the first exon. Of these, the cysteine rich and the arginine-rich regions are important in mediating neurotoxicity through cell surface interactions. The nuclear localizing domain (amino acids 48–56) is important in the gene transactivation properties of Tat
Immunological Responses to Tat
The immunological epitopes within Tat have not been studied in detail. Uninfected individuals have natural IgM antibodies directed against two portions of Tat (CTNCYCKKCCFH and GRKKRRQRRRPP), suggesting that this might be one basis for natural immunity against the virus (Rodman et al. 1992). Interestingly, following a period of post infection latency, the titers of natural antibodies decline and other Tat reactive antibodies do not rise (Zagury et al. 1998). This has made the use of Tat protein as an antigen for subunit vaccines extremely challenging. However, there is no information available regarding the antibody titers to Tat in patients with HIV-associated neurocognitive dysfunction. However, antibodies to the N or C terminal of Tat can form immune complexes that can bind to the NMDA receptor on neurons. These immune complexes can prevent activation of the receptor by Tat and even by other agonists of the receptor such as glutamate and quinolinic acid through stearic hinderance (Rumbaugh and Nath, unpublished observations).
Tat-Mediated HIV Transcription
HIV transcription is controlled primarily by Tat (Rosen et al. 1988). Transcription of the HIV provirus is characterized by an early, Tat-independent phase and a late, Tat-dependent phase. In the absence of Tat, a series of short transcripts are produced due to inefficient elongation by the recruited RNA pol II. However, inefficient, this process results in the synthesis of a small fraction of full length viral transcripts leading to the synthesis of the Tat protein (Yedavalli et al. 2003). Tat after binding to the hairpin loop (bulged RNA stem loop structure) TAR, that is present at the 5′-terminus of all viral transcripts, rapidly leads to the synthesis of more Tat and the establishment of a positive regulatory loop. The optimal activity of Tat is further dictated by its association with two classes of cellular proteins, Tat associated kinases (TAKs) (Deng et al. 2002) and Tat associated histone acetyl transferases (HAT) (Marcello et al. 2001). Association of Tat and p-TEFb (TAK) with TAR leads to phosphorylation of the RNAPII CTD (Deng et al. 2002). Phosphorylation of RNAPII CTD renders otherwise non processive RNAPII into productively elongating molecules. In addition to cellular kinases Tat also recruits cellular HATs. Two nuclear HATs, p300 and P/CAF, interact with and acetylate Tat on distinct lysine residues. The acetylation of the activator domain of Tat by P/CAF enhances the binding of Tat to the cellular factor CDK9/p-TEFb, whereas the acetylation of the TAR binding domain by p300 promotes its dissociation from TAR element during early transcriptional elongation, and both events increase the activation of transcription from LTR (D’Orso and Frankel 2009). Tat mediated LTR transactivation in astrocytes is unique and involves complex interactions with cellular transcription factors (Coyle-Rink et al. 2002) and may occur independent of TAR (Taylor et al. 1992).
Tat is Required for HIV Reverse Transcription
It has been shown that Tat is able to stimulate efficient reverse transcription (Harrich et al. 1997). Tat binding to TAR RNA may alter TAR structure such that the initiation of reverse transcription is enhanced. Mutations of the Tat gene decrease the initiation of reverse transcription of HIV-1 replication several thousand folds. Viruses lacking tat are also defective in endogenous assays of reverse transcription, although these viruses contain similar levels of reverse transcriptase. These results indicate that the Tat protein, in addition to regulating the level of gene expression, is also important for efficient HIV-1 reverse transcription.
Release and Uptake of Tat
Tat has been shown to be released by infected lymphoid (Ensoli et al. 1993), monocytic cells (Turchan et al. 2001), and glial cells (Tardieu et al. 1992) in vitro, by a leaderless but energy dependent pathway (Chang et al. 1997). Monocytic (Johnston et al. 2001) and astrocytic cells (Chauhan et al. 2003) stably expressing Tat also release Tat extracellularly (Bruce-Keller et al. 2003). Both forms of Tat, i.e., Tat formed by first exon only and that formed by first and second exon are released (Li et al. 2008; Malim and Cullen 1991). Tat release occurs most optimally in low serum conditions (Ensoli et al. 1993) such as that present in brain. Tat can be taken up readily by most cell types (Frankel and Pabo 1988). This property of Tat has been exploited to deliver other proteins conjugated to Tat derived peptides; a phenomenon called “protein transduction” (Ford et al. 2000; Snyder and Dowdy 2001). When full length Tat is taken up by cells it remains functionally active and can transactivate HIV expression. The basic domain that contains the arginine-rich region is critical for the nuclear localization of Tat (Vives et al. 1997). The region derived from the second exon of Tat is important for the uptake of Tat (Ma and Nath 1997). The mechanism of uptake involves interaction with the low-density lipoprotein receptor (Liu et al. 2000) and cell surface heparan sulfate proteoglycans (Tyagi et al. 2001). The transmembrane uptake of Tat is mediated through caveolar endocytosis (Fittipaldi et al. 2003) and clathrin-dependent endocytosis (Vendeville et al. 2004).
The cationic Tat peptide (amino acids 48–56), called the protein transduction domain (PTD) or cell penetrating peptide (CPP), is widely used as a vehicle for the intracellular delivery of macromolecules including oligonucleotides, peptides or proteins, low-molecular-mass drugs, nanoparticles, and liposomes (Schwarze et al. 1999; Snyder and Dowdy 2001). This approach has also been exploited for developing novel neuroprotective strategies (Spitere et al. 2008; Wei et al. 2008). The mechanisms of the CPP-cargo translocation include macropinocytosis, clathrin-mediated endocytosis, and caveolae/lipid-raft-mediated endocytosis (Kaplan et al. 2005; Richard et al. 2005).
Detection of Tat in Brain
Several groups have demonstrated the presence of Tat protein brains of patients with HIV encephalitis by immunostaining (Hofman et al. 1994; Hudson et al. 2000; Kruman et al. 1999; Liu et al. 2000; Valle et al. 2000). Additionally, mRNA levels for tat are also elevated in brain tissue of patients with HIV dementia (Hudson et al. 2000; Wesselingh et al. 1993; Wiley et al. 1996). In rhesus macaques with encephalitis due to a chimeric strain of HIV and the simian immunodeficiency virus, Tat has been demonstrated by immunostaining and western blot analysis (Hudson et al. 2000). Immunostaining patterns suggest that Tat can be found in cytoplasm of perivascular macrophages, microglial nodules, and in glial cells which likely represent HIV-infected cells. However, interestingly, Tat can also be found in the nuclei of some neurons (Liu et al. 2000) and oligodendroglia (Valle et al. 2000) which likely suggest that extracellular Tat was taken up by these cells. Tat can also be detected in the serum of patients with HIV infection in concentrations of about 1 ng/ml (Westendorp et al. 1995) and concentrations of 4 ng/ml have been reported in conditioned medium of HIV-infected cells (Albini et al. 1998). While most studies using recombinant Tat protein use nM concentrations, vast majority of the Tat protein in these preparations is polymerized or oxidized and hence are inactive. A recent study in which supernatants from Tat-transfected cells was directly used to cause neurotoxicity showed that low pM concentrations of Tat were sufficient to cause NMDA receptor activation and neurotoxicity (Li et al. 2008). In fact cell stably transfected with Tat when injected into the brain cause significant alterations in histological markers of inflammation, synaptic density, and most importantly in behavioral performance (Bruce-Keller et al. 2003; Chauhan et al. 2003).
Evolution of Tat in Brain
Tat sequences from the brain show distinct compartmentalization when compared to other tissues (Thomas et al. 2007). Non-synonymous/synonymous substitution (synonymous substitution is the substitution of one base for another in an exon of a gene coding for a protein, such that the amino acid sequence produced is not modified, while non-synonymous substitution leads to a change in the amino acid sequence) rates among the tat sequences derived from patients with HIV-associated neurocognitive impairment are significantly higher compared to the HIV-infected patient without neurocognitive impairment. The ratios of transversions (substitution of a purine for a pyrimidine or vice versa) to transitions (a mutation changing a purine to another purine nucleotide or a pyrimidine to another pyrimidine nucleotide) in the tat sequences are also significantly higher among the patients with HIV-associated neurocognitive impairment. Phylogenetic analyses show clustering of sequences from each clinical group among the brain-derived tat sequences (Bratanich et al. 1998). Comparison of matched brain- and spleen-derived tat sequences indicate that homology among brain-derived clones is greater than that between the brain- and spleen-derived clones. The brain-derived tat sequences are markedly heterogeneous in regions, which influence viral replication and intracellular transport (Mayne et al. 1998). Further, tat sequences from patients with HIV-associated neurocognitive impairment have different functional properties compared to those without associated neurocognitive impairment. For example the former are not as efficient at transactivation of the LTR, but suppress the expression of proapoptotic genes and have a differential effect on several other host genes, features that promote it’s neurotoxic potential (Boven et al. 2007). These studies indicate that differing selective forces act on the HIV tat gene in the brain, which may influence the development of neurocognitive impairment.
Transport of Tat Within the Brain
Tat can be transported in the brain along anatomical pathways both anterogradely and retrogradely. For example when Tat expressing cells were injected into the striatum, synaptic injury and gliosis was found in the substantia nigra and when the cells were injected into the hilus of the dentate gyrus, similar synaptic injury and gliosis was found in the CA3/4 region. Importantly, in both instances, Tat could be detected at the sites of synaptic injury clearly demonstrating the ability of Tat to be transported along these neurons (Bruce-Keller et al. 2003). Tat must have the ability to escape the proteolytic pathways within the cell and enter axonal transport systems. Consistent with the scenario, Tat can inhibit the proteolytic activity of the 20S proteasome (Huang et al. 2002; Seeger et al. 1997), which may explain its ability to escape proteolysis in neurons. Further, these data suggest that Tat can be transported both retrogradely (from the striatum to the pars compacta region of the substantia nigra) and anterogradely (from the hilus to CA3/4). These observations may have important implications for neuropathogenesis of retroviral infections suggesting that Tat-induced neuronal damage and glial cell activation may occur at sites distant from the cells infected with the virus.
Tat Receptors
In the past years, Tat entry into cells was thought to be receptorless, however, recent studies have demonstrated that Tat transduction is receptor mediated. Heparan sulfate is the receptor for Tat, present on almost all types of cells and helps localize Tat to the cell membrane. Another receptor the low-density lipoprotein receptor-related protein (LRP) helps to internalize Tat by an endocytic process. However, both heparan sulfate and LRP are necessary for Tat entry in neuronal cells (Liu et al. 2000). In neurons, LRP, postsynaptic density protein-95 (PSD-95), and the N-methyl-d-aspartic acid (NMDA) receptors form a macromolecular complex which is stimulated by Tat leading to neuronal injury (Eugenin et al. 2007). The core domain of Tat (aa 37–48) is directly involved in Tat interaction with LRP domains II, III, and IV. Tat can also bind directly to the NMDA receptor through Cys–Cys interactions with the extracellular domain of the NR-1 subunit (Li et al. 2008). Tat shows conserved amino acids corresponding to critical sequences of some chemokines. Synthetic Tat and a peptide (CysL24-51) encompassing the “chemokine-like” region of Tat induces a rapid and transient Ca2+ influx in monocytes and macrophages, analogous to beta-chemokines. Cross-desensitization studies indicate that Tat shares receptors with MCP-1, MCP-3, and eotaxin and can displace beta-chemokines from chemokine receptors CCR2 and CCR3, but not CCR1, CCR4, and CCR5 (Albini et al. 1998). Furthermore, Tat binds to chemokine receptor CXCR4, therefore it may act as an antagonist for virion binding to this receptor (Xiao et al. 2000). This directs pressure on HIV-1 CXCR4 strains to adapt to CCR5 tropic strains. However, neurons are not infected though they contain CXCR4 as well as CCR5. Thus Tat may disrupt neuronal function by interaction with CXCR4. Tat is also capable of inducing chemokine receptor expression. Tat can induce CXCR4 on both lymphocytes and monocytes/macrophages, whereas CCR5 and CCR3 is induced on monocytes/macrophages but not on lymphocytes (Huang et al. 1998; Secchiero et al. 1999; Weiss et al. 1999).
Intracellular Tat Binding Factors
HIV-Tat protein interacts intracellularly with host proteins which amplify or down modulate its effects on HIV transcription. A 110 kDa Tat interacting protein (Tip) in the nucleus amplifies the Tat effect on LTR transactivation (Liu et al. 2002). Another Tip 60 kDa also augments Tat dependent HIV-LTR transcription by interacting with various cellular transcription factors that belong to the nuclear histone acetyl transferase family. HIV-Tat also interacts with a 26S proteosome, through Tat binding protein-1 (Nelbock et al. 1990; Tanaka et al. 1997). Interestingly, another nuclear protein, pur alpha also interacts directly with Tat. Residues 49–72 of Tat are critical for binding with pur alpha (Wortman et al. 2000). Pur alpha is a single stranded DNA-binding protein, which binds to RNA, with much lower affinity. Pur alpha and Tat also synergistically stimulate the JC virus promoter (Tada et al. 1990).
Effect of Tat on Neurons
Intracerebral injection of Tat can be lethal to mice within hours of injection (Gourdou et al. 1990; Sabatier et al. 1991). In another animal model, Tat was shown to cause attenuation of spatial learning accompanied by suppression of long-term potentiation, the cellular basis of spatial learning in hippocampal slices (Li et al. 2004). In adult animals Tat affects pre-attentive processes and spatial memory (Fitting et al. 2008). In a Tat transgenic model there is marked glial cell activation and neuronal loss (Kim et al. 2003a). Tat causes loss of selective populations of neurons in vitro and in vivo (Hayman et al. 1993; Jones et al. 1998; Magnuson et al. 1995; Maragos et al. 2003). Regions particularly susceptible to Tat neurotoxicity include the striatum (Hayman et al. 1993), dentate gyrus, and the CA3 region of the hippocampus (Maragos et al. 2003). These regions are rich in NMDA receptors (Malva et al. 1998). Further, neuropathological studies from patients with HIV infection show a preferential loss of neurons in the dentate gyrus (Maragos et al. 2003) and striatum (Everall et al. 1995). Consistent with these observations, there is a reduction in evoked dopamine in the striatum of the Tat-treated animals suggesting a dysfunction of nerve terminals (Ferris et al. 2009). Specifically in dopaminergic neurons Tat mediated neurotoxicity can by blocked by antagonists of the D1 dopaminergic receptor (Silvers et al. 2007).
Tat produces dose-dependent depolarizations even in the presence of the sodium channel blocker tetrodotoxin, which suggests that Tat actions are independent of synaptic interactions (Magnuson et al. 1995; Perez et al. 2001). Tat also depolarizes the neuronal cell membrane when applied extracellularly to outside–out membrane patches providing strong evidence for direct excitation of neurons on the cell surface (Cheng et al. 1998). In contrast, neurotoxicity induced by gp120 is mediated primarily by indirect mechanisms. However, the polyamine site of the NMDA receptor (Prendergast et al. 2002) and Tat-induced phosphorylation of the NMDA receptor (Haughey et al. 2001) have also been implicated in Tat-mediated neurotoxicity. Further, Tat binds to the extracellular domain of the NR1 subunit of the NMDA receptor via Cys–Cys interactions, which suggest a novel mechanism for excitation of this receptor (Li et al. 2008). Another unique feature of the electrophysiological property of Tat is that it does not show any evidence of desensitization upon repetitive applications (Cheng et al. 1998). The degree of desensitization of glutamate receptors may be inversely predictive of agonist toxicity (Brorson et al. 1995; Garthwaite 1991; Jonas and Sakmann 1992). The non-desensitizing actions of Tat would cause the potentially deleterious actions to persist during the prolonged periods in which neurons in HIV-1 infected brain would be exposed to Tat.
Tat induces dramatic increases in levels of intracellular calcium in neurons. There is an initial brief burst of intracellular calcium release through IP-3 sensitive pools followed by prolonged increases in cytoplasmic calcium resulting from an influx of extracellular calcium (Haughey et al. 1999). This is followed by mitochondrial calcium uptake, inhibition of complex IV of the electron transport chain, generation of reactive oxygen species, activation of caspases, and eventually results in apoptosis (Kruman et al. 1998; New et al. 1997; Norman et al. 2007). Tat-induced neuronal cell death can be prevented by excitatory amino acid receptor antagonists (Brailoiu et al. 2008; Magnuson et al. 1995), inhibitors of nitric oxide synthase and caspases, antioxidants, and agents that stabilize mitochondrial membrane permeability and IP-3 pools of intracellular calcium (Haughey et al. 1999; Kruman et al. 1998; Perry et al. 2005) and inhibition of glycogen synthase kinase-3beta by lithium (Maggirwar et al. 1999; Sui et al. 2006). Down regulation of PTEN (phosphatase and tensin homolog deleted on chromosome 10), which is located upstream of glycogen synthase kinase-3beta also protects against Tat neurotoxicity (Zhao et al. 2007). Tat also induces rapid loss of calcium from endoplasmic reticulum mediated by the ryanodine receptor, followed by the unfolded protein response and pathologic dilatation of the endoplasmic reticulum in cortical neurons. These morphological features can also be seen in brain from patients with HIV encephalitis (Norman et al. 2008). Evidence of oxidative stress is also noted in vivo upon intrastriatal injections of Tat (Aksenov et al. 2001). Interestingly, Tat expression in glial cells has an antioxidative effect on the glial cells, however, upon release from these cells it causes oxidative stress and toxicity in neurons (Chauhan et al. 2003). Hence these toxic effects are specific for neuronal cells. Other mechanisms have also been implicated in Tat-induced neurotoxicity. It has been shown that Tat can induce the expression of SDF-1 in neurons, which in turn upon release can cause neurotoxicity in other neurons (Langford et al. 2002). Tat can synergize with other toxins such as gp120 (Bansal et al. 2000; Nath et al. 2000), glutamate (Wang et al. 1999), and drugs of abuse to cause neurotoxicity (Nath et al. 2002). Following a brief exposure to Tat, subsequent application of physiological levels of glutamate can cause massive derangement in intracellular calcium suggesting that the protein can sensitize the neurons to neurotoxic substances (Nath et al. 2000). In contrast, Tat may inhibit glutamate-mediated changes in intracellular calcium in astrocytes (Koller et al. 2001). The cysteine rich region and the basic domain seem to be critical for causing neurotoxicity (Gourdou et al. 1990; Nath et al. 1996; Weeks et al. 1995). Neurotrophic factors can also protect against Tat-induced neurotoxicity through the induction of anti-apoptotic genes (Ramirez et al. 2001). Another mechanism by which Tat may affect synaptic function is by dysregulation of selected microRNAs, particularly neuronal mir-128, in primary cortical neurons, which further inhibits expression of the pre-synaptic protein SNAP25 (Eletto et al. 2008).
Tat also induces marked aggregation of neurons and astrocytes in developing cultures and causes the neuritic processes to coalesce into fascicles. These effects have been mapped to the RGD (arginine–glycine–aspartic acid) sequence within the second exon (Kolson et al. 1993). These observations may be important not only for the developing brain but also in adults as it may impair neurogenesis and gliogenesis.
Effect of Tat on Glial Cell Function
Besides direct neurotoxicity, Tat can also cause neurotoxicity by the release of neurotoxic substances from glial cells and macrophages. Tat can also alter glial cell function, which leads to loss of support function for neurons. For example, intraventricular injection of Tat showed prominent glial cell activation and infiltration of perivascular macrophages (Jones et al. 1998). Tat also has a number of effects on glial cell function. It stimulates the production of pro-inflammatory cytokines in the brain (Chen et al. 1997) (Pulliam et al. 2007) and neurotoxins in these cells. Tat induces a milieu of cytokines and chemokines in macrophages and astrocytes (D’Aversa et al. 2004; Kutsch et al. 2000; Weiss et al. 1999). Significant among these are tumor necrosis factor-α (TNF-α), monocyte chemoattractant factor-1 (MCP-1)/CCL-2, and CXCL-10 (Eugenin et al. 2005; McManus et al. 2000). In fact, Tat is more potent than even lipopolysaccharide (LPS) in inducing TNF-α production (Chen et al. 1997) and may act on the TNF-α promoter (Darbinian et al. 2001). Cytokine induction in both cell types is NF-κB dependent (Conant et al. 1996). The epitope of Tat that induces TNF-α is different from the one that causes neurotoxicity (Buscemi et al. 2007). Tat-induced TNF-α can mediate neurotoxicity (New et al. 1998; Shi et al. 1998; Sui et al. 2007). MCP-1/CCL-2 is a highly potent chemoattractant for monocytes. Levels of this chemokine are elevated in the CSF and brain of patients with HIV dementia (Conant et al. 1998). Interestingly, MCP-1/CCL-2 may protect neurons against Tat-induced neurotoxicity suggesting that this chemokine may act as a double-edged sword (Eugenin et al. 2003). Tat-mediated MCP-1/CCL-2 production in astrocytes is mediated via kappa opiate receptors (Sheng et al. 2003). Tat can synergize with gamma interferon to produce CXCL-10 (Dhillon et al. 2008) and can also induce endothelin-1 in astrocytes (Chauhan et al. 2007). Tat also induces matrix metalloproteinases (MMP) expression in astrocytes, which also facilitates monocyte transmigration by degradation of the extracellular matrix (Johnston et al. 2001). Quinolinic acid production which is an excitotoxin can also be induced by macrophages by Tat (Smith et al. 2001). Together, these studies suggest that Tat may be an important mediator of the inflammatory response in the brain. In a Tat transgenic model it was shown that ginkgo biloba extract could attenuate glial cell activation (Zou et al. 2007).
Differences in neurotoxic potential have been shown between Tat derived from patients with HIV dementia when compared to those without dementia. For example macrophages expressing Tat from patients with HIV dementia exhibited elevated matrix metalloproteinase-2 and -7 releases and caused neurotoxicity, but cells expressing Tat from non-demented patients did not exhibit enhanced MMP expression or cause neurotoxicity (Johnston et al. 2001). Tat can synergize with gamma interferon to induce iNOS expression in microglia (Polazzi et al. 1999). It can also induce in astrocytes (Liu et al. 2002). Independent of its effects on nitric oxide, Tat can also potently decrease cyclic AMP levels in microglia. This effect was not noted in astrocytes (Patrizio et al. 2001).
Effect of Tat on Blood Brain Barrier
The blood brain barrier is formed of endothelial cells on the capillary luminal surface and astrocyte foot processes on the ablumenal surface. There is a basement membrane between the cells and the endothelial cells themselves are connected by tight junctions. Hence any compromise in the endothelial or astrocytic cell function could impair the blood brain barrier leading to influx of serum proteins and leukocytes. Tat is capable of crossing the intact blood brain barrier by a non-saturable mechanism with an unidirectional influx rate of about 0.490 microl/g/min. About 0.126% of an intravenous dose of Tat enters each gram of brain (Banks et al. 2005). Additionally, prolonged exposure of brain-derived endothelial cells to Tat may cause apoptosis (Kim et al. 2003b) or oxidative stress (Toborek et al. 2003). Tat can also induce IL-6 (Zidovetzki et al. 1998) and IL-8 (Hofman et al. 1999) expression on endothelial cells. Tat may also affect trafficking of leukocytes into the brain by inducing the expression of adhesion molecules, VCAM-1 and ICAM-1 in astrocytes (Woodman et al. 1999). Tat-induced cytokine dysregulation in endothelial cells can be prevented by agonists of nuclear receptors, the peroxisome proliferator–activated receptors (Huang et al. 2008) or by simvastatin (Andras et al. 2008). Tat can also decrease the expression of several tight junction proteins an effect that is mediated via calveolin-1 (Zhong et al. 2008). In contrast, cyclooxygenase-2 inhibitors attenuate Tat-induced alterations of occludin expression but have no effect on Tat-induced down regulation of zona occudens-1 expression or on increased blood brain barrier permeability (Pu et al. 2007). Importantly, Tat induces the expression of P-glycoprotein and multidrug resistance-associated protein-1 on brain endothelial cells and astrocytes which may have implications for delivery of antiretroviral drugs to the brain, since these efflux systems could prevent the CNS entry of these compounds (Hayashi et al. 2006; Hayashi et al. 2005). Further, as discussed above, some of the effects of Tat on chemokine expression, which may attract monocytes into the brain, matrix metalloproteinase production, which may degrade the extracellular matrix and alteration of astrocyte function may also impair the blood brain barrier. It remains unknown what is the net effect might be of these seemingly opposite effects, i.e., break down of the blood brain barrier and the upregulation of multidrug resistance-associated protein on drug delivery to the CNS.
Tat as a Chemoattractant
Beside the ability of Tat to induce the production of chemokines and chemokine receptors, Tat may itself have some chemoattractant properties particularly for monocytes (Albini et al. 1998). Significant sequence homology has been shown for Tat with several of the chemokines in key residues of functional importance in chemokines. These include a CCF motif, an SYXR motif, which determines CXC/CC chemokine cell type specificity (Lusti-Narasimhan et al. 1995) as well as a strongly conserved isoleucine. The greatest similarity is noted with the MCP/CCL family of chemokines (Albini et al. 1998). Mutations in CC motif of Tat, particularly Cys in position 31 as is found naturally in clade C Tat, impairs its chemotactic properties (Ranga et al. 2004). Consistent with this observation, it is able to bind to CCR2 and CCR3 (Albini et al. 1998).
Differences Between Tat from Various HIV Clades in Mediating Neuropathogenesis
HIV clade D found in Africa is highly virulent. Tat protein from this clade is a very potent transactivator of HIV replication and these functional changes are attributed to mutation in amino acids 61 S/G, 63 T/Q, and 67 S/D. These changes result in binding to TAR with higher affinity and prevent inactivation by a protein kinase called PKR. PKR can phosphorylate Tat and inhibit its binding to TAR (McMillan et al. 1995; Peloponese et al. 1999). Nuclear magnetic resonance structure shows that the major difference between clade B and D is the presence of a short alpha helix in clade D-Tat in region V, which is replaced by two beta turns in clade B-Tat (Gregoire et al. 2001). Several studies have compared clade B- and C-Tat for their neuropathogenic properties. HIV clade B is found mainly in North America, Western Europe, and Australia, while Clade C virus is found predominantly in Asia and Eastern Africa. Clade C Tat has a unique mutation at position 31 where a Cys is mutated to a Ser. This mutation is not found in other clades, however, as a result of this mutation there is attenuation of the chemotactic (Ranga et al. 2004) and neurotoxic (Mishra et al. 2008) properties of Tat, and its ability to directly bind and activate the NMDA receptor on neurons (Li et al. 2008). Further, mice injected with macrophages infected with HIV clade C virus also show less cognitive abnormalities compared to clade B virus (Rao et al. 2008).
Interaction of Tat with Drugs of Abuse in Mediating Neuronal Injury
Methamphetamine and cocaine synergize with Tat to cause increased neurotoxicity (Turchan et al. 2001) (Cai and Cadet 2008). An in vivo study demonstrated the synergism between methamphetamine and Tat (Maragos et al. 2002). Animals treated with methamphetamine alone showed only a 7% reduction in striatal dopamine levels and Tat-treated animals showed only an 8% decline, but animals treated with both methamphetamine and Tat demonstrated a 65% reduction in striatal dopamine. This study might be particularly relevant, because the doses of methamphetamine and Tat used were equivalent to what might be seen in human disease. Subsequent microdialysis studies in this same animal model showed that the synergistic reduction in striatal dopamine is accompanied by significant decrease in dopamine release from the striatum (Cass et al. 2003) and a decrease in dopamine transporter due to loss of dopamine terminals (Theodore et al. 2007, 2006b). Another possible mechanism for HIV-methamphetamine or cocaine interaction is via oxidative stress. Cocaine decreases mitochondrial respiration and increases the production of reactive oxygen species in animals (Boess et al. 2000). In one study (Flora et al. 2003), administration of either Tat or methamphetamine to mice increased markers of oxidative stress, including redox-regulated transcription factors, in cortical, striatal, and hippocampal brain regions. Furthermore, the DNA-binding activities of these transcription factors were greater in mice injected with both Tat and methamphetamine, than with either Tat or methamphetamine alone. This same study also suggested that Tat and methamphetamine may interact through changes in cell signaling and cytokine/chemokine expression. Mice treated with both agents had synergistic upregulation of intercellular adhesion molecule-1 (ICAM-1), tumor necrosis factor-alpha, and interleukin-1beta gene expression compared to mice treated with either agent alone. Interestingly, knock out animals lacking both receptors of tumor necrosis factor had no effect on dopamine levels when treated with Tat, suggesting that the Tat-mediated increase in tumor necrosis factor may contribute to the loss of dopaminergic terminals (Theodore et al. 2006a). Another study showed that Tat and methamphetamine interact to cause damage to calbindin-immunoreactive non-pyramidal neurons by dysregulating mitochondrial calcium metabolism, associated with increased levels of oxidative stress (Langford et al. 2004).
Morphine is the active metabolite of heroin and remarkable synergistic effects of Tat and morphine have been reported in glial cells. Sustained exposure to morphine and Tat causes dysfunction and death of both glial precursors and astrocytes, mediated by mu-opioid receptors through the activation of caspase-3 (Khurdayan et al. 2004). Similar changes has been reported in oligodendrocytes which also express the mu-opioid receptor (Hauser et al. 2009). Furthermore, recent studies have implicated astroglia as mediators for the proinflammatory effects of opiates in HIV-infected individuals. Combined opiate and Tat exposure synergistically destabilizes levels of intracellular calcium, increases reactive oxygen species, and causes massive release of proinflammatory chemokines in cultured striatal astroglia (El-Hage et al. 2005). The released chemokines include monocyte chemoattractant protein-1 (MCP-1) or CCL-2 and RANTES. MCP-1 triggers an influx of monocyte/macrophages and microglial activation.
Interplay Between HIV-Tat and Other Neurotropic Viruses
Herpes Viruses
Complex interactions occur with selective herpes viruses such as cytomegalovirus (CMV), human herpesvirus-6 (HHV-6), and human herpesvirus-8 (HHV-8), however, no effect has been reported with other herpes viruses such as HSV-1 and HSV-2. In brain infections with HIV, co-infection with other viruses has been seen and synergistic association for severity or attenuation of the disease is reported. CMV and HHV-6 infection stimulate HIV replication and transactivated the HIV-1 promoter (the long terminal repeat or LTR) in astrocytes (McCarthy et al. 1998). The level of this transactivation with immediate early genes, IE1/IE2, is similar to that seen following co-transfection with a Tat expression vector. Tat and HCMV IE1/IE2 have a synergistic effect on HIV-LTR transcription, however, Tat effect or synergistic effect is down regulated by another HCMV product, UL44. Co-infection of HIV and HHV-6 and its association with the severity of the disease has been seen in the brain. Tat upregulates HHV-6 replication by directly binding to the HHV-6 promoter or indirectly via activation of cellular factors.
JC Virus
Another viral infection seen with HIV is JCV, where Tat plays a prominent role in activation of JCV in glial cells. HIV-Tat can be detected in various JCV infected cells as well as in uninfected oligodendrocytes from patients with PML and HIV infection, supporting the earlier in vitro findings that secreted Tat from the infected cells can be localized in the neighboring uninfected cells. The presence of Tat in oligodendrocytes is particularly interesting as this protein can up-modulate JCV gene transcription and several key cell cycle regulatory proteins including cyclin E, Cdk2, and pRb (Valle et al. 2000). JCV contains sequences in the 5′ end of the late RNA species with an extensive homology to HIV TAR. Site directed mutagenesis studies show that critical G residues required for the function of HIV TAR that are conserved in the JCV TAR homolog play an important role in Tat activation of the JCV promoter. In addition, in vivo competition studies suggest that shared regulatory components mediate Tat activation of the JCV late and HIV-LTR promoters. These results suggest that the TAR homolog of the JCV late promoter is responsive to HIV-Tat induction and thus may participate in the overall activation of the JCV late promoter mediated by this transactivation (Chowdhury et al. 1992, 1990). Further, JCV activation at transcriptional level is mediated by interaction of several inducible regulatory proteins such as NF-κB, C Jun/Ap-1, and NF-1 (Amemiya et al. 1989, 1992; Wortman et al. 2000). These regulatory proteins can be induced by HIV-Tat protein in glial cells or by cytokines that are induced by HIV proteins in glial cells (Atwood et al. 1995; Chen et al. 1997; Conant et al. 1996). The ability of Tat protein to enter into other cells as well as to induce production of cytokines, may thus make the latent JCV in oligodendrocytes or astrocytes target for activation and may thus be involved in the neuropathogenesis of PML in patients with HIV infection.
Conversely, infection of human astrocytes with HIV and JCV show a decrease in the level of HIV replication in cells that are coinfected with JCV. The agnoprotein of JCV through its N-terminal domain associates with Tat and the interaction causes the suppression of Tat-mediated HIV replication (Kaniowska et al. 2006). This could potentially promote the development of a latent reservoir of HIV in the brain.
Neurotoxic Properties of Tat Protein from Other Retroviruses
The Tat protein derived from Maedi Visna virus that infects sheep has also been shown to be neurotoxic in a variety of in vitro and in vivo assays (Gourdou et al. 1990). Although this protein has not been studied as extensively as the HIV-Tat protein, similar mechanisms seem to be involved such as the stimulation of excitatory amino acid receptors (Starling et al. 1999), influx of extracellular calcium (Strijbos et al. 1995), and induction of nitric oxide synthase (Hayman et al. 1993). Similarly, the Tax protein of human T-cell leukemia virus type-I can induce the production of cytokines and chemokines in glial cells (Arai et al. 1998; Szymocha et al. 2000) and brain endothelial cells (Rott et al. 1993) and may result in neurotoxicity and destruction of myelin producing cells (Ohya et al. 1997).
Tat as a Target for Drug Development
Currently, available antiretroviral drugs have no effect on Tat production once the HIV proviral DNA is integrated. Hence there is a critical need to develop compounds that may antagonize Tat function (see Table 1). Merck Pharmaceuticals screened a panel of natural compounds and discovered durhamycin (DurA) as a potent Tat inhibitor (IC 50 = 4.8 nM) (Jayasuriya et al. 2002). Methods for its synthesis have recently been developed (Pragani and Roush 2008). Structural analogs of durhamycin were also isolated and evaluated for antagonism of Tat transactivation, with durhamycin B demonstrating one-tenth the potency as DurA and the DurA aglycone was inactive at 25 μM (Pragani and Roush 2008). However, it has a molecular mass of 1307 Da, and numerous saccharide moieties, which would likely make it too big and polar to cross the blood brain barrier. Another group has used a structure-based drug design or computer-aided drug discovery approach to generate a 2D-NMR structure of Tat and synthesized a series of compounds called TDS that bind to Tat and inhibit HIV replication. This family of compounds consists of a triphenylene aromatic ring substituted with at least one carbon chain bearing a succinimide group which could occupy the hydrophobic pocket of Tat (Montembault et al. 2004). However, toxicity in leukocytes of the lead compound TDS2 may require additional modifications to these compounds (Montembault et al. 2004). Polyarginine containing peptoid compounds mimicking the arginine-rich basic domain of Tat, such as CGP 74026 (Klimkait et al. 1998) bind potently to TAR with nanomolar potency and block Tat dependent LTR transactivation around 1 μM. Other prior approaches had included the development of a benzodiazepine derivative (Ro5-3335 and Ro24-7429) as a potential Tat inhibitor (Hsu et al. 1991, 1992), but it turned out that this compound was not binding to Tat but to cyclin T, a cellular co-factor essential for Tat (Hsu et al. 1993). Another study described a tetrahydropyrimidine derivative (THP A) able to bind to a polyarginine peptide (Lapidot et al. 1995). This polyarginine peptide binds to TAR but there was no evidence that tetrahydropyrimidine could bind to the basic region of Tat. Similarly, keto/enol epoxy steroids were found to act as Tat inhibitors, with potencies to block Tat-mediated LTR transactivation and HIV viral replication in the 2–3 μM range (Michne et al. 1995). Another group discovered that sulfonated stilbene derivatives (CGA137053) were capable of inhibiting Tat-TAR interactions in vitro at 3–61–10 nm concentrations, by binding to Tat protein with nanomolar potency (Hamy et al. 2000). However, low micromolar concentrations of CGA137053 were required to block Tat-mediated LTR transactivation and HIV infection in leukocytes and macrophages. However, a number of studies describe molecules that bind to TAR and act as Tat competitors. One of these studies (Lind et al. 2002) found compounds, such as prochlorperazine and acetylpromazine that bound to the TAR 5′ bulge and could block Tat-TAR interactions and Tat transactivation at low micromolar ranges. Another compound, CGP40336A, is an acridine derivative that competes with Tat for TAR binding at 22 nM, but requires 1.2 μM to block Tat mediated LTR transactivation (Hamy et al. 1998). The most interesting is a compound called TR87, which inhibits Tat-TAR interactions half maximally at 1 μM. It blocks Tat-mediated LTR transactivation at 1–5 μM, however, it inhibits HIV replication at high concentrations of 5 μM (Hwang et al. 2003). This same group synthesized a TR87 analog, squaryldiamide with a guanidine bioisostere with improved pharmacokinetic properties, but required concentrations of 12 μM to block HIV replication (Lee et al. 2005). These molecules can inhibit only the Tat-TAR interaction but have no effect on the other Tat functions. Another molecule bis-anthracycline WP631, which was developed as a DNA intercalator can also prevent Tat-mediated transcription but these type of compounds would not be expected to have any effect on the extracellular effects of Tat (Kutsch et al. 2004). An ideal compound would be one that binds directly to Tat itself and can also cross the blood brain barrier.
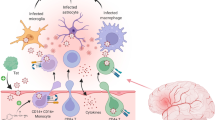
In summary, the Tat protein of HIV is critical for viral replication and survival. The protein has evolved to where it escapes surveillance by the immune system. It can assume various shapes and forms, hence is capable of interacting with a large number of cell surface and intracellular molecules. In brain it continues to evolve to develop potent glial and neuronal activating properties. While glial cell activation results in the induction of various detrimental cytokines and chemokines, neuronal activation results in impaired synaptic function, loss of neurites and eventually in cell death. These complex cascades of events may be self-perpetuating (Fig. 2) hence interruption of these cascades may be of therapeutic potential in patients at risk of developing HIV-associated neurocognitive impairment.

Tat-mediated cascades in HIV neuropathogenesis: Tat may cross the blood brain barrier or be released extracellularly from HIV-infected macrophages/microglia and astrocytes in the brain. Tat may activate uninfected macrophages/microglia or astrocytes to release cytokines, chemokines, or other toxins which may adversely affect neuronal function. Tat itself has chemotactic properties for macrophages which may set up a positive feedback loop. Additionally, Tat may directly interact with neurons causing neurodegeneration of axonal retraction. It may also be transported along neuronal pathways leading to synaptic injury and glial call activation at distant sites
References
Aksenov MY, Hasselrot U, Bansal AK, Wu G, Nath A, Anderson C, Mactutus CF, Booze RM (2001) Oxidative damage induced by the injection of HIV-1 Tat protein in the rat striatum. Neurosci Lett 305:5–8
Albini A, Ferrini S, Benelli R, Sforzini S, Giunciuglio D, Aluigi MG, Proudfoot AE, Alouani S, Wells TN, Mariani G, Rabin RL, Farber JM, Noonan DM (1998) HIV-1 Tat protein mimicry of chemokines. Proc Natl Acad Sci USA 95:13153–13158
Amemiya K, Traub R, Durham L, Major EO (1989) Interaction of a nuclear factor-1-like protein with the regulatory region of the human polyomavirus JC virus. J Biol Chem 264:7025–7032
Amemiya K, Traub R, Durham L, Major EO (1992) Adjacent nuclear factor-1 and activator protein binding sites in the enhancer of the neurotropic JC virus. A common characteristic of many brain-specific genes. J Biol Chem 267:14204–14211
Andras IE, Rha G, Huang W, Eum S, Couraud PO, Romero IA, Hennig B, Toborek M (2008) Simvastatin protects against amyloid beta and HIV-1 Tat-induced promoter activities of inflammatory genes in brain endothelial cells. Mol Pharmacol 73:1424–1433
Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE (2007) Updated research nosology for HIV-associated neurocognitive disorders. Neurology 69:1789–1799
Arai M, Ohashi T, Tsukahara T, Murakami T, Hori T, Uchiyama T, Yamamoto N, Kannagi M, Fujii M (1998) Human T-cell leukemia virus type 1 Tax protein induces the expression of lymphocyte chemoattractant SDF-1/PBSF. Virology 241:298–303
Atwood WJ, Wang L, Durham LC, Amemiya K, Traub RG, Major EO (1995) Evaluation of the role of cytokine activation in the multiplication of JC virus (JCV) in human fetal glial cells. J Neurovirol 1:40–49
Banks WA, Robinson SM, Nath A (2005) Permeability of the blood-brain barrier to HIV-1 Tat. Exp Neurol 193:218–227
Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM (2000) Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res 879:42–49
Barillari G, Gendelman R, Gallo RC, Ensoli B (1993) The Tat protein of HIV, a growth factor for AIDS Kaposi sarcoma and cytokine activated vascular cells, induces adhesion of the same cell types by using integrin receptors recognizing the RGD amino acid sequence. Proc Natl Acad Sci USA 90:7941–7945
Bilodeau PS, Domsic JK, Stoltzfus CM (1999) Splicing regulatory elements within tat exon 2 of human immunodeficiency virus type 1 (HIV-1) are characteristic of group M but not group O HIV-1 strains. J Virol 73:9764–9772
Boess F, Ndikum-Moffor FM, Boelsterli UA, Roberts SM (2000) Effects of cocaine and its oxidative metabolites on mitochondrial respiration and generation of reactive oxygen species. Biochem Pharmacol 60:615–623
Boven LA, Noorbakhsh F, Bouma G, van der Zee R, Vargas DL, Pardo C, McArthur JC, Nottet HS, Power C (2007) Brain-derived human immunodeficiency virus-1 Tat exerts differential effects on LTR transactivation and neuroimmune activation. J Neurovirol 13:173–184
Brailoiu GC, Brailoiu E, Chang JK, Dun NJ (2008) Excitatory effects of human immunodeficiency virus 1 Tat on cultured rat cerebral cortical neurons. Neuroscience 151:701–710
Bratanich AC, Liu C, McArthur JC, Fudyk T, Glass JD, Mittoo S, Klassen GA, Power C (1998) Brain-derived HIV-1 tat sequences from AIDS patients with dementia show increased molecular heterogeneity. J Neurovirol 4:387–393
Brorson JR, Manzolillo PA, Gibbons SJ, Miller RJ (1995) AMPA receptor desensitization predicts the selective vulnerability of cerebellar Purkinje cells to excitotoxicity. J Neurosci 15:4515–4524
Bruce-Keller AJ, Chauhan A, Dimayuga FO, Gee J, Keller JN, Nath A (2003) Synaptic transport of human immunodeficiency virus-Tat protein causes neurotoxicity and gliosis in rat brain. J Neurosci 23:8417–8422
Buscemi L, Ramonet D, Geiger JD (2007) Human immunodeficiency virus type-1 protein Tat induces tumor necrosis factor-alpha-mediated neurotoxicity. Neurobiol Dis 26:661–670
Cai NS, Cadet JL (2008) The combination of methamphetamine and of the HIV protein, Tat, induces death of the human neuroblastoma cell line, SH-SY5Y. Synapse 62:551–552
Cass WA, Harned ME, Peters LE, Nath A, Maragos WF (2003) HIV-1 protein Tat potentiation of methamphetamine-induced decreases in evoked overflow of dopamine in the striatum of the rat. Brain Res 984:133–142
Chang HC, Samaniego F, Nair BC, Buonaguro L, Ensoli B (1997) HIV-1 tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycan through its basic region. AIDS 11:1421–1431
Chauhan A, Turchan J, Pocernich C, Bruce-Keller A, Roth S, Butterfield DA, Major EO, Nath A (2003) Intracellular human immunodeficiency virus Tat expression in astrocytes promotes astrocyte survival but induces potent neurotoxicity at distant sites via axonal transport. J Biol Chem 278:13512–13519
Chauhan A, Hahn S, Gartner S, Pardo CA, Netesan SK, McArthur J, Nath A (2007) Molecular programming of endothelin-1 in HIV-infected brain: role of Tat in up-regulation of ET-1 and its inhibition by statins. FASEB J 21:777–789
Chen P, Mayne M, Power C, Nath A (1997) The Tat protein of HIV-1 induces tumor necrosis factor-a production: implications for HIV associated neurological diseases. J Biol Chem 272:22385–22388
Cheng J, Nath A, Knudsen B, Hochman S, Geiger JDMM, Magnuson DSK (1998) Neuronal excitatory properties of human immunodeficiency virus type 1 tat protein. Neuroscience 82:97–106
Chowdhury M, Taylor JP, Tada H, Rappaport J, Wong-Staal F, Amini S, Khalili K (1990) Regulation of the human neurotropic virus promoter by JCV-T antigen and HIV-1 tat protein. Oncogene 5:1737–1742
Chowdhury M, Taylor JP, Chang CF, Rappaport J, Khalili K (1992) Evidence that a sequence similar to TAR is important for induction of the JC virus late promoter by human immunodeficiency virus type 1 Tat. J Virol 66:7355–7361
Conant K, Ma M, Nath A, Major EO (1996) Extracellular HIV-1 Tat protein is associated with an increase in both NF-kappa B binding and protein kinase C activity in primary human astrocytes. J Virol 70:1384–1389
Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO (1998) Induction of monocyte chemotactic protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA 95:3117–3121
Coyle-Rink J, Sweet T, Abraham S, Sawaya B, Batuman O, Khalili K, Amini S (2002) Interaction between TGFbeta signaling proteins and C/EBP controls basal and Tat-mediated transcription of HIV-1 LTR in astrocytes. Virology 299:240–247
D’Aversa TG, Yu KO, Berman JW (2004) Expression of chemokines by human fetal microglia after treatment with the human immunodeficiency virus type 1 protein Tat. J Neurovirol 10:86–97
D’Orso I, Frankel AD (2009) Tat acetylation modulates assembly of a viral-host RNA-protein transcription complex. Proc Natl Acad Sci USA 106:3101–3106
Darbinian N, Sawaya BE, Khalili K, Jaffe N, Wortman B, Giordano A, Amini S (2001) Functional interaction between cyclin T1/cdk9 and Puralpha determines the level of TNFalpha promoter activation by Tat in glial cells. J Neuroimmunol 121:3–11
Deng L, Ammosova T, Pumfery A, Kashanchi F, Nekhai S (2002) HIV-1 Tat interaction with RNA polymerase II C-terminal domain (CTD) and a dynamic association with CDK2 induce CTD phosphorylation and transcription from HIV-1 promoter. J Biol Chem 277:33922–33929
Dhillon N, Zhu X, Peng F, Yao H, Williams R, Callen S, Ladner AO, Buch S, Qiu J (2008) Molecular mechanism(s) involved in the synergistic induction of CXCL10 by human immunodeficiency virus type 1 Tat and interferon-gamma in macrophages. J Neurovirol 14:196–204
Eletto D, Russo G, Passiatore G, Del Valle L, Giordano A, Khalili K, Gualco E, Peruzzi F (2008) Inhibition of SNAP25 expression by HIV-1 Tat involves the activity of mir-128a. J Cell Physiol 216:764–770
El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF (2005) Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia 50:91–106
Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan R, Wingfield P, Gallo R (1993) Release, uptake, and effects of extracellular human immunodeficiency virus type-1 Tat protein on cell growth and viral replication. J Virol 67:277–287
Eugenin EA, D’Aversa TG, Lopez L, Calderon TM, Berman JW (2003) MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem 85:1299–1311
Eugenin EA, Dyer G, Calderon TM, Berman JW (2005) HIV-1 tat protein induces a migratory phenotype in human fetal microglia by a CCL2 (MCP-1)-dependent mechanism: possible role in NeuroAIDS. Glia 49:501–510
Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW (2007) HIV-tat induces formation of an LRP-PSD-95-NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci USA 104:3438–3443
Everall I, Barnes H, Spargo E, Lantos P (1995) Assessment of neuronal density in the putamen in human immunodeficiency virus (HIV) infection. Application of stereology and spatial analysis of quadrats. J Neurovirol 1:126–129
Ferris MJ, Frederick-Duus D, Fadel J, Mactutus CF, Booze RM (2009) In vivo microdialysis in awake, freely moving rats demonstrates HIV-1 Tat-induced alterations in dopamine transmission. Synapse 63:181–185
Fitting S, Booze RM, Mactutus CF (2008) Neonatal intrahippocampal injection of the HIV-1 proteins gp120 and Tat: differential effects on behavior and the relationship to stereological hippocampal measures. Brain Res 1232:139–154
Fittipaldi A, Ferrari A, Zoppe M, Arcangeli C, Pellegrini V, Beltram F, Giacca M (2003) Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem 278:34141–34149
Flora G, Lee YW, Nath A, Hennig B, Maragos W, Toborek M (2003) Methamphetamine potentiates HIV-1 Tat protein-mediated activation of redox-sensitive pathways in discrete regions of the brain. Exp Neurol 179:60–70
Ford KG, Darling D, Souberbielle B, Farzaneh F (2000) Protein transduction: a new tool for the study of cellular ageing and senescence. Mech Ageing Dev 121:113–121
Frankel AD, Pabo CO (1988) Cellular uptake of the tat protein from human immunodeficiency virus. Cell 55:1189–1193
Frankel A, Bredt D, Pabo C (1988) Tat protein from immunodeficiency virus forms a metal-linked dimer. Science 240:70–73
Garthwaite J (1991) Glutamate, nitric oxide and cell–cell signalling in the nervous system. Trends Neurosci 14:60–67
Gourdou I, Mabrouk K, Harkiss G, Marchot P, Watt N, Hery F, Vigne R (1990) Neurotoxicity in mice due to cysteine-rich parts of visna virus and HIV-1 Tat proteins. C R Acad Sci III 311:149–155
Gregoire C, Peloponese JM Jr, Esquieu D, Opi S, Campbell G, Solomiac M, Lebrun E, Lebreton J, Loret EP (2001) Homonuclear (1)H-NMR assignment and structural characterization of human immunodeficiency virus type 1 Tat Mal protein. Biopolymers 62:324–335
Hamy F, Brondani V, Florsheimer A, Stark W, Blommers MJ, Klimkait T (1998) A new class of HIV-1 Tat antagonist acting through Tat-TAR inhibition. Biochemistry 37:5086–5095
Hamy F, Gelus N, Zeller M, Lazdins JL, Bailly C, Klimkait T (2000) Blocking HIV replication by targeting Tat protein. Chem Biol 7:669–676
Harrich D, Ulich C, Garcia-Martinez LF, Gaynor RB (1997) Tat is required for efficient HIV-1 reverse transcription. EMBO J 16:1224–1235
Haughey NJ, Holden CP, Nath A, Geiger JD (1999) Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J Neurochem 73:1363–1374
Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD (2001) HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem 78:457–467
Hauser KF, Hahn YK, Adjan VV, Zou S, Buch SK, Nath A, Bruce-Keller AJ, Knapp PE (2009) HIV-1 Tat and morphine have interactive effects on oligodendrocyte survival and morphology. Glia 57:194–206
Hayashi K, Pu H, Tian J, Andras IE, Lee YW, Hennig B, Toborek M (2005) HIV-Tat protein induces P-glycoprotein expression in brain microvascular endothelial cells. J Neurochem 93:1231–1241
Hayashi K, Pu H, Andras IE, Eum SY, Yamauchi A, Hennig B, Toborek M (2006) HIV-TAT protein upregulates expression of multidrug resistance protein 1 in the blood-brain barrier. J Cereb Blood Flow Metab 26:1052–1065
Hayman M, Arbuthnott G, Harkiss G, Brace H, Filippi P, Philippon V, Thomson D, Vigne R, Wright A (1993) Neurotoxicity of peptide analogues of the transactivating protein tat from Maedi-Visna virus and human immunodeficiency virus. Neuroscience 53:1–6
Hofman FM, Dohadwala MM, Wright AD, Hinton DR, Walker SM (1994) Exogenous tat protein activates central nervous system-derived endothelial cells. J Neuroimmunol 54:19–28
Hofman FM, Chen P, Incardona F, Zidovetzki R, Hinton DR (1999) HIV-1 tat protein induces the production of interleukin-8 by human brain-derived endothelial cells. J Neuroimmunol 94:28–39
Hsu MC, Schutt AD, Holly M, Slice LW, Sherman MI, Richman DD, Potash MJ, Volsky DJ (1991) Inhibition of HIV replication in acute and chronic infections in vitro by a Tat antagonist. Science 254:1799–1802
Hsu MC, Schutt AD, Holly M, Slice LW, Sherman MI, Richman DD, Potash MJ, Volsky DJ (1992) Discovery and characterization of an HIV-1 Tat antagonist. Biochem Soc Trans 20:525–531
Hsu MC, Dhingra U, Earley JV, Holly M, Keith D, Nalin CM, Richou AR, Schutt AD, Tam SY, Potash MJ et al (1993) Inhibition of type 1 human immunodeficiency virus replication by a tat antagonist to which the virus remains sensitive after prolonged exposure in vitro. Proc Natl Acad Sci USA 90:6395–6399
Huang L, Bosch I, Hofmann W, Sodroski J, Pardee AB (1998) Tat protein induces human immunodeficiency virus type 1 (HIV-1) co-receptors and promotes infection with both macrophage-tropic and T-lymphotropic HIV-1 strains. J Virol 72:8952–8960
Huang X, Seifert U, Salzmann U, Henklein P, Preissner R, Henke W, Sijts AJ, Kloetzel PM, Dubiel W (2002) The RTP site shared by the HIV-1 Tat protein and the 11S regulator subunit alpha is crucial for their effects on proteasome function including antigen processing. J Mol Biol 323:771–782
Huang W, Rha GB, Han MJ, Eum SY, Andras IE, Zhong Y, Hennig B, Toborek M (2008) PPARalpha and PPARgamma effectively protect against HIV-induced inflammatory responses in brain endothelial cells. J Neurochem 107:497–509
Hudson L, Liu J, Nath A, Narayan O, Male D, Jones M, Everall I (2000) Detection of human immunodeficiency virus regulatory protein Tat in CNS tissues. J Neurovirol 6:145–155
Hwang S, Tamilarasu N, Kibler K, Cao H, Ali A, Ping YH, Jeang KT, Rana TM (2003) Discovery of a small molecule Tat-transactivation-responsive RNA antagonist that potently inhibits human immunodeficiency virus-1 replication. J Biol Chem 278:39092–39103
Jayasuriya H, Lingham RB, Graham P, Quamina D, Herranz L, Genilloud O, Gagliardi M, Danzeisen R, Tomassini JE, Zink DL, Guan Z, Singh SB (2002) Durhamycin A, a potent inhibitor of HIV Tat transactivation. J Nat Prod 65:1091–1095
Jeang KT, Xiao H, Rich EA (1999) Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J Biol Chem 274:28837–28840
Johnston JB, Zhang K, Silva C, Shalinsky DR, Conant K, Ni W, Corbett D, Yong VW, Power C (2001) HIV-1 Tat neurotoxicity is prevented by matrix metalloproteinase inhibitors. Ann Neurol 49:230–241
Jonas P, Sakmann B (1992) Glutamate receptor channels in isolated patches from CA1 and CA3 pyramidal cells of rat hippocampal slices. J Physiol (Lond) 455:143–171
Jones M, Olafson K, Del Bigio MR, Peeling J, Nath A (1998) Intraventricular injection of human immunodeficiency virus type 1 (HIV-1) Tat protein causes inflammation, gliosis, apoptosis, and ventricular enlargement. J Neuropathol Exp Neurol 57:563–570
Kaniowska D, Kaminski R, Amini S, Radhakrishnan S, Rappaport J, Johnson E, Khalili K, Del Valle L, Darbinyan A (2006) Cross-interaction between JC virus agnoprotein and human immunodeficiency virus type 1 (HIV-1) Tat modulates transcription of the HIV-1 long terminal repeat in glial cells. J Virol 80:9288–9299
Kaplan IM, Wadia JS, Dowdy SF (2005) Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control Release 102:247–253
Khurdayan VK, Buch S, El-Hage N, Lutz SE, Goebel SM, Singh IN, Knapp PE, Turchan-Cholewo J, Nath A, Hauser KF (2004) Preferential vulnerability of astroglia and glial precursors to combined opioid and HIV-1 Tat exposure in vitro. Eur J Neurosci 19:3171–3182
Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ (2003a) Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol 162:1693–1707
Kim TA, Avraham HK, Koh YH, Jiang S, Park IW, Avraham S (2003b) HIV-1 Tat-mediated apoptosis in human brain microvascular endothelial cells. J Immunol 170:2629–2637
Klimkait T, Felder ER, Albrecht G, Hamy F (1998) Rational optimization of a HIV-1 Tat inhibitor: rapid progress on combinatorial lead structures. Biotechnol Bioeng 61:155–168
Koller H, Schaal H, Freund M, Garrido SR, von Giesen HJ, Ott M, Rosenbaum C, Arendt G (2001) HIV-1 protein Tat reduces the glutamate-induced intracellular Ca2+ increase in cultured cortical astrocytes. Eur J Neurosci 14:1793–1799
Kolson DL, Buchhalter J, Collman R, Hellmig B, Farrell CF, Debouck C, Gonzalez-Scarano F (1993) HIV-1 Tat alters normal organization of neurons and astrocytes in primary rodent brain cell cultures: RGD sequence dependence. AIDS Res Hum Retroviruses 9:677–685
Kruman I, Nath A, Mattson MP (1998) HIV protein Tat induces apoptosis by a mechanism involving mitochondrial calcium overload and caspase activation. Exp Neurol 154:276–288
Kruman II, Nath A, Maragos WF, Chan SL, Jones M, Rangnekar VM, Jakel RJ, Mattson MP (1999) Evidence that Par-4 participates in the pathogenesis of AIDS dementia. Am J Pathol 155:39–46
Kuppuswamy M, Subramanian T, Srinivasan A, Chinnadurai G (1989) Multiple functional domains of Tat, the trans-activator of HIV-1, defined by mutational analysis. Nucleic Acids Res 17:3551–3561
Kutsch O, Oh J, Nath A, Benveniste EN (2000) Induction of the chemokines interleukin-8 and IP-10 by human immunodeficiency virus type 1 tat in astrocytes. J Virol 74:9214–9221
Kutsch O, Levy DN, Bates PJ, Decker J, Kosloff BR, Shaw GM, Priebe W, Benveniste EN (2004) Bis-anthracycline antibiotics inhibit human immunodeficiency virus type 1 transcription. Antimicrob Agents Chemother 48:1652–1663
Langford D, Sanders VJ, Mallory M, Kaul M, Masliah E (2002) Expression of stromal cell-derived factor 1alpha protein in HIV encephalitis. J Neuroimmunol 127:115–126
Langford D, Grigorian A, Hurford R, Adame A, Crews L, Masliah E (2004) The role of mitochondrial alterations in the combined toxic effects of human immunodeficiency virus Tat protein and methamphetamine on calbindin positive-neurons. J Neurovirol 10:327–337
Lapidot A, Ben-Asher E, Eisenstein M (1995) Tetrahydropyrimidine derivatives inhibit binding of a Tat-like, arginine-containing peptide, to HIV TAR RNA in vitro. FEBS Lett 367:33–38
Lee CW, Cao H, Ichiyama K, Rana TM (2005) Design and synthesis of a novel peptidomimetic inhibitor of HIV-1 Tat-TAR interactions: squaryldiamide as a new potential bioisostere of unsubstituted guanidine. Bioorg Med Chem Lett 15:4243–4246
Li ST, Matsushita M, Moriwaki A, Saheki Y, Lu YF, Tomizawa K, Wu HY, Terada H, Matsui H (2004) HIV-1 inhibits long-term potentiation and attenuates spatial learning. Ann Neurol 55:362–371
Li W, Huang Y, Reid R, Steiner J, Malpica-Llanos T, Darden TA, Shankar SK, Mahadevan A, Satishchandra P, Nath A (2008) NMDA receptor activation by HIV-Tat protein is clade dependent. J Neurosci 28:12190–12198
Lind KE, Du Z, Fujinaga K, Peterlin BM, James TL (2002) Structure-based computational database screening, in vitro assay, and NMR assessment of compounds that target TAR RNA. Chem Biol 9:185–193
Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ (2000) Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med 6:1380–1387
Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K (2002) Human immunodeficiency virus type-1 (HIV-1) Tat induces nitric oxide synthase in human astroglia. J Biol Chem 277(42):39312–39319
Lusti-Narasimhan M, Power CA, Allet B, Alouani S, Bacon KB, Mermod JJ, Proudfoot AE, Wells TN (1995) Mutation of Leu25 and Val27 introduces CC chemokine activity into interleukin-8. J Biol Chem 270:2716–2721
Ma M, Nath A (1997) Molecular determinants for cellular uptake of Tat protein of human immunodeficiency virus type 1 in brain cells. J Virol 71:2495–2499
Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S (1999) HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J Neurochem 73:578–586
Magnuson DS, Knudsen BE, Geiger JD, Brownstone RM, Nath A (1995) Human immunodeficiency virus type 1 tat activates non-N-methyl-d-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann Neurol 37:373–380
Malim MH, Cullen BR (1991) HIV-1 structural gene expression requires the binding of multiple Rev monomers to the viral RRE: implications for HIV-1 latency. Cell 65:241–248
Malva JO, Carvalho AP, Carvalho CM (1998) Kainate receptors in hippocampal CA3 subregion: evidence for a role in regulating neurotransmitter release. Neurochem Int 32:1–6
Maragos WF, Young KL, Turchan JT, Guseva M, Pauly JR, Nath A, Cass WA (2002) Human immunodeficiency virus-1 Tat protein and methamphetamine interact synergistically to impair striatal dopaminergic function. J Neurochem 83:955–963
Maragos WF, Tillman P, Jones M, Bruce-Keller AJ, Roth S, Bell JE, Nath A (2003) Neuronal injury in hippocampus with human immunodeficiency virus transactivating protein, Tat. Neuroscience 117:43–53
Marcello A, Zoppe M, Giacca M (2001) Multiple modes of transcriptional regulation by the HIV-1 Tat transactivator. IUBMB Life 51:175–181
Mayne M, Bratanich AC, Chen P, Rana F, Nath A, Power C (1998) HIV-1 tat molecular diversity and induction of TNF-alpha: implications for HIV-induced neurological disease. Neuroimmunomodulation 5:184–192
McCarthy M, Auger D, He J, Wood C (1998) Cytomegalovirus and human herpesvirus-6 transactivate the HIV-1 long terminal repeat via multiple response regions in human fetal astrocytes. J Neurovirol 4:495–511
McManus CM, Weidenheim K, Woodman SE, Nunez J, Hesselgesser J, Nath A, Berman JW (2000) Chemokine and chemokine-receptor expression in human glial elements: induction by the HIV protein, Tat, and chemokine autoregulation. Am J Pathol 156:1441–1453
McMillan NA, Chun RF, Siderovski DP, Galabru J, Toone WM, Samuel CE, Mak TW, Hovanessian AG, Jeang KT, Williams BR (1995) HIV-1 Tat directly interacts with the interferon-induced, double-stranded RNA-dependent kinase, PKR. Virology 213:413–424
Michne WF, Schroeder JD, Bailey TR, Neumann HC, Cooke D, Young DC, Hughes JV, Kingsley SD, Ryan KA, Putz HS et al (1995) Keto/enol epoxy steroids as HIV-1 Tat inhibitors: structure-activity relationships and pharmacophore localization. J Med Chem 38:3197–3206
Mishra M, Vetrivel S, Siddappa NB, Ranga U, Seth P (2008) Clade-specific differences in neurotoxicity of human immunodeficiency virus-1 B and C Tat of human neurons: significance of dicysteine C30C31 motif. Ann Neurol 63:366–376
Montembault M, Vo-Thanh G, Deyine A, Fargeas V, Villieras M, Adjou A, Dubreuil D, Esquieu D, Gregoire C, Opi S, Peloponese JM, Campbell G, Watkins J, de Mareuil J, Aubertin AM, Bailly C, Loret E, Lebreton J (2004) A possible improvement for structure-based drug design illustrated by the discovery of a Tat HIV-1 inhibitor. Bioorg Med Chem Lett 14:1543–1546
Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD (1996) Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol 70:1475–1480
Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD (2000) Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol 47:186–194
Nath A, Hauser KF, Wojna V, Booze RM, Maragos W, Prendergast M, Cass W, Turchan JT (2002) Molecular basis for interactions of HIV and drugs of abuse. J Acquir Immune Defic Syndr 31(suppl 2):S62–S69
Nelbock P, Dillon PJ, Perkins A, Rosen CA (1990) A cDNA for a protein that interacts with the human immunodeficiency virus Tat transactivator. Science 248:1650–1653
New DR, Ma M, Epstein LG, Nath A, Gelbard HA (1997) Human immunodeficiency virus type 1 Tat protein induces death by apoptosis in primary human neuron cultures. J Neurovirol 3:168–173
New DR, Maggirwar SB, Epstein LG, Dewhurst S, Gelbard HA (1998) HIV-1 Tat induces neuronal death via tumor necrosis factor-alpha and activation of non-N-methyl-d-aspartate receptors by a NFkappaB-independent mechanism. J Biol Chem 273:17852–17858
Norman JP, Perry SW, Kasischke KA, Volsky DJ, Gelbard HA (2007) HIV-1 trans activator of transcription protein elicits mitochondrial hyperpolarization and respiratory deficit, with dysregulation of complex IV and nicotinamide adenine dinucleotide homeostasis in cortical neurons. J Immunol 178:869–876
Norman JP, Perry SW, Reynolds HM, Kiebala M, De Mesy Bentley KL, Trejo M, Volsky DJ, Maggirwar SB, Dewhurst S, Masliah E, Gelbard HA (2008) HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS ONE 3:e3731
Ohya O, Tomaru U, Yamashita I, Kasai T, Morita K, Ikeda H, Wakisaka A, Yoshiki T (1997) HTLV-I induced myeloneuropathy in WKAH rats: apoptosis and local activation of the HTLV-I pX and TNF-alpha genes implicated in the pathogenesis. Leukemia 11(suppl 3):255–257
Patrizio M, Colucci M, Levi G (2001) Human immunodeficiency virus type 1 Tat protein decreases cyclic AMP synthesis in rat microglia cultures. J Neurochem 77:399–407
Peloponese JM Jr, Collette Y, Gregoire C, Bailly C, Campese D, Meurs EF, Olive D, Loret EP (1999) Full peptide synthesis, purification, and characterization of six Tat variants. Differences observed between HIV-1 isolates from Africa and other continents. J Biol Chem 274:11473–11478
Perez A, Probert AW, Wang KK, Sharmeen L (2001) Evaluation of HIV-1 Tat induced neurotoxicity in rat cortical cell culture. J Neurovirol 7:1–10
Perry SW, Norman JP, Litzburg A, Zhang D, Dewhurst S, Gelbard HA (2005) HIV-1 transactivator of transcription protein induces mitochondrial hyperpolarization and synaptic stress leading to apoptosis. J Immunol 174:4333–4344
Polazzi E, Levi G, Minghetti L (1999) Human immunodeficiency virus type 1 Tat protein stimulates inducible nitric oxide synthase expression and nitric oxide production in microglial cultures. J Neuropathol Exp Neurol 58:825–831
Pragani R, Roush WR (2008) Studies on the synthesis of durhamycin A: stereoselective synthesis of a model aglycone. Org Lett 10:4613–4616
Prendergast MA, Rogers DT, Mulholland PJ, Littleton JM, Wilkins LH Jr, Self RL, Nath A (2002) Neurotoxic effects of the human immunodeficiency virus type-1 transcription factor Tat require function of a polyamine sensitive-site on the N-methyl-d-aspartate receptor. Brain Res 954:300–307
Pu H, Hayashi K, Andras IE, Eum SY, Hennig B, Toborek M (2007) Limited role of COX-2 in HIV Tat-induced alterations of tight junction protein expression and disruption of the blood-brain barrier. Brain Res 1184:333–344
Pulliam L, Sun B, Rempel H, Martinez PM, Hoekman JD, Rao RJ, Frey WHII, Hanson LR (2007) Intranasal tat alters gene expression in the mouse brain. J Neuroimmune Pharmacol 2:87–92
Ramirez SH, Sanchez JF, Dimitri CA, Gelbard HA, Dewhurst S, Maggirwar SB (2001) Neurotrophins prevent HIV Tat-induced neuronal apoptosis via a nuclear factor-kappaB (NF-kappaB)-dependent mechanism. J Neurochem 78:874–889
Ranga U, Shankarappa R, Siddappa NB, Ramakrishna L, Nagendran R, Mahalingam M, Mahadevan A, Jayasuryan N, Satishchandra P, Shankar SK, Prasad VR (2004) Tat protein of human immunodeficiency virus type 1 subtype C strains is a defective chemokine. J Virol 78:2586–2590
Rao VR, Sas AR, Eugenin EA, Siddappa NB, Bimonte-Nelson H, Berman JW, Ranga U, Tyor WR, Prasad VR (2008) HIV-1 clade-specific differences in the induction of neuropathogenesis. J Neurosci 28:10010–10016
Richard JP, Melikov K, Brooks H, Prevot P, Lebleu B, Chernomordik LV (2005) Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem 280:15300–15306
Robert-Guroff M, Popovic M, Gartner S, Markham P, Gallo RC, Reitz MS (1990) Structure and expression of tat-, rev-, and nef-specific transcripts of human immunodeficiency virus type 1 in infected lymphocytes and macrophages. J Virol 64:3391–3398
Rodman TC, Pruslin FH, To SE, Winston R (1992) Human immunodeficiency virus (HIV) Tat-reactive antibodies present in normal HIV-negative sera and depleted in HIV-positive sera. Identification of the epitope. J Exp Med 175:1247–1253
Rosen CA, Terwilliger E, Dayton A, Sodroski JG, Haseltine WA (1988) Intragenic cis-acting art gene-responsive sequences of the human immunodeficiency virus. Proc Natl Acad Sci USA 85:2071–2075
Rott O, Tontsch U, Fleischer B, Cash E (1993) Interleukin-6 production in “normal” and HTLV-1 tax-expressing brain-specific endothelial cells. Eur J Immunol 23:1987–1991
Ruben S, Perkins A, Purcell R, Joung K, Sia R, Burghoff R, Haseltine WA, Rosen CA (1989) Structural and functional characterization of human immunodeficiency virus tat protein. J Virol 63:1–8
Sabatier JM, Vives E, Marbrouk K et al (1991) Evidence for neurotoxicity of tat from HIV. J Virol 65:961–967
Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF (1999) In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285:1569–1572
Secchiero P, Zella D, Capitani S, Gallo RC, Zauli G (1999) Extracellular HIV-1 tat protein up-regulates the expression of surface CXC-chemokine receptor 4 in resting CD4+ T cells. J Immunol 162:2427–2431
Seeger M, Ferrell K, Frank R, Dubiel W (1997) HIV-1 tat inhibits the 20S proteasome and its 11S regulator-mediated activation. J Biol Chem 272:8145–8148
Sheng WS, Hu S, Lokensgard JR, Peterson PK (2003) U50, 488 inhibits HIV-1 Tat-induced monocyte chemoattractant protein-1 (CCL2) production by human astrocytes. Biochem Pharmacol 65:9–14
Shi B, Raina J, Lorenzo A, Busciglio J, Gabuzda D (1998) Neuronal apoptosis induced by HIV-1 Tat protein and TNF-alpha: potentiation of neurotoxicity mediated by oxidative stress and implications for HIV-1 dementia. J Neurovirol 4:281–290
Silvers JM, Aksenova MV, Aksenov MY, Mactutus CF, Booze RM (2007) Neurotoxicity of HIV-1 Tat protein: involvement of D1 dopamine receptor. Neurotoxicology 28:1184–1190
Smith DG, Guillemin GJ, Pemberton L, Kerr S, Nath A, Smythe GA, Brew BJ (2001) Quinolinic acid is produced by macrophages stimulated by platelet activating factor, Nef and Tat. J Neurovirol 7:56–60
Snyder EL, Dowdy SF (2001) Protein/peptide transduction domains: potential to deliver large DNA molecules into cells. Curr Opin Mol Ther 3:147–152
Spitere K, Toulouse A, O’Sullivan DB, Sullivan AM (2008) TAT-PAX6 protein transduction in neural progenitor cells: a novel approach for generation of dopaminergic neurones in vitro. Brain Res 1208:25–34
Starling I, Wright A, Arbuthnott G, Harkiss G (1999) Acute in vivo neurotoxicity of peptides from Maedi Visna virus transactivating protein Tat. Brain Res 830:285–291
Stauber RH, Pavlakis GN (1998) Intracellular trafficking and interactions of the HIV-1 Tat protein. Virology 252:126–136
Strijbos PJ, Zamani MR, Rothwell NJ, Arbuthnott G, Harkiss G (1995) Neurotoxic mechanisms of transactivating protein Tat of Maedi-Visna virus. Neurosci Lett 197:215–218
Sui Z, Sniderhan LF, Fan S, Kazmierczak K, Reisinger E, Kovacs AD, Potash MJ, Dewhurst S, Gelbard HA, Maggirwar SB (2006) Human immunodeficiency virus-encoded Tat activates glycogen synthase kinase-3beta to antagonize nuclear factor-kappaB survival pathway in neurons. Eur J Neurosci 23:2623–2634
Sui Z, Sniderhan LF, Schifitto G, Phipps RP, Gelbard HA, Dewhurst S, Maggirwar SB (2007) Functional synergy between CD40 ligand and HIV-1 Tat contributes to inflammation: implications in HIV type 1 dementia. J Immunol 178:3226–3236
Szymocha R, Brisson C, Bernard A, Akaoka H, Belin MF, Giraudon P (2000) Long-term effects of HTLV-1 on brain astrocytes: sustained expression of Tax-1 associated with synthesis of inflammatory mediators. J Neurovirol 6:350–357
Tada H, Rappaport J, Lashgari M, Amini S, Wong-Staal F, Khalili K (1990) Trans-activation of the JC virus late promoter by the tat protein of type 1 human immunodeficiency virus in glial cells. Proc Natl Acad Sci USA 87:3479–3483
Tanaka T, Nakamura T, Takagi H, Sato M (1997) Molecular cloning and characterization of a novel TBP-1 interacting protein (TBPIP): enhancement of TBP-1 action on Tat by TBPIP. Biochem Biophys Res Commun 239:176–181
Tardieu M, Hery C, Peudenier S, Boespflug O, Montagnier L (1992) Human immunodeficiency virus type 1-infected monocytic cells can destroy human neural cells after cell-to-cell adhesion. Ann Neurol 32:11–17
Taylor JP, Pomerantz R, Bagasra O, Chowdhury M, Rappaport J, Khalili K, Amini S (1992) TAR-independent transactivation by Tat in cells derived from the CNS: a novel mechanism of HIV-1 gene regulation. EMBO J 11:3395–3403
Theodore S, Cass WA, Nath A, Steiner J, Young K, Maragos WF (2006a) Inhibition of tumor necrosis factor-alpha signaling prevents human immunodeficiency virus-1 protein Tat and methamphetamine interaction. Neurobiol Dis 23:663–668
Theodore S, Stolberg S, Cass WA, Maragos WF (2006b) Human immunodeficiency virus-1 protein tat and methamphetamine interactions. Ann N Y Acad Sci 1074:178–190
Theodore S, Cass WA, Nath A, Maragos WF (2007) Progress in understanding basal ganglia dysfunction as a common target for methamphetamine abuse and HIV-1 neurodegeneration. Curr HIV Res 5:301–313
Thomas ER, Dunfee RL, Stanton J, Bogdan D, Kunstman K, Wolinsky SM, Gabuzda D (2007) High frequency of defective vpu compared with tat and rev genes in brain from patients with HIV type 1-associated dementia. AIDS Res Hum Retroviruses 23:575–580
Toborek M, Lee YW, Pu H, Malecki A, Flora G, Garrido R, Hennig B, Bauer HC, Nath A (2003) HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. J Neurochem 84:169–179
Turchan J, Anderson C, Hauser KF, Sun Q, Zhang J, Liu Y, Wise PM, Kruman I, Maragos W, Mattson MP, Booze R, Nath A (2001) Estrogen protects against the synergistic toxicity by HIV proteins, methamphetamine and cocaine. BMC Neurosci 2:3
Tyagi M, Rusnati M, Presta M, Giacca M (2001) Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem 276:3254–3261
Valle LD, Croul S, Morgello S, Amini S, Rappaport J, Khalili K (2000) Detection of HIV-1 Tat and JCV capsid protein, VP1, in AIDS brain with progressive multifocal leukoencephalopathy. J Neurovirol 6:221–228
Vendeville A, Rayne F, Bonhoure A, Bettache N, Montcourrier P, Beaumelle B (2004) HIV-1 Tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Mol Biol Cell 15:2347–2360
Vives E, Brodin P, Lebleu B (1997) A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem 272:16010–16017
Wang P, Barks JD, Silverstein FS (1999) Tat, a human immunodeficiency virus-1-derived protein, augments excitotoxic hippocampal injury in neonatal rats. Neuroscience 88:585–597
Weeks BS, Lieberman DM, Johnson B, Roque E, Green M, Lowenstein P, Oldfield EH, Kleinman HK (1995) Neurotoxicity of the human immunodeficiency virus type 1 Tat transactivator to PC12 cells requires the Tat amino acid 49–58 basic domain. J Neurosci Res 42:34–40
Wei X, Miou Z, Baudry M (2008) Neuroprotection by cell permeable TAT-mGluR1 peptide in ischemia: synergy between carrier and cargo sequences. Neuroscientist 14:409–414
Weiss JM, Nath A, Major EO, Berman JW (1999) HIV-Tat induces MCP-1 mediated monocyte transmigration and upregulates CCR5 expression on human monocytes. J Immunol 163:2953–2959
Wesselingh SL, Power C, Glass JD, Tyor WR, McArthur JC, Farber JM, Griffin JW, Griffin DE (1993) Intracerebral cytokine messenger RNA expression in acquired immunodeficiency syndrome dementia. Ann Neurol 33:576–582
Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH (1995) Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375:497–500
Wiley CA, Baldwin M, Achim CL (1996) Expression of regulatory and structural mRNA in the central nervous system. AIDS 10:943–947
Woodman SE, Benveniste EN, Nath A, Berman JW (1999) Human immunodeficiency virus type 1 TAT protein induces adhesion molecule expression in astrocytes. J Neurovirol 5:678–684
Wortman MJ, Krachmarov CP, Kim JH, Gordon RG, Chepenik LG, Brady JN, Gallia GL, Khalili K, Johnson EM (2000) Interaction of HIV-1 Tat with Puralpha in nuclei of human glial cells: characterization of RNA-mediated protein–protein binding. J Cell Biochem 77:65–74
Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT (2000) Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci USA 97:11466–11471
Yedavalli VS, Benkirane M, Jeang KT (2003) Tat and trans-activation-responsive (TAR) RNA-independent induction of HIV-1 long terminal repeat by human and murine cyclin T1 requires Sp1. J Biol Chem 278:6404–6410
Zagury JF, Sill A, Blattner W, Lachgar A, Le Buanec H, Richardson M, Rappaport J, Hendel H, Bizzini B, Gringeri A, Carcagno M, Criscuolo M, Burny A, Gallo RC, Zagury D (1998) Antibodies to the HIV-1 Tat protein correlated with nonprogression to AIDS: a rationale for the use of Tat toxoid as an HIV-1 vaccine [see comments]. J Hum Virol 1:282–292
Zhao T, Adams MH, Zou SP, El-Hage N, Hauser KF, Knapp PE (2007) Silencing the PTEN gene is protective against neuronal death induced by human immunodeficiency virus type 1 Tat. J Neurovirol 13:97–106
Zhong Y, Smart EJ, Weksler B, Couraud PO, Hennig B, Toborek M (2008) Caveolin-1 regulates human immunodeficiency virus-1 Tat-induced alterations of tight junction protein expression via modulation of the Ras signaling. J Neurosci 28:7788–7796
Zidovetzki R, Wang JL, Chen P, Jeyaseelan R, Hofman F (1998) Human immunodeficiency virus Tat protein induces interleukin 6 mRNA expression in human brain endothelial cells via protein kinase C- and cAMP-dependent protein kinase pathways. AIDS Res Hum Retroviruses 14:825–833
Zou W, Kim BO, Zhou BY, Liu Y, Messing A, He JJ (2007) Protection against human immunodeficiency virus type 1 Tat neurotoxicity by Ginkgo biloba extract EGb 761 involving glial fibrillary acidic protein. Am J Pathol 171:1923–1935
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, W., Li, G., Steiner, J. et al. Role of Tat Protein in HIV Neuropathogenesis. Neurotox Res 16, 205–220 (2009). https://doi.org/10.1007/s12640-009-9047-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-009-9047-8