
Abstract
Purpose
We examined potential causes of nutrient release from the sediments of a shallow polymictic reservoir that, unlike many other shallow lakes, had not received large amounts of nutrient loading. It was hypothesised that the sediments released greater amounts of nutrients under anoxic rather than oxic conditions and that nitrogen (N) and phosphorus (P) release was limited by the availability of different types of organic substrate.
Materials and methods
Sediment incubation experiments under oxic and anoxic conditions were conducted in summer and in winter. One anoxic incubation experiment with the addition of different types of dissolved organic substrate (glucose, acetate, formate and propionate) was carried out. Concentrations of nitrite and nitrate (NOx −-N), ammonium (NH4 +-N), soluble reactive P (srP) and iron (Fe(II)) were monitored in the supernatant of the incubation chambers over 28 days. Thermal stratification was monitored in situ for 17 months and sediment composition was analysed.
Results and discussion
NOx −-N concentration increased by 5.67 ± 0.33 mg N l−1 in anoxic conditions but was much lower in oxic treatments. Release of srP was low in anoxic and oxic treatments (0.004 ± 0.001 mg P l−1) but was stimulated by 0.027 ± 0.015 mg P l−1 under additions of formate. Fe(II) release was mostly stimulated by glucose additions (25.59 ± 4.28 mg Fe l−1). P release was much lower than observed in eutrophic lakes. P release does not appear to be as a result of the reduction of Fe minerals and concurrent dissolution of orthophosphate, often reported as the most prevalent pathway for P release from anaerobic lake sediments.
Conclusions
Iron and P flux from lake sediments can be independent from each other and can be controlled by dissolved organic C. Further, the study confirms the role of oxygen availability in controlling nutrient release from the sediments.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Water quality in shallow lakes has suffered worldwide in recent decades. This is often due to internal phosphorus (P) storage which can support extensive phytoplankton growth when released into the water column (Søndergaard et al. 2001, 2003, 2013). The release of nitrogen (N) has been examined less often, but, as a major algal nutrient, N is also decisive for water quality. Sediments can play an important role as a nitrate sink through denitrification (Saunders and Kalff 2001), but they can also be a source of ammonium (NH4 +-N), with release rates exceeding external input (Burger et al. 2007). This warrants closer examination of N as well as P release from the sediments of shallow lakes.
Microbially mediated transformations and chemical sorption and dissolution processes in the sediments lead to the release of N and P into the water column. Processes underlying these releases depend on various environmental conditions but oxygen availability is often an important driver. Under anoxic conditions, release rates of P (Andersen and Ring 1999; Malecki et al. 2004; Nowlin et al. 2005; Ahlgren et al. 2011), N (Malecki et al. 2004; Beutel et al. 2008) and iron (Fe) (Andersen and Ring 1999) can be significantly higher compared with oxic conditions. Inversely, the transport of inorganic dissolved P out of the sediments can be hindered if the surface layer of the sediments is oxidised (Penn et al. 2000; Christophoridis and Fytianos 2006). It is often suggested that the mechanism behind oxygen-controlled P release is the reduction of Fe(III)oxyhydroxides resulting in the dissolution and release of orthophosphate (Boström et al. 1988; Amirbahman et al. 2003; Christophoridis and Fytianos 2006; Loh et al. 2013). Nitrogen may be released as ammonium in the remineralisation of organic matter. In oxygenated parts of lake sediments, ammonium may be oxidised to nitrate by nitrifying bacteria. In anoxic parts of the sediments, nitrate may in turn be reduced to nitrite and eventually dinitrogen gas in the denitrification process facilitated by anaerobic bacteria. Both nitrate and ammonium may accumulate and diffuse into the overlying water column where they may enhance primary productivity.
Generally, the amount of organic matter deposited in the sediments determines bacterial abundance (Schallenberg and Kalff 1993) and bacterial activity (Goedkoop et al. 1997; Törnblom and Rydin 1998). In turn, the release of ammonium and orthophosphate from the sediments can increase with organic matter content (Watts 2000; Autio et al. 2003). Specifically, the availability and the type of organic substrate affects the bacterial community composition (Mitchell 2002; Findlay et al. 2003; Torres et al. 2011) and can control rates and pathways of nutrient cycles in the sediments (Mitchell 2002; Mitchell et al. 2005). Nitrogen and P release processes can be stimulated by additional macrophyte material (Watts 2000), formate (Mitchell et al. 2005) or glucose (Ahlgren et al. 2011) availability, respectively. Further, additions of glucose (Bradley et al. 1992, 1995; Wang et al. 2007), acetate (Wang et al. 2007), starch (Wang et al. 2007) and propionate (Mitchell 2002) can enhance denitrification and thus remove bioavailable N from the sediments. Aside from catalysing redox reactions leading to the release of P, sediment bacteria can act as a polyphosphate store and release P depending on redox conditions (Gächter and Meyer 1993; Hupfer et al. 2007).
The water column of shallow lakes is often well mixed and thus oxygenated. Nutrients are often re-introduced into the water column through sediment resuspension (Kristensen et al. 1992; Reddy et al. 1996; Holmroos et al. 2009), but they may also diffuse from the sediments despite the overlying water being aerated (Jensen and Andersen 1992; Fisher et al. 2005). However, there may be periods when the water at the sediment interface becomes anoxic. It has been demonstrated that anoxia plays an important role for re-introduction of nutrients into the water column in shallow lakes (Reddy et al. 1996; Nowlin et al. 2005; Wilhelm and Adrian 2008). Stable weather conditions, i.e., low wind speeds, may lead to persistent thermal stratification of the water column during hotter periods. Organic matter mineralisation can consume the dissolved oxygen in bottom waters and result in anoxia and potentially increase nutrient release. These periods of stratification can lead to accumulation of dissolved inorganic nutrients in bottom waters followed by large nutrient pulses into the euphotic zone and intensified phytoplankton growth (Wilhelm and Adrian 2008). Most shallow lakes, in which anoxic nutrient release and internal loading has been investigated, have often experienced external nutrient loading over long periods, sometimes decades (Christophoridis and Fytianos 2006; Burger et al. 2007; Spears et al. 2007). Less information exists on the nutrient release of sediments of shallow lakes that have not received large amounts of nutrient loading.
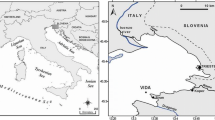
We focused our sediment examinations on Grahamstown Dam, a shallow mesotrophic drinking water reservoir on the coast of New South Wales, Australia. The reservoir was constructed for the purpose of drinking water supply in 1965 and has not received large amounts of agricultural or industrial run off. Its large surface area in combination with prevailing coastal weather patterns (such as frequent windy days) is assumed to lead to a well-mixed water column at most times. There are limited data suggesting that periods of thermal stratification may occur for several days, mainly in summer (Cole and Williams 2011) and anoxia of bottom waters is likely to accompany these periods. Phytoplankton growth is often co-limited by N and P (Müller and Mitrovic 2015) which necessitates gaining insight into release patterns and mechanisms of both nutrients from these sediments. Thus, the purpose of this paper was to determine: (i) to what extent persistent thermal stratification occured in the reservoir; (ii) if anoxic conditions increased the release of N and P; and (iii) how organic substrate affected this. To test the hypotheses that (1) the sediments released higher amounts of nutrients under anoxic conditions than under oxic conditions and (2) that under anoxic conditions N and P release would be limited by the availability of different types of organic substrate, we conducted a series of sediment incubation experiments.
2 Materials and methods
2.1 Study site
Grahamstown Dam is a shallow reservoir located 20 km north of Newcastle on the coast of New South Wales, Australia. The lake is nominally well-mixed because of its large surface area of (28 km2), which provides a large fetch, and its shallow mean depth (7 m). The catchment comprises 73 km2 which at times may not be sufficient to maintain the necessary level for drinking water supply. To compensate, water levels can be increased by pumping water from a weir pool on the nearby Williams River through a canal (Balickera Canal) into the northern end of the lake. This procedure also increases nutrient levels in the reservoir as the Williams River carries a higher nutrient concentration than the lake. Concentrations of nitrate and nitrite (NOx −-N) and soluble reactive phosphorus (srP) can be up to nine and six times, respectively, higher in the river than in the lake. Between 1 April 2000 and 1 April 2009, concentrations varied between 3 and 100 μg N l−1 and 1 to 58 μg P l−1 in Grahamstown Dam and 3 to 740 μg N l−1 and 1 to 170 μg P l−1 at Seaham Weir (Hunter Water monitoring data) from where water is pumped into the dam via Balickera Canal. Currently, the lake has been classified as mesotrophic according to OECD guidelines (Cole and Williams 2011).
Three different sites in the lake were chosen for their individual environmental characteristics. Site 3, close to Balickera Canal, is the most protected and shallowest (4–6 m, depending on capacity of the lake) while site 2, closest to the centre of the lake, is the most wind exposed and deepest (9–10 m). Site 1, close to the drinking water off take, is between 7 and 9 m deep. Each site is permanently marked with a buoy.
2.2 Sediment composition
Sediment was collected with an Ekman grab at each of the three sites. Fresh samples were stored at −20 °C until further processing unless mentioned otherwise. Loss-on-ignition (LOI) was carried out to determine the organic matter content of the sediments. LOI was determined by dry combustion at 550 °C and calculated as the percentage of the dry weight of the sediment before combustion on three independent sediment samples from each site.
Concentrations of pore water nutrients in comparison with concentrations in the overlying water column are an indicator for the diffusion gradient for nutrient release into the water column. The focus was on N species (NH4 +-N and NOx −-N) to elucidate details of the N cycle in Grahamstown Dam. Three independent 50 ml sediment samples from each site were centrifuged at 3500 rpm for at least 10 min on the day of sampling. The supernatant was filtered (0.45 μm) and stored at −10 °C until analysis. Fe(II) and both N species were analysed in all pore water samples. As there was not enough supernatant from some samples, srP was only measured in one sample from site 1 and two samples from site 2. Concentrations of srP and N species were determined photometrically (QuikChem 8500 Lachat nutrient analyser, Loveland, USA) following APHA (1995) methods. Fe(II) was analysed photometrically (Varian Cary 50 Bio UV spectrophotometer, Santa Clara, USA ) using the ferrozine method (Stookey 1970; Lovley and Philips 1986).
Total carbon (TC) indicates the amount of C that is potentially available as electron donor. Total N (TN) and total P (TP) content indicate the amount of these nutrients that could potentially be remobilised. Analysis of TC, TN and TP was conducted on two samples from sites 1 and 3 each and three samples from site 2 collected on 8 April 2013, and also on sediments that had been used in the oxic treatments of the nutrient release experiment in June 2010 and stored at −20 °C until analysis (five replicates from sites 1 and 2 and four from site 3). Analysis of TC and TN was carried out with a TruSpec CN Determinator (Leco Corporation, St Joseph, USA). Total P was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES) after acid digestion by Envirolab Services Pty Ltd, Sydney, Australia.
Major elemental composition of the sediments (Si, Al, Fe, Mn, Mg, Ca, Na, K, P and S) was determined by X-ray fluorescence spectrometry (XRF) on three independent samples from sites 1 and 2 each and one sample from site 3. Spectra were determined with a Bruker-AXS S4 Pioneer X-ray Fluorescence Spectrometer (Billerica, USA) by the Advanced Analytical Centre at James Cook University, Townsville, Australia.
2.3 Sediment incubation under oxic and anoxic conditions
To test the nutrient release under oxic and anoxic conditions, laboratory sediment incubation experiments were conducted in July 2010 (winter experiment) and in March 2011 (summer experiment). Each experiment lasted for 28 days. Sediment was collected from the three sites in Grahamstown Dam on day 0 (17 June 2010 and 10 February 2011, respectively). Treatments included incubation under anoxic and oxic conditions with five replicates per treatment. The release of TN, TP, srP, NOx −-N, NH4 +-N and Fe(II) was monitored on days 0, 1, 4, 7, 11, 15, 21 and 28.
Plastic containers (2 l) with a lid that allowed airtight sealing, a sampling port (a butyl rubber septum) that allowed sampling with a syringe and hypodermic needle and a device for pressure equalisation (constructed according to Baldwin et al. (2005)) served as incubation chambers for anoxic treatments. Oxic treatment chambers were similar but were aerated for the duration of the experiment, did not have the pressure equalisation device and were only sealed airtight for transport. Oxic chambers were kept in water up to their lids and anoxic chambers were fully submersed under water. Both were kept in a dark temperature-controlled environment at 16 °C in winter and 23 °C in summer.
Sediments were collected from Grahamstown Dam using an Ekman grab sampler. An amount of 0.5 l of sediment was immediately transferred to each incubation chamber and topped up with ultrapure water (conductivity adjusted with concentrated sodium chloride solution to match lake water). Before sealing, pH, temperature and dissolved oxygen were measured. Ultrapure water used for anoxic treatments had also been degassed by bubbling helium gas through it until dissolved oxygen concentrations reached 2.0 mg l−1. Degassing was omitted in the summer experiment as dissolved oxygen concentrations decreased from 8.5 to below 1.0 mg l−1 in the supernatant after addition to the incubation chambers of the oxic treatments. Dissolved oxygen, pH and temperature were measured in the anoxic treatments in a freshly taken sample and on day 28 after opening the chambers. Oxic treatments were monitored on the sampling days. Dissolved nutrient samples and Fe(II) samples were filtered (0.45 μm). Fe(II) samples were preserved in 10 % ultrapure concentrated hydrochloric acid (0.1 ml per 1 ml sample). Concentrations of srP, NOx −-N, NH4 +-N, TN and TP were determined photometrically (QuikChem 8500 Lachat nutrient analyser, Loveland, USA) following APHA (1995) methods. Fe(II) was analysed photometrically (Varian Cary 50 Bio UV spectrophotometer, Santa Clara, USA) using the ferrozine method (Stookey 1970; Lovley and Philips 1986).
2.4 Sediment incubation with additional dissolved organic carbon
To test the effect of additional C sources on the release of nutrients from the sediments, one laboratory incubation experiment was conducted in October 2011 (C limitation experiment). As release patterns in the first two experiments were very similar between sites, sediments were collected from one site only on day 0 (27 October 2011). Methods described above were applied for collection, filling of incubation chambers, measurement of water quality parameters and incubation, and the airtight incubation chambers used for the anoxic treatment were used. Sediments were incubated at 20 °C for 28 days.
Solutions of electron donors in the form of short-chain aliphatic carboxylic acids and glucose in ultrapure water were added to each chamber after filling with sediment. The four treatments were acetate, formate, propionate and glucose (n = 4). One control contained ultrapure water and sediment (n = 4). Solutions were made from reagent grade chemicals to achieve a concentration of 120 mg C l−1, i.e. 410 mg l−1 acetate, 680 mg l−1 formate, 320 mg l−1 propionate and 300 mg l−1 glucose, and degassed. Environmental concentrations are usually lower than 120 mg C l−1 (5–12 mg l−1 of total organic C at site 1 according to Hunter Water monitoring data) but the concentration was chosen to ensure levels of C were high enough to be able to record a response to C additions in a batch experiment.
2.5 Thermal stratification
Temperature data loggers (TidbiT v2 Water Temperature Data Logger, Onset, Bourne, USA) were deployed at each of the three sites. Data loggers were positioned at approximately 0.5 m above the sediment surface and at 1- to 2-m intervals throughout the water column for 17 months (September 2011–February 2013). Water temperatures were recorded at 30-min intervals. Thermal stratification data were smoothed with a rolling mean using the function rollmean of R’s zoo package before plotting.
2.6 Data analysis
Analysis of Similarities (ANOSIM) was performed to illustrate qualitative similarities in nutrient release between the three different sites for both assays. As there were four replicates for srP in the winter assay, it was not included in ANOSIM. The analysis was carried out with the statistics software R (version 2.13.0, R Development Core Team (2011)) using the functions anosim and metaMDS (with Euclidean distances) from the vegan package.
3 Results
3.1 Thermal stratification
Several periods of persistent thermal stratification occurred at the three sites throughout the 17-month measuring period. Events of persistent thermal stratification were observed mostly during the spring and summer months, while the water column was well mixed in winter. The longest and most pronounced period of stratification was recorded in November 2011. It was most obvious at site 2 with temperature differences of up to 7.80 °C between surface (0.5 m) and bottom water (9 m) lasting for up to 23 days (Fig 1. Diurnal stratification affecting the surface to a depth of ∼3 m occurred frequently, sometimes temperature differences between warm surface layer and colder bottom layer were up to 4.64 °C.

Temperature profile of the water column at site 2 in Grahamstown Dam and temperature difference between surface and 9 m during the period of the most pronounced stratification between September 2011 and February 2013. Shaded areas highlight temperature differences >0.15 °C (according to Sherman et al. (1998), persistent thermal stratification occurs when the difference between water surface and bottom water is >0 °C) and lasting for more than 24 h. Data were smoothed with a rolling mean with a window of ten observations
3.2 Sediment composition
Sediments at site 1 were patchy and consisted of either sand or silty grey brown material interspersed with some small roots and other plant matter. The silty sediments were visually similar to the sediments found at the other two sites. The content of TC, TP and TN, different minerals and organic matter in the sediments as well as concentrations of NH4 +-N, NOx −-N, Fe(II) and srP in the pore water are summarized for all three sites in Table 1. The main constituent of sediments from all three sites was SiO2, followed by organic matter (measured by LOIXRF), Al2O3, TC and Fe2O3T. Pore water contained up to 6.50 ± 1.29 mg l−1 Fe(II) and 1.65 ± 0.51 mg N l−1 in the form of NH4 +-N, while concentrations of NOx −-N and srP were below 0.014 mg P l−1 and 0.01 mg N l−1, respectively. Concentrations of N species in the pore water and proportions of SiO2 were higher, and proportions of Al2O3 were lower at site 1 compared with the other two sites.
3.3 Nutrient release under oxic and anoxic conditions
The presence of oxygen had a large impact on the type and amount of nutrients released in both experiments. Sediments from all three sites released N in both experiments and there was a distinct difference in the release of N species from oxic and anoxic treatments (Figs. 2 and 3). Concentrations of TP decreased independently from oxygen presence during the course of both experiments (Figs. 2 and 3). A release of srP and Fe(II) was detected at all three sites in the summer experiment (Fig. 3).

Nitrogen (N) and phosphorus (P) species and iron (Fe(II)) concentration in both treatments during the incubation experiment in winter 2010. Concentrations were measured as mg N l−1, mg P l−1 and mg Fe l−1 and are averages of five replicates from the three sites. Note that the graph for Fe(II) has a logarithmic y-axis. Error bars are one standard error from the mean, n = 15, except for srP where n = 12

Nitrogen (N) and phosphorus (P) species and iron (Fe(II)) concentration in both treatments during the incubation experiment in summer 2011. Concentrations were measured as mg N l−1, mg P l−1 and mg Fe l−1 and are averages of five replicates from the three sites. Note that the graph for Fe(II) has a logarithmic y-axis. Error bars are one standard error from the mean, n = 15
ANOSIM analysis indicated that site location exerted small influences on nutrient release in both experiments (winter experiment: global R = 0.0140, p = 0.014; summer experiment: global R = 0.121, p = 0.001). As site did not have a big impact on nutrient release, concentrations of nutrients were averaged over the three sites and concentrations in the following text are the mean ± standard error of all three sites (n = 15, except srP in the winter experiment where n = 12).
3.3.1 Nitrogen release
The largest release of N occurred in the form of NOx −-N in the anoxic treatments in both experiments. Up to 5.67 ± 0.33 mg N l−1 in the form of NOx −-N was released by day 7 in the winter experiment (Fig. 2) and by day 4 in the summer experiment (Fig. 3). Subsequently, concentrations declined until almost initial values were reached (0.21 ± 0.02 mg N l−1 in winter, 0.05 ± 0.08 mg N l−1 in summer). In the oxic treatments, NOx −-N release was several orders of magnitude smaller and also slower compared with the anoxic treatments. Maximum concentrations were reached on day 28 in the winter experiment (0.08 ± 0.05 mg N l−1) and day 21 in the summer experiment (0.05 ± 0.03 mg N l−1).
Up to 1.36 ± 0.030 mg N l−1 NH4 +-N was released rapidly by day 7 in the anoxic treatments in the winter experiment (Fig. 2) and up to 0.86 ± 0.02 mg N l−1 by day 4 in the summer experiment (Fig. 3). Subsequently, release rates slowed down but concentrations continued to increase to a maximum of 2.12 ± 0.03 mg N l−1 on day 21 in winter and 2.14 ± 0.09 mg N l−1 on day 21 in summer until the end of the experiment. In the oxic treatments, NH4 +-N was released more slowly and nearly linearly during the course of both experiments. In the oxic treatments, concentrations increased to 1.80 ± 0.05 mg N l−1 between day 0 and 28 in the winter experiment and 2.20 ± 0.03 mg N l−1 between day 0 and 21 in the summer experiment.
The release of TN followed a pattern similar to the release of NOx in both treatments albeit TN concentrations being higher. When added, concentrations of NOx −-N and NH4 +-N were nearly the same as TN concentrations in both treatments and both experiments (Figs. 2 and 3). Similar to the release of NOx −-N, the release of TN was much slower and smaller in the oxic treatment than in the anoxic one.
3.3.2 Phosphorus release
Concentrations of srP varied within a range of <0.001 and 0.003 mg P l−1 (method detection limit: 0.7 μg P l−1 determined following US EPA methods (DNR 1996) but neither a clear increase nor decrease was discernible in any treatment in the winter experiment (Fig. 2). During the summer experiment, concentrations of srP were <0.001 mg P l−1 and increased by 0.004 ± 0.001 mg P l−1 in the anoxic treatments and by 0.002 mg P l−1 in the oxic treatments (Fig. 3).
Concentration of TP decreased in both treatments in both experiments (Figs. 2 and 3). In the winter experiment, TP decreased by 0.173 ± 0.040 mg P l−1 in the anoxic treatments and by 0.132 ± 0.028 mg P l−1 in the oxic ones by day 7. In the summer experiment, the decrease was 0.081 ± 0.040 mg P l−1 in the anoxic treatments and 0.077 ± 0.028 mg P l−1 in the oxic treatments by day 4.
3.3.3 Iron (Fe(II)) release
Fe(II) was only released between day 15 and 28 in the anoxic treatments in the summer experiments (Fig. 3). Concentrations increased from 0.02 ± 0.003 by 0.95 ± 0.36 mg Fe l−1. In the winter experiment, Fe(II) concentrations varied within a range of 0.01 and 0.27 mg Fe l−1 but neither a clear increase nor decrease was discernible in either treatment (Fig. 2).
3.3.4 Dissolved oxygen concentrations
In the winter experiment, dissolved oxygen concentrations were 0.4 mg l−1 in the supernatant of the oxic treatments and 0.3 mg l−1 in the anoxic treatments within a few minutes after addition of the ultrapure water on day 0. On day 28, dissolved oxygen concentrations were 10.0 mg l−1 in the oxic and 0.9 mg l−1 in the anoxic treatments. In the summer experiment, concentrations decreased rapidly from 8.8 to 4.0 mg l−1 in the supernatant of the oxic treatments and to 3.2 mg l−1 in the anoxic treatments. On day 28, concentrations were 9.2 mg l−1 in the oxic and 0.4 mg l−1 in the anoxic treatments.
3.4 Nutrient release with additional dissolved organic carbon sources
Addition of different C sources increased the release of Fe(II) and srP from the sediments, while the release of NH4 +-N and NOx −-N was not stimulated (Fig. 4). Concentrations in the following are the mean of four replicates ± standard error for each treatment.

Nitrogen (N) and phosphorus (P) species and iron (Fe(II)) concentration in all treatments during the carbon (C) limitation experiment in 2011. Concentrations were measured as mg N l−1, mg P l−1 and mg Fe l−1. Error bars are one standard error from the mean, n = 4
3.4.1 Iron (Fe(II)) release
The concentration of Fe(II) increased in all treatments and the control from day 0 until the end of the experiment (Fig. 4). The highest increase was stimulated by additions of glucose, followed by propionate, acetate, formate and the control. In the glucose treatment, net Fe(II) increase was 25.59 ± 4.28 mg Fe l−1 on day 21. In the control, the net increase was 2.55 ± 1.68 mg Fe l−1 on day 28.
3.4.2 Phosphorus release
The concentration of srP increased in all treatments from day 0 until day 14 (Fig. 4). The highest increase was stimulated by the additions of formate. Concentrations increased by 0.027 ± 0.015 mg P l−1 in the formate treatment, by 0.011 ± 0.003 mg P l−1 in the acetate treatment and by ≤0.008 ± 0.003 mg P l−1 in the remaining treatments by day 14. This was followed by a decrease in all treatments and the control except the formate treatment where srP concentration remained stable until the end of the experiment.
The concentration of TP increased in the formate treatment by 0.043 ± 0.009 mg P l−1 between days 0 and 14. Subsequently, it decreased to 0.034 ± 0.006 mg P l−1 on day 28. In the remaining treatments and the control, mean TP concentrations were variable between 0.012 and 0.028 mg P l−1 throughout the experiment.
3.4.3 Nitrogen release
The concentrations of NH4 +-N increased by 1.01 ± 0.04 mg N l−1 in the acetate, formate and propionate treatments and in the control between days 0 and 4 (Fig. 4). After day 4, NH4 +-N release slowed down. The concentration of NH4 +-N in the glucose treatment was lowest and increased only after day 4. By day 28, concentrations in all treatments and controls were similar (2.15 ± 0.12 mg N l−1).
The concentration of NOx −-N decreased from 0.23 ± 0.02 mg N l−1 on day 0 to less than 0.01 mg N l−1 until day 28 in all treatments and the control. Release of TN followed a pattern similar to NH4 +-N release. TN concentrations in all treatments and the control were lower than NH4 +-N concentrations between days 4 and 28.
3.4.4 Dissolved oxygen concentrations
After degassing, dissolved oxygen concentrations in all of the C solutions were <0.9 mg l−1. Dissolved oxygen concentrations were <1.0 mg l−1 in the supernatant after addition of the degassed C solutions. On day 28, incubation chambers of all treatments had <0.6 mg l−1 dissolved oxygen in the supernatant.
4 Discussion and conclusions
Monitoring data showed the water column in Grahamstown Dam was subject to persistent thermal stratification for periods up to 23 days in summer. Sediment organic matter content was high and dissolved oxygen concentrations in the supernatant in oxic treatments decreased rapidly (within minutes), suggesting that anoxic conditions would set in soon after thermal stratification became stable. The winter and summer experiments revealed that N release was highest within the first 4 to 7 days of reduced oxygen availability to the sediments, implying that short periods of thermal stratification would be enough to increase nutrient concentration in the overlying water column.
Periods of persistent thermal stratification were frequently interrupted by mixing of the entire water column. This may increase nutrient availability in the euphotic zone and may promote phytoplankton growth in a similar way as described by Wilhelm and Adrian (2008).
Our results show that sediments of Grahamstown Dam were a source of dissolved inorganic N, particularly when the overlying water column was depleted of oxygen. Detecting a large release of NOx −-N in the absence of oxygen, while comparatively little was released in the aerated treatments, was unexpected. As denitrification and nitrification rates were not monitored and nitrite and nitrate concentrations were not measured independently, the following explanations for this observation remain speculative.
Although dependent on oxygen, the oxidation of ammonium to nitrite, the first step in nitrification, can occur under oxygen concentrations as low as 0.5 mg l−1 (Bernet et al. 2001). Dissolved oxygen concentrations were between 0.3 and 3.2 mg l−1 in the anoxic treatments at the beginning of the summer and winter experiments and between 0.4 and 0.9 mg l−1at the end. Thus, it is possible that dissolved oxygen concentrations may not have prevented nitrification, and it may have been responsible for the increase in NOx −-N in the anoxic treatments of summer and winter experiment.
It has been observed that denitrifying bacteria start accumulating nitrite when they are cut off from a continuous nitrate supply (Xu and Enfors 1996; Liu et al. 2013). An alternative explanation for the increase in NOx −-N after day 1 in the anoxic treatments may be an accumulation of nitrite. This scenario would be based on the following assumptions: (1) denitrification and nitrification offset each other in the sediments so that NOx −-N was not detectable in the supernatant in the incubation chambers at the beginning of the experiments, and (2) the addition of degassed supernatant may have slowed the nitrification process leading to a decrease in nitrate supply which in turn may have caused an accumulation of nitrite.
Denitrification rates are higher than nitrification rates in many sediments (Seitzinger 1988), and denitrification has also been observed in aerobic conditions (Davies et al. 1989; Lloyd 1993; Chen et al. 2006). Thus, it is likely that nitrification and denitrification were balanced to the extent that free NOx −-N was not detectable. This would also explain the lack of NOx −-N in the supernatant in the oxic treatments. Further, our observations are in agreement with Rysgaard et al. (1994) who found that if the overlying water column was oversaturated with oxygen, nitrate from nitrification within the sediments was the source of nitrate for denitrification. They suggest that high oxygen concentrations would stimulate the denitrification of nitrate produced within the sediments especially in environments with low water column nitrate concentrations.
The release of NH4 +-N under oxic and anoxic conditions in our study is not unusual because organic N can be mineralised under both conditions. Similar release rates of NH4 +-N from oxic and anoxic sediments have been reported previously (e.g. Rysgaard et al. 1994; Nowlin et al. 2005). The flux of organic N from the sediment was comparatively small as the concentrations and release patterns of the three N species (TN, NOx −-N and NH4 +-N) in the summer and winter experiments suggested. TN mainly consisted of dissolved inorganic N, i.e. the type of N that can be utilised by phytoplankton. In the C limitation experiment, problems with the persulfate digestion of samples occurred and recovery was about 80 % of original concentrations, leading to TN concentrations being lower than NOx −-N and NH4 +-N concentrations.
Nitrogen release was largely unaffected by the different C substrates, in contrast to Fe and P dynamics. Unlike the NOx −-N release in the anoxic treatments in the summer and winter experiments, NOx −-N was not detected in any of the treatments or the control of the C limitation experiment. This suggests that the additional C sources may have enhanced denitrification rates. Increased denitrification rates due to additional dissolved organic C have been reported previously (Bradley et al. 1992, 1995; Mitchell 2002; Wang et al. 2007). Both Fe cycling (Lovley and Philips 1986; Lovley 1991; Mitchell 2002; Mitchell et al. 2005) and P cycling (Watts 2000; Mitchell 2002; Mitchell et al. 2005) have previously been shown to be enhanced by additions of dissolved organic C. The stimulation of Fe and P release by bioavailable C indicates that P and Fe cycling was controlled by microbial processes and not solely by lack of oxygen resulting from microbial activity. This result supports the notion that mechanisms involved in P cycling are more complex than merely the absence of oxygen and resulting reductive conditions (Hupfer and Lewandowski 2008) but that bacteria play an important role in actively controlling the cycling of P (Davelaar 1993; Gächter and Meyer 1993). Our results also suggest that P release was not caused by the reduction of Fe minerals and concurrent dissolution of orthophosphate as often suggested as a mechanism of anoxic P release (Boström et al. 1988; Amirbahman et al. 2003; Christophoridis and Fytianos 2006; Loh et al. 2013) but rather that cycling of Fe and P occurs independently in the sediments of Grahamstown Dam. Each added C source, except formate, enhanced the release of Fe but glucose caused by far the highest release. In contrast, P release was mostly stimulated by formate additions and to a small extent by acetate additions. While Fe was released until day 21 or 28 in some treatments of the C limitation experiment, P release stopped after day 14 and appeared to be taken up by the sediments. Fermenting bacteria have been shown to use Fe(III) as an electron acceptor (Lovley 1991). As glucose is a main substrate for fermenters, fermentation would explain the increased release of Fe in the glucose treatment. Sediment pore water concentrations of Fe were exceeded by release of Fe in the glucose, propionate and acetate treatments. Similarly, srP concentrations released in the formate treatment, but not the other treatments or the control, were higher than pore water concentrations. This substantiates the claim that Fe and P release were due to additional bioavailable C. Had the concentration of Fe or P increased in the supernatant solely due to diffusion of Fe and P dissolute in the pore water prior to the experiment, concentrations in the supernatant in the incubation chambers would not have exceeded pore water concentrations.
The apparent decoupling of the P and Fe cycles in the C limitation experiment are in agreement with the results from Mitchell et al. (2005) who examined the effect of different C sources (acetate, formate, lactate, propionate and glucose) on microbial communities and microbial metabolite cycling in wetland sediments. It was observed that glucose stimulated the highest Fe release (6.72 mg per 1-l microcosm) while formate stimulated the highest P release (0.93 mg per 1-l microcosm) from the sediments. Acetate additions also lead to a small release of P (0.31 mg per 1-l microcosm). Moreover, an inverse pattern was also recorded; formate led to Fe uptake and glucose to P uptake by the sediments.
Unlike as reported by Mitchell et al. (2005), the amount of P the sediments released seems to be comparatively small. The highest net release of srP was 0.027 mg l−1 in the formate treatment after 14 days of incubation. Amounts of srP recorded by Mitchell (2002) were approximately 30 times higher, for example. Even without additional bioavailable C, P release from the sediments under anoxic conditions is often much higher than measured in this study. Burger et al. (2007) reported srP concentrations between 0.2 and 0.6 mg l−1 after 24 to 48 h of in situ sediment incubations in Lake Rotorua, New Zealand, and Malecki et al. (2004) found concentrations of srP release between 0.5 and 2.0 mg l−1 in sediment cores from the St. Johns River, USA, in 25-day incubation experiments. Obviously, the size of the P pool in the sediments influences how much P can potentially be remobilised. Accordingly, sediments from the St. Johns River and Lake Rotorua sediments contained two to three times as much TP (between 0.88 and 1.34 g kg−1 (Malecki et al. 2004) and around 1.1 to 1.7 g kg−1 (Hickey and Gibbs 2009), respectively) than the ones at site 1 in Grahamstown Dam (0.42 ± 0.01 g kg−1, Table 1). Content of P205 was 0.13 % in the sediments examined by Mitchell (2002) while 0.09 % was found in Grahamstown Dam at site 1 (Table 1). In contrast to Lake Rotorua or the St. Johns River, Grahamstown Dam has not received high external nutrient loads in the past decades and thus it is not surprising that the P content in its sediments is lower.
Aside from the amount of P in the sediments, it is possible that the form it is bound in might have inhibited a greater release. Other than Fe oxyhydroxides which are a main binding partner in the classical Fe and P cycling model, P may adsorb to Al, Mn or calcium minerals or be present in organic form. In contrast to Fe oxyhydroxides, Al hydroxide is not redox sensitive (Ulrich and Pöthig 2000) and it has been shown that molar ratios of Al/Fe >3 or Al/P >25 prevent the release of P from the sediments (Kopáček et al. 2005; Lake et al. 2007). Ratios of Al/Fe were <3 for all three sites sampled in Grahamstown Dam but Al/P were 105, 86, and 130 for sites 1, 2 and 3, respectively, indicating that Al precipitates may inhibit the release of P in Grahamstown Dam. In conclusion, a combination of low P content, high Al content and C limitation of bacterial P regeneration processes is likely to be responsible for the small amount of P release from Grahamstown Dam sediments. However, unless additional C substrate becomes available, it is not likely that P would be released from the sediments in greater quantities despite thermal stratification. A potential source of organic C may be found in the water pumped into the lake from the Williams River weir pool. High inputs of dissolved organic C, as commonly associated with high river flows and floods (Buffam et al. 2001; Westhorpe and Mitrovic 2012), may accumulate in the weir pool due to flooding upstream in the Williams River. If pumped across into the reservoir, these C sources might stimulate P release from the sediments.
References
Ahlgren J, Reitzel K, De Brabandere H, Gogoll A, Rydin E (2011) Release of organic P forms from lake sediments. Water Res 45:565–572
Amirbahman A, Pearce AR, Bouchard RJ, Norton SA, Kahl JS (2003) Relationship between hypolimnetic phosphorus and iron release from eleven lakes in Maine, USA. Biogeochemistry 65:369–386
Andersen FØ, Ring P (1999) Comparison of phosphorus release from littoral and profundal sediments in a shallow, eutrophic lake. Hydrobiologia 408:175–183
APHA (ed) (1995) Standard methods for the examination of water and wastewater, 19th edn. American Public Health Association/American Water Works Association/Water Pollution Control Federation, Washington
Autio L, Mäkelä K, Lethonen KK, Kuparinen J, Haahti H (2003) Effects of algal sedimentation and Monoporeia affinis on nutrient fluxes, pore water profiles and denitrification in sediment microcosms. Boreal Environ Res 8:229–243
Baldwin DS, Rees GN, Mitchell AM, Watson G (2005) Spatial and temporal variability of nitrogen dynamics in an upland stream before and after a drought. Mar Freshwater Res 56:457–464
Bernet N, Dangcong P, Delgenès J, Moletta R (2001) Nitrification at low oxygen concentration in biofilm reactor. J Environ Eng 12:266–271
Beutel MW, Horne AJ, Taylor WD, Losee RF, Whitney RD (2008) Effects of oxygen and nitrate on nutrient release from profundal sediments of a large, oligo-mesotrophic reservoir, Lake Mathews, California. Lake Res Manag 24:18–29
Boström B, Andersen JM, Fleischer S, Jansson M (1988) Exchange of phosphate across the sediment-water interface. Hydrobiologia 170:229–244
Bradley PM, Fernandez M, Chapelle FH (1992) Carbon limitation of denitrification rates in an anaerobic groundwater system. Environ Sci Technol 26:2377–2381
Bradley PM, McMahon PB, Chapelle FH (1995) Effects of carbon and nitrate on denitrification in bottom sediments of an effluent-dominated river. Water Resour Res 31:1063–1068
Buffam I, Galloway JN, Blum LK, McGlathery KJ (2001) A stormflow/baseflow comparison of dissolved organic matter concentrations and bioavailability in an Appalachian stream. Biogeochemistry 53:269–306
Burger DF, Hamilton DP, Pilditch CA, Gibbs MM (2007) Benthic nutrient fluxes in a eutrophic, polymictic lake. Hydrobiologia 584:13–25
Chen F, Xia Q, Ju LK (2006) Competition between oxygen and nitrate respirations in continuous culture of Pseudomonas aeruginosa performing aerobic denitrification. Biotechnol Bioeng 93:1069–1078
Christophoridis C, Fytianos K (2006) Conditions affecting the release of phosphorus from surface lake sediments. J Environ Qual 35:1181–1192
Cole B, Williams B (2011) Grahamstown Dam water quality & aquatic ecological functioning, a compilation of scientific and adaptive management studies. Hunter Water Corporation, 36 Honeysuckle Drive, Newcastle, NSW 2300, Australia
Davelaar D (1993) Ecological significance of bacterial polyphosphate metabolism in sediments. Hydrobiologia 253:179–192
Davies KJP, Lloyd D, Boddy L (1989) The effect of oxygen on denitrification in Paracoccus denitrificans and Pseudomonas aeruginosa. J Gen Microbiol 135:2445–2451
DNR (1996) Analytical detection guidance & Laboratory guide for determining method detection limits. Wisconsin Department of Natural Resources, Laboratory certification program, PUBL-TS-056-96, Wisconsin, USA
Findlay SEG, Sinsabaugh RL, Sobczak W, Hoostal M (2003) Metabolic and structural response of hyporheic microbial communities to variations in supply of dissolved organic matter. Limnol Oceanogr 48:1608–1617
Fisher MM, Reddy KR, James RT (2005) Internal nutrient loads from sediments in a shallow, subtropical lake. Lake Res Manag 21:338–349
Gächter R, Meyer JS (1993) The role of microorganisms in mobilization and fixation of phosphorus in sediments. Hydrobiologia 253:103–121
Goedkoop W, Gullberg KR, Johnson RK, Ahlgren I (1997) Microbial response of a freshwater benthic community to a simulated diatom sedimentation event: interactive effects of benthic fauna. Microbial Ecol 34:131–143
Hickey CW, Gibbs MM (2009) Lake sediment phosphorus release management—decision support and risk assessment framework. New Zeal J Mar Fresh 43:819–856
Holmroos H, Niemistö J, Weckström K, Horppila J (2009) Seasonal variation of resuspension-mediated aerobic release of phosphorus. Boreal Environ Res 14:937–946
Hupfer M, Lewandowski J (2008) Oxygen controls the phosphorus release from lake sediments—a long-lasting paradigm in limnology. Int Rev Hydrobiol 93:415–432
Hupfer M, Gloess S, Grossart HP (2007) Polyphosphate-accumulating microorganisms in aquatic sediments. Aquat Microb Ecol 47:299–311
Jensen HS, Andersen FØ (1992) Importance of temperature, nitrate, and pH for phosphate release from aerobic sediments of four shallow, eutrophic lakes. Limnol Oceanogr 37:577–589
Kopáček J, Borovec J, Hejzlar J, Ulrich KU, Norton S, Amirbahman A (2005) Aluminum control of phosphorus sorption by lake sediments. Environ Sci Technol 39:8784–8789
Kristensen P, Søndergaard M, Jeppesen E (1992) Resuspension in a shallow eutrophic lake. Hydrobiologia 228:101–109
Lake BA, Coolidge KM, Norton SA, Amirbahman A (2007) Factors contributing to the internal loading of phosphorus from anoxic sediments in six Maine, USA, lakes. Sci Total Environ 373:534–541
Liu B, Mao Y, Bergaust L, Bakken LR, Frostegård A (2013) Strains in the genus Thauera exhibit remarkably different denitrification regulatory phenotypes. Environ Microbiol 15:2816–2828
Lloyd D (1993) Aerobic denitrification in soils and sediments: from fallacies to facts. Trends Ecol Evol 8:352–356
Loh PS, Molot LA, Nurnberg GK, Watson SB, Ginn B (2013) Evaluating relationships between sediment chemistry and anoxic phosphorus and iron release across three different water bodies. Inland Water 3:105–118
Lovley DR (1991) Dissimilatory Fe(III) and Mn(IV) reduction. Microbiol Rev 55:259–287
Lovley DR, Philips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments. Appl Environ Biol 51:683–689
Malecki LM, White JR, Reddy KR (2004) Nitrogen and phosphorus flux rates from sediment in the Lower St. Johns River Estuary. J Environ Qual 33:1545–1555
Mitchell AM (2002) Anaerobic nutrient cycles in freshwater sediments. PhD thesis, School of Science and Technology, Charles Sturt University, Australia
Mitchell AM, Baldwin DS, Rees GN (2005) Alterations to potential phosphorus release processes from anaerobic freshwater sediments with additions of different species of labile carbon. In: Serrano L, Golterman H (eds) Phosphates in sediments, Backhuys Publishers, Kerkwerve, The Netherlands, pp 43–45
Müller S, Mitrovic SM (2015) Phytoplankton co-limitation by nitrogen and phosphorus in a shallow reservoir: progressing from the phosphorus limitation paradigm. Hydrobiologia 744:255–269
Nowlin WH, Evarts JL, Vanni MJ (2005) Release rates and potential fates of nitrogen and phosphorus from sediments in a eutrophic reservoir. Freshwater Biol 50:301–322
Penn MR, Auer MT, Doerr SM, Driscoll CT, Brooks CM, Effler SW (2000) Seasonality in phosphorus release rates from the sediments of a hypereutrophic lake under a matrix of pH and redox conditions. Can J Fish Aquat Sci 57:1033–1041
R Development Core Team (2011) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, URL http://www.Rproject.org/, ISBN 3-900051-07-0
Reddy KR, Fisher MM, Ivanoff D (1996) Resuspension and diffusive flux of nitrogen and phosphorus in a hypereutrophic lake. J Environ Qual 25(2):363–371
Rysgaard S, Risgaard-Petersen N, Sloth NP, Jensen K, Nielsen LP (1994) Oxygen regulation of nitrification and denitrification in sediments. Limnol Oceanogr 39(7):1643–1652
Saunders DL, Kalff J (2001) Nitrogen retention in wetlands, lakes and rivers. Hydrobiologia 443:205–211
Schallenberg M, Kalff J (1993) The ecology of sediment bacteria in lakes and comparisons with other aquatic ecosystems. Ecology 74:919–934
Seitzinger SP (1988) Denitrification in freshwater and coastal marine ecosystems: ecological and geochemical significance. Limnol Oceanogr 33:702–724
Sherman BS, Webster IT, Jones GJ, Oliver RL (1998) Transitions between Aulacoseira and Anabaena dominance in a turbid river weir pool. Limnol Oceanogr 43:1902–1915
Søndergaard M, Jensen JP, Jeppesen E (2001) Retention and internal loading of phosphorus in shallow eutrophic lakes. Scientific World Journal 1:427
Søndergaard M, Jensen JP, Jeppesen E (2003) Role of sediment and internal loading of phosphorus in shallow lakes. Hydrobiologia 506:135–145
Søndergaard M, Bjerring R, Jeppesen E (2013) Persistent internal phosphorus loading during summer in shallow eutrophic lakes. Hydrobiologia 710:95–107
Spears BM, Carvalho L, Perkins R, Kirika A, Paterson DM (2007) Sediment phosphorus cycling in a large shallow lake: spatio-temporal variation in phosphorus pools and release. Hydrobiologia 584:37–48
Stookey LL (1970) Ferrozine—a new spectrophotometric reagent for iron. Anal Chem 42:779–781
Törnblom E, Rydin E (1998) Bacterial and phosphorus dynamics in profundal Lake Erken sediments following the deposition of diatoms: a laboratory study. Hydrobiologia 364:55–63
Torres IC, Inglett KS, Reddy KR (2011) Heterotrophic microbial activity in lake sediments: effects of organic electron donors. Biogeochemistry 104:165–181
Ulrich KU, Pöthig R (2000) Precipitation of aluminium and phosphate affected by acidification. Acta Hydrochim Hydrobiol 28:313–322
Wang D, Huang Q, Wang C, Ma M, Wang Z (2007) The effects of different electron donors on anaerobic nitrogen transformations and denitrification processes in Lake Taihu sediments. Hydrobiologia 581:71–77
Watts CJ (2000) The effect of organic matter on sedimentary phosphorus release in an Australian reservoir. Hydrobiologia 431:13–25
Westhorpe DP, Mitrovic SM (2012) Dissolved organic carbon mobilisation in relation to variable discharges and environmental flows in a highly regulated lowland river. Mar Freshwater Res 63:1218–1230
Wilhelm S, Adrian R (2008) Impact of summer warming on the thermal characteristics of a polymictic lake and consequences for oxygen, nutrients and phytoplankton. Freshwater Biol 53:226–237
Xu B, Enfors S-O (1996) Influence of nitrate starvation on nitrite accumulation during denitrification by Pseudomonas stutzeri. Appl Microbiol Biotechnol 45:229–235
Acknowledgments
We would like to thank Hunter Water for funding this project under the Grahamstown Dam Adaptive Management Strategy, in particular Bruce Cole for management of the project and Ian Graham, Stuart Bailey, Jim Carter, Greg Mason and Paul for their help with boat operations and sampling. Further, we are grateful for the assistance of Martin Pfeil in the field.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Brian Kronvang
Rights and permissions
About this article

Cite this article
Müller, S., Mitrovic, S.M. & Baldwin, D.S. Oxygen and dissolved organic carbon control release of N, P and Fe from the sediments of a shallow, polymictic lake. J Soils Sediments 16, 1109–1120 (2016). https://doi.org/10.1007/s11368-015-1298-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-015-1298-9