
Abstract
The insufficient hazard thresholds of specific individual aromatic hydrocarbon compounds (AHCs) with diverse structures limit their ecological risk assessment. Thus, herein, quantitative structure–activity relationship (QSAR) models for estimating the hazard threshold of AHCs were developed based on the hazardous concentration for 5% of species (HC5) determined using the optimal species sensitivity distribution models and on the molecular descriptors calculated via the PADEL software and ORCA software. Results revealed that the optimal QSAR model, which involved eight descriptors, namely, Zagreb, GATS2m, VR3_Dzs, AATSC2s, GATS2c, ATSC2i, ω, and Vm, displayed excellent performance, as reflected by an optimal goodness of fit (R2adj = 0.918), robustness (Q2LOO = 0.869), and external prediction ability (Q2F1 = 0.760, Q2F2 = 0.782, and Q2F3 = 0.774). The hazard thresholds estimated using the optimal QSAR model were approximately close to the published water quality criteria developed by different countries and regions. The quantitative structure–toxicity relationship demonstrated that the molecular descriptors associated with electrophilicity and topological and electrotopological properties were important factors that affected the risks of AHCs. A new and reliable approach to estimate the hazard threshold of ecological risk assessment for various aromatic hydrocarbon pollutants was provided in this study, which can be widely popularised to similar contaminants with diverse structures.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Unreasonable petrochemical pollutant discharge, frequent oil spill accidents, and biomass fuel combustion have caused serious ecological losses from petroleum hydrocarbon pollutants during the rapid development of industrialisation and urbanisation (Boehm and Page 2007; Margesin and Schinner 2001; Othman et al. 2023; Zhao et al. 2020). Among these oil pollutants, aromatic hydrocarbon compounds (AHCs) have attracted widespread attention owing to their high biotoxicity, severe negative biological effects, and strong persistence in the environment (Abdel-Shafy and Mansour 2016; Head et al. 2006; Liu et al. 2022). AHCs affect the physiological processes and biological functions of environmental organisms through various methods such as volatilisation, migration, ingestion, and breathing, causing serious damages to their biological organs and tissues and thus disturbing endocrine, nervous, reproductive, and other metabolic processes (Cousin and Cachot 2014; Diggs et al. 2011; Gamboa et al. 2008; Lewtas 2007; Lotufo and Fleeger 1997; Zhao et al. 2020). Thus, it is imperative to accurately and rapidly assess the ecological risks of AHCs. However, the accurate ecological risk assessment (ERA) of AHCs remains a challenge owing to the lack of hazard thresholds of specific individual AHCs because of their abundance and diverse structures in the environment. Therefore, it would be an effective solution to develop a reliable approach to directly estimate the hazard thresholds of AHCs to improve the ERA for AHCs.
Biotoxicity is an important and immediate indicator of ERA, as recommended by numerous authoritative international environmental organisations such as Organisation for Economic Co-operation and Development (OECD), European Centre for Ecotoxicology and Toxicology of Chemicals (ECETOC), and United States Environmental Protection Agency (US EPA) (ECETOC 1993; OECD 1992; USEPA 1992). However, biotoxicity testing procedures are usually complicated, expensive, and time-consuming (OECD 1984a, 1984b) and often cause potential debate concerning animal ethics, especially for threatened and endangered species (Ha et al. 2019; Wang et al. 2014; Zvinavashe et al. 2008). Quantitative structure–activity relationship (QSAR) can serve as an important alternative for biotoxicity testing, as it can establish mathematical models for biotoxicity estimation based on the quantitative relations between molecular structure and biotoxicity (Bo et al. 2023; Hamadache et al. 2016; Redl et al. 1974; Tropsha 2010). In the past decades, the associations between toxicity effects and molecular structures have been examined for various pollutants using QSAR models, which have been proved as an effective solution for biotoxicity estimation. Various mathematical and statistical techniques, including multiple linear regression (MLR), artificial neural network, and support vector machine, have been applied in the QSAR development (Lei and Shiverdecker 2019; Liu and Long 2009; Xu et al. 2011). A series of molecular descriptors have been successfully used to develop QSAR models for toxicity estimation (Cao et al. 2018; Hao et al. 2019, 2020; Singh et al. 2023), such as the octanol–water partition coefficient (Log Kow), energy of the highest/lowest unoccupied molecular orbital (Ehomo/Elumo), electrophilicity index (ω), and average centred Broto–Moreau autocorrelation index (AATSC0p), that relate to lipophilicity and electronic and topological properties, which have been identified as important factors affecting biotoxicity (Pandey et al. 2020; Wang et al. 2023; Yang et al. 2020). High-quality QSAR models have been applied to correctly estimate the toxicity of polycyclic aromatic hydrocarbons (PAHs) in rats (Rattus norvegicus) (Sun et al. 2021), benzo-triazoles in fish (Oncorhynchus mykiss) (Cassani et al. 2013), pesticides in honeybees (Apis mellifera L.) (Hamadache et al. 2018), and algae (Skeletonema costatum) (Yang et al. 2021) and emerging contaminants such as pharmaceuticals and personal care products and endocrine-disrupting chemicals in crustaceans (Dugesia japonica) (Önlü and Saçan 2018). Most of the existing QSAR models were developed based on acute or chronic toxicity endpoints such as LC50, EC50, LD50, and NOEC of individual species. However, it remains a challenge to develop QSAR models to directly obtain the hazard threshold concentration of pollutants with diverse structures.
Defining appropriate hazard threshold is critical for the accurate risk assessment of pollutants. The hazardous concentration for 5% of species (HC5) derived from the species sensitivity distribution (SSD) models was often used to characterise the allowable hazard threshold to protect 95% of species in a community from the significant impacts of pollutants (Aldenberg and Slob 1993; Maltby et al. 2005; Posthuma et al. 2002; Vighi et al. 2006). HC5 describes the biological effects at a community or ecosystem level rather than at an individual level and can aid in forming a rapid response of early warning to organismal damages, thereby providing adequate safety information for an ecosystem (Ding et al. 2018; Fedorenkova et al. 2010; Maltby et al. 2009). HC5 has become an important and sensitive indicator for establishing environmental hazard thresholds and assessing the ecological risks of pollutants in numerous countries and regions (Luit et al. 2003; Margesin and Schinner 2001; USEPA 1985). For example, the effective acute HC5 was successfully applied in proposing the sediment quality criteria for nonionic organic chemicals (Di Toro and McGrath 2000; USEPA 1993) and the acute water quality criteria for dibutyltin dilaurate (Zhang et al. 2017). The HC5 derived from chronic toxicity data was also used to quantitatively characterise the ecological risk of nonylphenol in coastal waters of China using the assessment factor and risk quotient methods (Gao et al. 2014). However, little attention was paid to investigate the quantitative relationship between the molecular structures and hazard thresholds of AHCs and develop QSAR models for the direct estimation of the hazard thresholds (such as HC5).
Herein, QSAR models between the molecular structures and hazard threshold concentrations of AHCs were initially developed based on the HC5 derived using the SSD models and the molecular structure quantified via the PADEL software (Yap 2011) and ORCA software (Neese 2022). Then, the hazard threshold concentrations estimated using the developed QSAR models were compared with the published water quality criteria. The objectives of this study were as follows: (1) to develop QSAR models with high accuracy in directly estimating the hazard threshold of AHCs for risk assessment improvement and (2) to investigate the quantitative relationship between the molecular structures and hazard thresholds of AHCs for an in-depth understanding of the toxicity mechanism.
Materials and methods
Toxicity data collection of AHCs
Herein, all the existing acute toxicity concentrations of all the studied toxicity endpoints (e.g. inhibition of growth, reproduction and development, dysfunction of physiology and biochemistry, and mortality) of AHCs, including the median lethal concentration (LC50) and median effective concentration (EC50), were collected from ECOTOX knowledgebase (https://cfpub.epa.gov/ecotox/index.cfm) and other published literatures. The collected toxicity data involved multiple toxicity test organisms, such as various aquatic and terrestrial toxicity organisms at multiple trophic levels, including phytoplankton, crustaceans, fish, worms, molluscs, and insects/spiders (Table S1). The collected toxicity data were used to derive HC5 concentration in this study, as shown in Table 1.
A wide variety of AHCs with diverse structures including benzenoid aromatic hydrocarbons (monocyclic, polycyclic and biphenyl), non-benzenoid aromatic hydrocarbons (pyridine, quinolone, dibenzofuran and acridine), and their derivatives were involved in this study. Detailed chemical information including chemical names, abbreviations, CAS numbers, and chemical formulas of these AHCs are shown in Table S2, while their molecular structures are shown in Fig. 1.

Two-dimensional molecular structure diagrams of 40 AHCs
HC5 of AHCs
HC5 is usually used to characterise the allowable hazard threshold to protect 95% of species in the ecosystem from significant negative impacts, characterising the sensitivity of biological communities to chemicals (Traas et al. 2002; Korsman et al. 2016; Jesus et al. 2022). It has provided an important basis for establishing the environmental limits of pollutants in numerous countries and regions, such as Europe, USA, and China (Eduljee 2000; Kemmlein et al. 2009; Wu et al. 2015). Herein, the HC5 of AHCs was derived using the SSD models, and it covered at least three trophic levels, including primary consumers, secondary consumers, and producers, as per the US EPA requirements for an excellent SSD model (Lu et al. 2020, 2018; Wang et al. 2021). To improve the accuracy of HC5, the toxicity concentrations of different endpoints that meet the requirements were used to build the SSD models (USEPA 1985). The SSD curve of AHCs was fitted considering the logarithm of the toxicity concentration as the dependent variable and the cumulative probability as the independent variable using six prior distribution models (normal, logistic, triangular, Gumbel, Weibull, and Burr) and four fitting methods (maximum likelihood [ML], moment estimators [MO], linearisation [GR], and metropolis hastings [MH]) through the SSD Toolbox software. The proportion of the simulated discrepancy statistics (P) was used to evaluate the quality of the SSD models, which described the goodness of fit between the empirical and parametric cumulative distribution functions. The closer the P-value is to 1, the better the goodness of fit (Posthuma et al. 2002). The HC5 of the AHCs was finally obtained from their optimal SSD curve with the best goodness of fit. The obtained HC5 were subsequently log-normalised to facilitate statistical analysis, which were negatively correlated with the risk of AHCs.
Molecular structure quantification of AHCs
In total, 1468 molecular descriptors were used to characterise the molecular structure of the AHCs. First, the two-dimensional molecular structures of the AHCs were determined using ChemDraw 20.0 (Fig. 1) and initial geometric optimisations were performed based on the molecular mechanics method to minimise to their lowest energy conformation using Chem3D 20.0. Then, 1440 molecular descriptors (e.g. autocorrelation descriptors, constitutional descriptors, and topological descriptors) were obtained using the PADEL software. Twenty quantum chemical descriptors (e.g. electrical descriptors and thermodynamic descriptors) were directly acquired using the ORCA software at the B3LYP/6-311G + + (d, p) level based on the density functional theory. χ, η, S, ω, α, and Qii were calculated according to the formulas shown in Table S3. Octanol–water partition coefficient (Log Kow) and molecular weight (MW) of AHCs were directly calculated using the EPI Suite 4.1 according to their isomeric SMILES and CAS number.
QSAR model development and validation
Herein, QSAR models were developed using MLR analysis based on ordinary least squares approach and principal component regression (PCR) analysis via the Statistical Package for the Social Sciences (SPSS) 26 software. MLR is commonly used in QSAR linear modelling which is applicable for a small amount of data. Before QSAR model development, a screening process was conducted for all the obtained molecular descriptors to remove the inter-correlated ones, avoiding overfitting in the QSAR modelling. First, the molecular descriptors with missing values were manually excluded. Then, the Pearson correlation coefficients between the remaining molecular descriptors were analysed using the SPSS 26 software and the molecular descriptors with an absolute value of Pearson correlation coefficient of > 0.95 were removed to eliminate multicollinearity (Cai et al. 2022; Hamadache et al. 2016). After screening, 238 molecular descriptors (212 PADEL descriptors and 26 quantum chemical descriptors) that characterised the electrical effects, geometric structures, and thermodynamic properties of AHCs were used to develop the QSAR models (Table S4). The specific modelling procedures were as follows.
First, the logHC5 of the AHCs were divided into training set and testing sets with a best ratio of 4:1 after modelling exercises with different proportion divisions using a random distribution approach. This ensures the uniform selection of the dataset and wide distribution of the training set. The training set was used to develop the QSAR models to reduce bias in model performance and enhance the fitting and generalisation capabilities of the developed models. Meanwhile, the testing set was used to initially evaluate the external prediction ability of the developed QSAR model. Stepwise regression was performed via MLR analysis to automatically eliminate variables with complete multicollinearity and to gradually remove the unimportant molecular descriptors according to the absolute value of standardised coefficients and the significance of t-test until the significance level of < 0.05 (Zhang et al. 2007). An initial regression model was obtained after mathematical substitution via PCR analysis following the principle of orthogonal linear transformation to achieve the dimensionality reduction of the data and the further elimination of multicollinearity. The descriptors with tolerance (T > 0.1) and variance inflation factors (VIF < 5) indicated no collinearity (Xu and Zhang 2001; Yang et al. 2021; Zhang et al. 2007). The goodness of fit of the QSAR models was characterised by the coefficient of determination (R2), adjusted R2 (R2adj), root-mean-square error (RMSE), and mean absolute error (MAE). The QSAR model was considered highly reliable if R2 and R2adj of the regression model were close to 1 with a smaller RMSE and MAE value. It is suggested that MAE should be ≤ 0.1 multiplied by the range of the training set in a reliable QSAR model (Roy et al. 2016).
Then, the leave-one-out (LOO) method was used for double cross-validation (internal validation and external validation) to evaluate the stability and reliability of the developed QSAR models (Baumann and Baumann 2014). Internal stability was assessed using the LOO cross-validation coefficient (Q2LOO) and consistency correlation coefficient (CCCCV). The external predictability was estimated using the validation parameters (Q2F1, Q2F2, and Q2F3) and coordination correlation coefficient (CCCEXT). The calculation equations for these statistical validation parameters were summarised in Table S3. The limits of these validation parameters were in line with the OECD Requirements Guidelines for QSAR modelling (Golbraikh and Tropsha 2002; OECD 2014; Tropsha 2010), as shown in Table 3.
Finally, after the double cross-validation, y-randomisation tests were performed 20 times to further justify the chance correlation between the original descriptor matrix and the scrambled vector of response (logHC5). The average coefficients of the y-randomisation models (R2yrand and Q2yrand) of the developed models within the threshold value (R2yrand < 0.4 and Q2yrand < 0.05) were considered as non-accidental modelling.
According to the procedures described above, a final QSAR model for HC5 estimation was developed, whose general form was shown as follows:
where endpoint (yi) is described with the estimated logHC5; independent variables (x1, x2…, xn) are the parameters of the most relevant molecular descriptors; k1, k2…, kn are their regression coefficients of the molecular descriptors; and k0 is the constant term.
Application domain analysis of the developed QSAR models
The leverage approach combined with the ratio of the residual to the standardised residual was adopted to describe the application domain (AD), as per the OECD guideline for QSAR model development (OECD 2014). Then, the AD was visualised using the Williams plot to evaluate the reliability of the developed QSAR model (Singh et al. 2023). AD was defined by the normalised residual/standard residual outliers (± 3) and the warning leverage value (h*). A chemical with a leverage value (hi) higher than h* was identified as a structural outlier that is beyond the AD of the developed models (Gramatica et al. 2013). A chemical with a ratio of the residual to the standardised residual of > 3 or < -3 was identified as a response outlier of the QSAR model. The hi and h* of the chemicals were calculated as shown in the equations below.
where xi is the row vector of the molecular descriptor matrix for the ith chemical, X is the matrix of molecular descriptors for the training set, k is the number of molecular descriptors involved in the QSAR model, and m is the number of the compounds in the training set.
Accuracy of the estimated hazard thresholds using the developed QSAR models
In total, six AHCs, including benzo[a]pyrene (BaP), cumene (CUM), o-xylene (OX), m-xylene (MX), p-xylene (PX), and naphthalene (NAP), were selected to verify the accuracy of the estimated hazard thresholds using the developed QSAR models. The estimated HC5 of these AHCs were transferred to the acute water quality criteria (AWQC) and then compared with the published water quality standards of different countries and regions to evaluate the estimation accuracy of the hazard thresholds via the developed QSAR models. The AWQC that were within the same order of magnitude as the published water quality criterion was considered as the developed QSAR model, with high accuracy in estimating the hazard thresholds of AHCs, as proposed by Dyer et al. (2008).
The AWQC were calculated as shown as follows:
where the AF was considered 5 according to the ‘worst case scenario’ of ecological risk (Sun et al. 2017).
Quantitative relationship analysis between molecular structure and hazard threshold
Herein, the standardised coefficients of the molecular descriptors involved in the developed QSAR models were used to characterise the quantitative relationship between the molecular structure and hazard threshold of the AHCs. The standardised coefficients were calculated by standardising the regression coefficients of the involved molecular descriptors in Eq. (1). The standardised coefficients reflected the influence of these molecular descriptors on the hazard thresholds of AHCs. The higher the weight of the standardised coefficient of a molecular descriptor, the greater the influence of the molecular descriptor on the hazard threshold of AHCs.
Results and discussion
Toxicity of the AHCs
Acute and chronic toxicity concentrations of 40 AHCs in a total of 225 species (Table S1) were collected and screened to obtain the HC5 for the QSAR model development of AHCs (Table 1). From the EC50 or LC50 values, the acute toxicity was noted to vary considerably with different AHCs. The mean EC50 or LC50 values of the 40 AHCs varied 5 orders of magnitude (from 0.2 to 1049.5 mg/L). BaP and pyrene (PYR) were determined as the most toxic among the studied AHCs, with the minimum EC50 or LC50 (0.0006 and 0.0009 mg/L) to the algae C.fusca and the crustaceans A.bahia, respectively. In contrast to BaP and PYR, pyridine (PYD) was the least toxic to amphibians X. laevis, having the highest EC50 or LC50 concentration (9550.0 mg/L).
The species tested for the toxicity of AHCs included planktonic algae, invertebrates (e.g. crustacean, worms, molluscs, and insects/spiders), and vertebrates (e.g. fish, amphibians, and reptiles) (Table S1). A majority of crustaceans (e.g., D. magna) and algae (e.g., R. subcapitata) were deemed to be the most sensitive, while fish (e.g. P. promelas) and amphibians (e.g. A. bahia) were most tolerant to the AHCs at maximum and minimum EC50 or LC50 (Table 1), indicating that AHCs were more likely to endanger invertebrates than vertebrates. Consistent results were also reported by previous studies, stating that benthic invertebrates (e.g. D. magna and A. salina) were more susceptible to PAHs than fish (e.g. D. rerio and O. latipes) with high mobility and metabolic capacity; this might be attributed to differences in ingestion pathways, toxic characteristics, metabolic capacity, and habitats (Honda and Suzuki 2020).
HC5 of the AHCs
Herein, the species of the SSD models included at least ‘three phyla and eight families’, with priority given but not limited to the toxicity concentrations from three trophic levels (algae, daphnia, and fish). The best fitting method, distribution models, and P-values are shown in Table S5. The P-values of 34 AHCs ranged from 0.8 to 1.0. The remaining six AHCs used the metropolis hastings algorithm to fit the SSD curves, with P-values ranging from 0.4 to 0.6. In this situation, the closer the P-value is to 0.5, the better goodness of fit for the metropolis hastings algorithm.
The slope of the SSD curve reflected the difference in species sensitivity to toxic substances (Beiras and Schönemann 2021). As can be seen from the optimal SSD curves demonstrated in Fig. 2, sensitivities among the species varied with different AHCs. For monocyclic aromatic hydrocarbons (MAHs), no significant difference in the species sensitivity was observed at low logHC5 concentration (< 1 mg/L); however, species sensitivity gradually increased with increasing logHC5 concentration (Fig. 2a). Nevertheless, PAHs such as NAP, acenaphthene (ACE), and fluorene (FLU) were usually more sensitive to the selected organism species (Fig. 2b and d) than MAHs (Fig. 2a).

Optimal SSD curves fitted by SSD Toolbox of monocyclic aromatic hydrocarbons (MAHs) (a), polycyclic aromatic hydrocarbon (PAHs) (b), derivatives of MAHs (c), and derivatives of PAHs (d)
As shown in Fig. 3, the HC5 of the AHCs were widely distributed (from 0.0009 to 44.119 mg/L), ranging 6 orders of magnitude. The benzene ring numbers of AHCs significantly affected the HC5 in the following order: BEN > NAP > ANT > PYR > BaP. The HC5 of the AHCs with three or five benzene rings were generally lower than that with one or two benzene rings, indicating that the ecological risk of AHCs increased with increasing number of benzene rings. Previous studies have reported that increase in the number of benzene rings may lead to high hydrophobicity and strong persistence, thus exacerbating the risks of PAHs (Jesus et al. 2022; Mackay et al. 1992). Additionally, the HC5 of aromatic hydrocarbons were generally lower than that of their derivatives. The hazard levels of AHCs were as follows: PAHs > derivatives of PAHs > MAHs > derivatives of MAHs. Taking MAHs as an example, lower HC5 and higher risk were observed for xylene (OX, MX, and PX) than alcohols (phenol [PHN] and phenylmethanol [PHM]) and phenols (o-cresol [OC], m-cresol [MC], and p-cresol [PC]). A higher risk of MAHs compared with their derivatives may be due to their high hydrophobicity and degradability, rendering them easier to combine with biological cells (Rorije et al. 1998).

Derived HC5 (mg/L) from the optimal SSD curves of 40 AHCs
Developed QSAR models
Herein, three highly reliable QSAR models were developed using MLR and PCR analyses, considering the logHC5 as the dependent variable and the molecular descriptor as the independent variable. As shown in Table 2, models (1), (2), and (3) were developed based on the PADEL descriptors, quantum chemical descriptors, and both of them, respectively. High R2 (0.998, 0.907, and 0.937) and R2adj (0.908, 0.871, and 0.918) and low RMSE (0.395, 0.468, and 0.375) and MAE (0.151, 0.370, and 0.284) were observed in Table 3, indicating that the developed QSAR models have excellent fitting performance. R2 estimates the proportion of the changes of dependent variable explained via regression. The closer R2 is to 1, the better the goodness of fit for the developed models. The threshold value of MAE for models (1), (2), and (3) was 0.571, 0.485, and 0.535, respectively, which were calculated according to the range of the logHC5 of the AHCs in the training sets (Table S6). The MAE (0.017, 0.266, and 0.214) of the three QSAR models in this study were lower than the MAE threshold value (Table 3), suggesting that the developed three models have good metrics performance and high prediction accuracy.
The three QSAR models developed from different combinations of molecular descriptors have demonstrated acceptable internal stability and external predictability. They explained > 85% variance of the training set with R2LOO (0.998, 0.885, and 0.937) and 60% variance of the testing set with R2test (0.616, 0.706, and 0.759). The R2LOO of the three models was comparable with the corresponding Q2LOO (0.939, 0.860, and 0.869). Additionally, high CCCtrain (0.971, 0.928, and 0.927) and low RMSE (0.308, 0.500, and 0.165) have also indicated reliable internal fitting ability and robustness of the developed models. The external validation parameters of the three models demonstrated good predictive performance on the HC5 of AHCs, with Q2F1 (0.625, 0.707, and 0.760), Q2F2 (0.616, 0.706, and 0.782), and Q2F3 (0.744, 0.750, and 0.774), meeting the criteria thresholds of OECD principles (Q2F1~F3 > 0.6). High CCCtest (0.7998, 0.861, and 0.914) further verified the excellent predictive ability of the three models. As per the Williams plot analysis shown in Fig. 4a, two response outliers (benzoquinone (BEQ) and 4-tert-octylphenol (4-TEO)) were detected whose normalised residual/standard residual (3.357 and 4.082, respectively) were outside the AD (± 3) of model (1). The residuals between the experimental and estimated logHC5 of the two outliers were determined to range from 1.3 to 1.6, could probably owing to the underrated experimental data rather than to molecular descriptors according to previous studies (Hamadache et al. 2018; Kar and Roy 2012). As shown in Fig. 4 b and c, almost all the AHCs were within the AD. The leverage values hi for all the AHCs were concentrated between 0 and 0.5 and below the warning leverage values h* (2.531, 0.656, 0.844), indicating no structural outliers and response outliers for the developed models. The average coefficients of y-randomisation models further indicated that the developed models were not accidental modelling, with R2yrand (0.169, 0.188, and 0.192) and Q2yrand (0.109, − 0.024, and 0.014) significantly lower than the threshold value (R2yrand < 0.4 and Q2yrand < 0.05) (Table 3).

Williams plots for model (1) (a), model (2) (b), and model (3) (c), and the correlation between experimental and estimated logHC5 values of model (1) (d), model (2) (e), and model (3) (f)
Model (3) was identified to be the best developed QSAR model for the hazard threshold estimation of AHCs in this study owing to its best fitting parameters, internal and external validation parameters, as well as the best prediction performance. The results of significance analysis revealed that there was no significant difference between the experimental and estimated logHC5 with a variance significance of 0.845 (> 0.05) of Levene test for variance equality and equalisation significance of 0.903 (> 0.05) of t-test for equality of means (Table 2), confirming the optimal fitting relationship and generalisation ability of model (3). Visually, as shown in Fig. 4 d, e, and f, the best agreement was observed for model (3) between the experimental and estimated logHC5 (Table S7) for both the training and testing sets.
The hazard thresholds of six AHCs estimated using model (3) in this study were compared with the published criterion of Environmental Quality Standards for Surface Water (GB3838-2002) of China and Environmental Quality Standards for Priority Substances of the European Union. As shown in Fig. 5a, the AWQC values of the six AHCs were approximate to the published water quality limits. For example, the estimated logAWQC of xylene (2.94, 2.97, and 2.80) were within an order of magnitude with the criterion GB3838-2002 (2.70) (Dyer et al. 2008). The estimated logAWQC (2.3) of PAHs such as NAP was considerably approximate to the criterion from the European Union (2.1). These results have well supported the idea that the developed QSAR Model (3) is highly accurate in estimating the hazard thresholds.

Comparison of acute water quality criteria of AHCs with relevant standards among different countries and regions (a). Weights of the standardized coefficients of molecular descriptors in QSAR Model (3) (b)
Underlying toxicity mechanism of the quantitative structure–hazard threshold relationship
As shown in model (3) (Table 2), three topological descriptors (Zagreb, GATS2m, and Vm), four electrotopological descriptors (VR3_Dzs, AATSC2s, GATS2c and ATSC2i), and one electrophilic descriptor (ω) were significantly associated with the hazard thresholds of AHCs. The detailed definitions of these descriptors are shown in Table S8. The values of the eight molecular descriptors varied significantly among the different AHCs (Table S9). The ratio of the maximum and the minimum values of ω, VR3_Dzs, and Zagreb was > 5. The maximum values of Vm, GATS2c, and GATS2m were 2.96, 2.73, and 1.85 times the minimum values, respectively. The values of ATSC2i and AATSC2s varied extensively from − 13.6294 to 16.4054 and from − 0.2755 to 0.3765, respectively. These results indicate that the AHCs used for developing the QSAR model (3) varied significantly with regard to spatial topological structure, electrotopological state and electrophilic properties. The AHCs with various molecular structures effectively supported the development of the QSAR model and the investigation of quantitative relationship between molecular structure and hazard threshold.
The electrotopological descriptors (VR3_Dzs and GATS2c) and topological descriptor (Vm) were determined to be positively correlated with the logHC5, whereas the topological descriptors (Zagreb and GATS2m), electrophilic descriptor (ω), and electrotopological descriptors (AATSC2s and ATSC2i) were negatively correlated (Fig. 5b). The quantitative structure–hazard threshold relationship in model (3) demonstrated the important influence of molecular structure on ecological risks wherein it was deemed beneficial for understanding the toxicity mechanisms of AHCs.
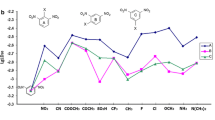
Topological descriptors including Zagreb, GATS2m, and Vm were found to be the most important molecular descriptors affecting the hazard thresholds of AHCs, as they have the highest influence weight (54%) in all the influencing molecular descriptors. Zagreb was a graph theoretical topological descriptor that measured the number and types of connections between atoms in a molecule. It was identified to be the maximal influencing factor (accounted for 31.3% of the influencing weight) affecting the hazard threshold of AHCs (Fig. 5b). Herein, AHCs with a higher Zagreb value (e.g. BaP, FLT, and PYR with Zagreb values of 120, 94, and 94, respectively) were usually more toxic and had a lower logHC5 concentration (− 2.999, − 2.118, and − 2.705) (Fig. 6). Significant positive correlation between hyper-Zagreb and cytotoxicity was also found in natural compounds such as vitamin E and caffeic acid (Parvathi and Dodoala 2022). A higher Zagreb value indicated a greater number and complexity of interatomic connections in the chemical molecule, making the chemical more difficult to be metabolised in and cleared from the biological system, thus increasing its toxicity and potential risks (Janežič et al. 2017). GATS2m, which is defined as the Geary autocorrelation − lag 2/weighted by mass and is known to encode the topological distribution of atomic mass along with the spatial molecular graph, was determined to be another important influencing factor (accounted for 16.9% of the influencing weight) affecting the hazard threshold of AHCs. As shown in Fig. 6, lower logHC5 values (0.015 and − 0.462, respectively) were observed for the AHCs such as NAP and BIP that showed a higher GATS2m values (1.02 and 1.07, respectively). The negative effect of GATS2m on the hazard threshold of AHCs is most likely due to the antioxidant activity (Saber et al. 2019). Previous studies have shown that PAHs could undergo biotransformation reactions after being taken up by organisms, which subsequently stimulate the production of reactive oxygen species and induce oxidative damage to increase toxicity and risk (Hannam et al. 2010; Livingstone 1991; Valavanidis et al. 2006). Vm, which is the van der Waals volume of molecule, was also an influential descriptor (accounted for 6.1% of the influencing weight) affecting the hazard threshold of AHCs. Molecular volume has been proved to be related to hydrophobicity, thus affecting the toxicity and risks of chemicals (Di Marzio et al. 2001; Wang et al. 2023). The Vm of a chemical was usually observed to negatively influence toxicity (Zhu et al. 2010). Similar results that AHCs with lower Vm values (e.g., BEQ and QUL with Vm values of 87.102 and 90.896, respectively) showed lower logHC5 values (− 3.142 and − 0.066, respectively) were also found in this study (Fig. 6). Generally, chemicals with lower Vm can easily cross through the cytoderm or cytomembrane and enter organisms, thereby giving functional incapacitation of cells and organs (Ding et al. 2011; Wang et al. 2022).

Positive and negative contributions of important molecular descriptors on the logHC5 for some specific compounds in model (3); molecular descriptors highlighted with ↑ and blue colour, positive contribution; molecular descriptors highlighted with ↓ and red colour, negative contribution
Electrotopological descriptors including VR3_Dzs, AATSC2s, GATS2c, and ATSC2i have showed the significant importance of electronic information on the hazard thresholds of AHCs, accounting for 31.7% of the weight in all the influencing molecular descriptors. VR3_Dzs, the logarithmic Randic-like eigenvector-based index from Barysz matrix/weighted by I-state, has significantly affected the hazard threshold of AHCs (accounted for 10.5% of the influencing weight). The hazard threshold (logHC5) of the AHCs with a higher VR3_Dzs value (e.g. BaP, with a value of 13.193) was usually observed to be higher (Fig. 6). A higher VR3_Dzs value is explained as more partitioning of the effect of non-σ electrons throughout the σ bonds starting from the atom in question, which made it more difficult for electrons of the chemical to interact between molecules, thus reducing the toxicity and risk of AHCs (Önlü and Saçan 2018). AATSC2s, GATS2c, and ATSC2i are two-dimensional sub-group autocorrelation descriptors determined by the corresponding number and specific weighting scheme at the real of ‘lag’, which accounted for 8.8%, 7.3%, and 5.1% of the weight in all of the influencing molecular descriptors, respectively. As shown in Fig. 6, the AHCs (e.g. CAF), which had the higher ATSC2i (16.405) and AATSC2s (0.377) values and the lower GATS2c value (0.825), presented a relatively higher hazard threshold (logHC5). AATSC2s and GATS2c have described the properties of atomic mass, polarisability, and electronegativity and reflected the attribute distribution of specific atoms that explained the ability of electron recovery and release (Adeniji et al. 2020). The difference between AATSC2s and GATS2c is that they were weighted by I-state and charges, respectively. ATSC2i was generally considered to be the absolute value of Ehomo, which could determine the possibility of attack reaction to radical attack, for example, OH radical affecting molecular properties (Cvetnic et al. 2019; Kušić et al. 2009). Electrotopological descriptors encoding electronic and topological characteristics of chemicals were usually found to significantly influence the toxicity and risk of aromatics (Cvetnic et al. 2019). Chemicals with positively charged atoms and higher ionisation potential have led to increased toxicity and higher risk (Khan and Roy 2017). Electrotopological descriptors could influence the interactions at the active sites and form hydrogen bonds, thereby potentially creating the risk of AHCs (Barzegar et al. 2017).
Electrophilic descriptor ω was also observed to be an important molecular descriptor as it affects the hazard thresholds of AHCs, accounting for 14.1% of the weight in all the influencing molecular descriptors. ω measured the global electrophilic power of the molecule and the ability of a chemical to accept electrons (Parthasarathi et al. 2004). Herein, AHCs with higher ω value such as BaP, FLT, and PYR (0.157, 0.158 and 0.137) usually showed a lower logHC5 value (Fig. 6). Higher electrophilicity was proved to enhance the toxicity of PAHs and amines and trigger mutations of nitroaromatic compounds (Huang et al. 2021; Roy et al. 2006). A chemical with higher ω value was more prone to electrophilic–nucleophilic reactions at nucleophilic sites to form covalent bonds, thus irreversibly influencing the normal functions of DNA, enzymes, structural proteins, and other biomacromolecules and subsequently increasing the toxicity and risk of the chemical (LoPachin et al. 2019).
Conclusion
Herein, an effective QSAR method was developed to estimate the hazard thresholds of AHCs to improve the ERA and investigate the quantitative relationship between the molecular structure and risk thresholds of AHCs.
-
(1)
Three effective QSAR models were developed to estimate the hazard thresholds of AHCs. Model (3), which was developed by combining the PADEL descriptors and quantum chemical descriptors, was identified as the optimal QSAR model, characterised by good fitness, excellent internal stability, external predictability, and wide applicability domain.
-
(2)
The eight molecular descriptors involved in model (3) demonstrated the importance of electrophilicity and topological and electrotopological properties affecting the hazard thresholds of AHCs. Topological descriptors (Zagreb and GATS2m), electrophilic descriptor (ω), and electrotopological descriptors (AATSC2s and ATSC2i) were negatively correlated with the hazard thresholds, whereas electrotopological descriptors (VR3_Dzs and GATS2c) and topological descriptor (Vm) were positively related to the hazard thresholds.
-
(3)
The AWQC derived from the hazard thresholds estimated using model (3) were approximate to the safety limits of AHCs as per the published water quality standards.
Data Availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Abdel-Shafy HI, Mansour MSM (2016) A review on polycyclic aromatic hydrocarbons: source, environmental impact, effect on human health and remediation. Egypt J Pet 25(1):107–123. https://doi.org/10.1016/j.ejpe.2015.03.011
Adeniji SE, Uba S, Uzairu A (2020) Theoretical modeling for predicting the activities of some active compounds as potent inhibitors against Mycobacterium tuberculosis using GFA-MLR approach. J King Saud Univ Sci 32(1):575–586. https://doi.org/10.1016/j.jksus.2018.08.010
Aldenberg T, Slob W (1993) Confidence limits for hazardous concentrations based on logistically distributed NOEC toxicity data. Ecotox Environ Safe 25(1):48–63. https://doi.org/10.1006/eesa.1993.1006
Barzegar A, Jafari MS, Hamidi H, Sadeghi M (2017) 2D-QSAR study of fullerene nanostructure derivatives as potent HIV-1 protease inhibitors. Physica E Low Dimens Syst Nanostruct 93:324–331. https://doi.org/10.1016/j.physe.2017.06.016
Baumann D, Baumann K (2014) Reliable estimation of prediction errors for QSAR models under model uncertainty using double cross-validation. J Cheminf 6(1):47. https://doi.org/10.1186/s13321-014-0047-1
Beiras R, Schönemann AM (2021) Water quality criteria for selected pharmaceuticals and personal care products for the protection of marine ecosystems. Sci Total Environ 758:143589. https://doi.org/10.1016/j.scitotenv.2020.143589
Bo T, Lin Y, Han J, Hao Z, Liu J (2023) Machine learning-assisted data filtering and QSAR models for prediction of chemical acute toxicity on rat and mouse. J Hazard Mater 452:131344. https://doi.org/10.1016/j.jhazmat.2023.131344
Boehm PD, Page DS (2007) Exposure elements in oil spill risk and natural resource damage assessments: a review. Hum Ecol Risk Assess 13(2):418–448. https://doi.org/10.1080/10807030701226293
Cai Z, Zafferani M, Akande OM, Hargrove AE (2022) Quantitative structure–activity relationship (QSAR) study predicts small-molecule binding to RNA structure. J Med Chem 65(10):7262–7277. https://doi.org/10.1021/acs.jmedchem.2c00254
Cao L, Zhu P, Zhao Y, Zhao J (2018) Using machine learning and quantum chemistry descriptors to predict the toxicity of ionic liquids. J Hazard Mater 352:17–26. https://doi.org/10.1016/j.jhazmat.2018.03.025
Cassani S, Kovarich S, Papa E, Roy PP, van der Wal L, Gramatica P (2013) Daphnia and fish toxicity of (benzo)triazoles: Validated QSAR models, and interspecies quantitative activity-activity modelling. J Hazard Mater 258–259:50–60. https://doi.org/10.1016/j.jhazmat.2013.04.025
Cousin X, Cachot J (2014) PAHs and fish-exposure monitoring and adverse effects-from molecular to individual level. Environ Sci Pollut Res 21(24):13685–13688. https://doi.org/10.1007/s11356-014-3161-8
Cvetnic M, Juretic Perisic D, Kovacic M, Ukic S, Bolanca T, Rasulev B, Kusic H, Loncaric Bozic A (2019) Toxicity of aromatic pollutants and photooxidative intermediates in water: a QSAR study. Ecotox Environ Safe 169:918–927. https://doi.org/10.1016/j.ecoenv.2018.10.100
Di Toro DM, McGrath JA (2000) Technical basis for narcotic chemicals and polycyclic aromatic hydrocarbon criteria. II. Mixtures and sediments. Environ Toxicol Chem 19(8):1971–1982. https://doi.org/10.1002/etc.5620190804
Di Marzio W, Galassi S, Todeschini R, Consolaro F (2001) Traditional versus WHIM molecular descriptors in QSAR approaches applied to fish toxicity studies. Chemosphere 44(3):401–406. https://doi.org/10.1016/S0045-6535(00)00353-2
Diggs DL, Huderson AC, Harris KL, Myers JN, Banks LD, Rekhadevi PV, Niaz MS, Ramesh A (2011) Polycyclic aromatic hydrocarbons and digestive tract cancers: a perspective. J Env Sci Heal C-Tox 29(4):324–357. https://doi.org/10.1080/10590501.2011.629974
Ding F, Guo J, Song W, Hu W, Li Z (2011) Comparative quantitative structure–activity relationship (QSAR) study on acute toxicity of triazole fungicides to zebrafish. Chem Ecol 27(4):359–368. https://doi.org/10.1080/02757540.2011.585780
Ding C, Ma Y, Li X, Zhang T, Wang X (2018) Determination and validation of soil thresholds for cadmium based on food quality standard and health risk assessment. Sci Total Environ 619–620:700–706. https://doi.org/10.1016/j.scitotenv.2017.11.137
Dyer SD, Versteeg DJ, Belanger SE, Chaney JG, Raimondo S, Barron MG (2008) Comparison of species sensitivity distributions derived from interspecies correlation models to distributions used to derive water quality criteria. Environ Sci Technol 42(8):3076–3083. https://doi.org/10.1021/es702302e
ECETOC (1993) Environmental hazard assessment of substances. ECETOC Technical Report. European Center for Ecotox-icology and Toxicology of Chemicals, Brussels
Eduljee GH (2000) Trends in risk assessment and risk management. Sci Total Environ 249(1):13–23. https://doi.org/10.1016/S0048-9697(99)00507-0
Fedorenkova A, Vonk JA, Lenders HJR, Ouborg NJ, Breure AM, Hendriks AJ (2010) Ecotoxicogenomics: bridging the gap between genes and populations. Environ Sci Technol 44(11):4328–4333. https://doi.org/10.1021/es9037287
Gamboa RT, Gamboa AR, Bravo AH, Ostrosky WP (2008) Genotoxicity in child populations exposed to polycyclic aromatic hydrocarbons (PAHs) in the air from Tabasco, Mexico. Int J Environ Res Public Health 5(5):349–355. https://doi.org/10.3390/ijerph5050349
Gao P, Li Z, Gibson M, Gao H (2014) Ecological risk assessment of nonylphenol in coastal waters of China based on species sensitivity distribution model. Chemosphere 104:113–119. https://doi.org/10.1016/j.chemosphere.2013.10.076
Golbraikh A, Tropsha A (2002) Beware of q2! J Mol Graph Model 20(4):269–276. https://doi.org/10.1016/S1093-3263(01)00123-1
Gramatica P, Chirico N, Papa E, Cassani S, Kovaric S (2013) QSARINS: a new software for the development, analysis, and validation of QSAR MLR models. J Comput Chem 34(24):2121–2132. https://doi.org/10.1002/jcc.23361
Ha H, Park K, Kang G, Lee S (2019) QSAR study using acute toxicity of Daphnia magna and Hyalella azteca through exposure to polycyclic aromatic hydrocarbons (PAHs). Ecotoxicology 28(3):333–342. https://doi.org/10.1007/s10646-019-02025-1
Hamadache M, Benkortbi O, Hanini S, Amrane A, Khaouane L, Si Moussa C (2016) A quantitative structure activity relationship for acute oral toxicity of pesticides on rats: validation, domain of application and prediction. J Hazard Mater 303:28–40. https://doi.org/10.1016/j.jhazmat.2015.09.021
Hamadache M, Benkortbi O, Hanini S, Amrane A (2018) QSAR modeling in ecotoxicological risk assessment: application to the prediction of acute contact toxicity of pesticides on bees (Apis mellifera L.). Environ Sci Pollut Res 25(1):896–907. https://doi.org/10.1007/s11356-017-0498-9
Hannam ML, Bamber SD, John Moody A, Galloway TS, Jones MB (2010) Immunotoxicity and oxidative stress in the Arctic scallop Chlamys islandica: effects of acute oil exposure. Ecotox Environ Safe 73(6):1440–1448. https://doi.org/10.1016/j.ecoenv.2010.06.012
Hao Y, Sun G, Fan T, Sun X, Liu Y, Zhang N, Zhao L, Zhong R, Peng Y (2019) Prediction on the mutagenicity of nitroaromatic compounds using quantum chemistry descriptors based QSAR and machine learning derived classification methods. Ecotox Environ Safe 186:109822. https://doi.org/10.1016/j.ecoenv.2019.109822
Hao Y, Sun G, Fan T, Tang X, Zhang J, Liu Y, Zhang N, Zhao L, Zhong R, Peng Y (2020) In vivo toxicity of nitroaromatic compounds to rats: QSTR modelling and interspecies toxicity relationship with mouse. J Hazard Mater 399:122981. https://doi.org/10.1016/j.jhazmat.2020.122981
Head IM, Jones DM, Röling WFM (2006) Marine microorganisms make a meal of oil. Nat Rev Microbiol 4(3):173–182. https://doi.org/10.1038/nrmicro1348
Honda M, Suzuki N (2020) Toxicities of polycyclic aromatic hydrocarbons for aquatic animals. Int J Environ Res Public Health 17(4):1363. https://doi.org/10.3390/ijerph17041363
Huang T, Sun G, Zhao L, Zhang N, Zhong R, Peng Y (2021) Quantitative structure–activity relationship (QSAR) studies on the toxic effects of nitroaromatic compounds (NACs): a systematic review. Int J Mol Sci 22(16):8557. https://doi.org/10.3390/ijms22168557
Janežič D, Miličević A, Nikolić S, Trinajstić N (2017) Topological complexity of molecules. Encyclopedia of Complexity and Systems Science. Springer, Berlin, Heidelberg pp 1–21. https://doi.org/10.1007/978-3-642-27737-5_554-3
Jesus F, Pereira JL, Campos I, Santos M, Ré A, Keizer J, Nogueira A, Gonçalves FJM, Abrantes N, Serpa D (2022) A review on polycyclic aromatic hydrocarbons distribution in freshwater ecosystems and their toxicity to benthic fauna. Sci Total Environ 820:153282. https://doi.org/10.1016/j.scitotenv.2022.153282
Kar S, Roy K (2012) First report on development of quantitative interspecies structure–carcinogenicity relationship models and exploring discriminatory features for rodent carcinogenicity of diverse organic chemicals using OECD guidelines. Chemosphere 87(4):339–355. https://doi.org/10.1016/j.chemosphere.2011.12.019
Kemmlein S, Herzke D, Law RJ (2009) Brominated flame retardants in the European chemicals policy of REACH-Regulation and determination in materials. J Chromatogr A 1216(3):320–333. https://doi.org/10.1016/j.chroma.2008.05.085
Khan K, Roy K (2017) Ecotoxicological modelling of cosmetics for aquatic organisms: A QSTR approach. SAR QSAR Environ Res 28(7):567–594. https://doi.org/10.1080/1062936X.2017.1352621
Korsman JC, Schipper AM, Hendriks AJ (2016) Dietary toxicity thresholds and ecological risks for birds and mammals based on species sensitivity distributions. Environ Sci Technol 50(19):10644–10652. https://doi.org/10.1021/acs.est.6b01258
Kušić H, Rasulev B, Leszczynska D, Leszczynski J, Koprivanac N (2009) Prediction of rate constants for radical degradation of aromatic pollutants in water matrix: a QSAR study. Chemosphere 75(8):1128–1134. https://doi.org/10.1016/j.chemosphere.2009.01.019
Lei PW, Shiverdecker LK (2019) Performance of estimators for confirmatory factor analysis of ordinal variables with missing data. Struct Equ Modeling 27(4):584–601. https://doi.org/10.1080/10705511.2019.1680292
Lewtas J (2007) Air pollution combustion emissions: characterization of causative agents and mechanisms associated with cancer, reproductive, and cardiovascular effects. Mutat Res-Rev Mutat Res 636(1):95–133. https://doi.org/10.1016/j.mrrev.2007.08.003
Liu M, Zheng H, Chen M, Liang J, Duan M, Du H, Chen M, Ke H, Zhang K, Cai M (2022) Dissolved PAHs impacted by air-sea interactions: net volatilization and strong surface current transport in the Eastern Indian Ocean. J Hazard Mater 431:128603. https://doi.org/10.1016/j.jhazmat.2022.128603
Liu P, Long W (2009) Current mathematical methods used in QSAR/QSPR studies. Int J Mol Sci 10(5):1978–1998. https://doi.org/10.3390/ijms10051978
Livingstone DR (1991) Organic xenobiotic metabolism in marine invertebrates. Advances in Comparative and Environmental Physiology, volume 7. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-75897-3_2
LoPachin RM, Geohagen BC, Nordstroem LU (2019) Mechanisms of soft and hard electrophile toxicities. Toxicology 418:62–69. https://doi.org/10.1016/j.tox.2019.02.005
Lotufo GR, Fleeger JW (1997) Effects of sediment-associated phenanthrene on survival, development and reproduction of two species of meiobenthic copepods. Mar Ecol Prog Ser 151:91–102. https://doi.org/10.3354/meps151091
Lu C, Yang S, Yan Z, Ling J, Jiao L, He H, Zheng X, Jin W, Fan J (2018) Deriving aquatic life criteria for PBDEs in China and comparison of species sensitivity distribution with TBBPA and HBCD. Sci Total Environ 640–641:1279–1285. https://doi.org/10.1016/j.scitotenv.2018.06.002
Lu BQ, Liu SS, Wang ZJ, Xu YQ (2020) Conlecs: a novel procedure for deriving the concentration limits of chemicals outside the criteria of human drinking water using existing criteria and species sensitivity distribution based on quantitative structure-activity relationship prediction. J Hazard Mater 384:121380. https://doi.org/10.1016/j.jhazmat.2019.121380
Luit RJ, Beems J, Benthem J, Bodar CWM, Engelen JGM, Hulzebos EM, Loveren H, Maslankiewicz L, Pronk MEJ (2003) Inventory of revisions in the EC Technical Guidance Documents (TGDs) on risk assessment of chemicals. Dutch National Institute for Public Health and the Environment (RIVM)
Mackay D, Shiu WY, Ma KC (1992) Illustrated handbook of physical-chemical properties and environmental fate for organic chemicals Polynuclear Aromatic Hydrocarbons. Lewis Publishers, Chelsea, MI, USA
Maltby L, Brock TCM, van den Brink PJ (2009) Fungicide risk assessment for aquatic ecosystems: importance of interspecific variation, toxic mode of action, and exposure regime. Environ Sci Technol 43(19):7556–7563. https://doi.org/10.1021/es901461c
Maltby L, Blake N, Brock TCM, Van den Brink PJ (2005) Insecticide species sensitivity distributions: importance of test species selection and relevance to aquatic ecosystems. Environ Toxicol Chem 24(2). https://doi.org/10.1897/04-025R.1
Margesin R, Schinner F (2001) Biodegradation and bioremediation of hydrocarbons in extreme environments. Appl Microbiol Biot 56(5):650–663. https://doi.org/10.1007/s002530100701
Neese F (2022) Software update: the orca program system—version 5.0. Wiley Interdiscip Rev: Comput Mol Sci 12(5):e1606. https://doi.org/10.1002/wcms.1606
OECD (1984a) OECD guidelines for the testing of chemicals no. 201. Freshwater alga and cyanobacteria, growth inhibition test. OECD, Paris, France
OECD (1984b) OECD guidelines for the testing of chemicals, no. 202 Daphnia sp. acute immobilization test and reproduction test. OECD, Paris, France
OECD (1992) OECD Guidelines for the testing of chemicals, section 2-effects on biotic systems. OECD, Paris, France
OECD (2014) Guidance document on the validation of (quantitative) structure–activity relationship [(Q)SAR] models. OECD Series on Testing and Assessment, No. 69. OECD, Paris, France
Önlü S, Saçan MT (2018) Toxicity of contaminants of emerging concern to Dugesia japonica: QSTR modeling and toxicity relationship with Daphnia magna. J Hazard Mater 351:20–28. https://doi.org/10.1016/j.jhazmat.2018.02.046
Othman HB, Pick FR, Hlaili AS, Leboulanger C (2023) Effects of polycyclic aromatic hydrocarbons on marine and freshwater microalgae—a review. J Hazard Mater 441:129869. https://doi.org/10.1016/j.jhazmat.2022.129869
Pandey SK, Ojha PK, Roy K (2020) Exploring QSAR models for assessment of acute fish toxicity of environmental transformation products of pesticides. Chemosphere 252:126508. https://doi.org/10.1016/j.chemosphere.2020.126508
Parthasarathi R, Subramanian V, Roy DR, Chattaraj PK (2004) Electrophilicity index as a possible descriptor of biological activity. Bioorg Med Chem 12(21):5533–5543. https://doi.org/10.1016/j.bmc.2004.08.013
Parvathi S, Dodoala STS (2022) A statistical comparison between Zagreb indices for correlation with toxicity predictions of natural products. Int J Pharm Res Alli 13:121–125. https://doi.org/10.26452/ijrps.v13i1.32
Posthuma L, Suter WG, Trass PT (2002) Species sensitivity distributions in ecotoxicology. Boca Raton, pp 616. https://doi.org/10.1201/9781420032314
Redl G, Cramer-tert RD, Berkoff CE (1974) Quantitative drug design. Chem Soc Rev 3(3). https://doi.org/10.1039/cs9740300273
Rorije E, Peijnenburg WJGM, Klopman G (1998) Structural requirements for anaerobic biodegradation of organic chemicals: a fragment model analysis. Environ Toxicol Chem 17(10):1943–1950. https://doi.org/10.1002/etc.5620171008
Roy DR, Sarkar U, Chattaraj PK, Mitra A, Padmanabhan J, Parthasarathi R, Subramanian V, Van Damme S, Bultinck P (2006) Analyzing toxicity through electrophilicity. Mol Divers 10(2):119–131. https://doi.org/10.1007/s11030-005-9009-x
Roy K, Das RN, Ambure P, Aher RB (2016) Be aware of error measures. Further studies on validation of predictive QSAR models. Chemometr Intell Lab 152:18–33. https://doi.org/10.1016/j.chemolab.2016.01.008
Saber S, Mohammad H, Aziz M (2019) A quantitative structure–antioxidant relationship (QSAR) model for 1,3,4-oxadiazole derivatives using PLS regression. Zanco J Pure Appl Sci 31:31. https://doi.org/10.21271/zjpas.31.s4.17
Singh A, Kumar S, Kapoor A, Kumar P, Kumar A (2023) Development of reliable quantitative structure–toxicity relationship models for toxicity prediction of benzene derivatives using semiempirical descriptors. Toxicol Mech Method 33(3):222–232. https://doi.org/10.1080/15376516.2022.2118092
Sun H, Giesy JP, Jin X, Wang J (2017) Tiered probabilistic assessment of organohalogen compounds in the Han River and Danjiangkou Reservoir, central China. Sci Total Environ 586:163–173. https://doi.org/10.1016/j.scitotenv.2017.01.194
Sun G, Zhang Y, Pei L, Lou Y, Mu Y, Yun J, Li F, Wang Y, Hao Z, Xi S, Li C, Chen C, Zhao L, Zhang N, Zhong R, Peng Y (2021) Chemometric QSAR modeling of acute oral toxicity of polycyclic aromatic hydrocarbons (PAHs) to rat using simple 2D descriptors and interspecies toxicity modeling with mouse. Ecotox Environ Safe 222:112525. https://doi.org/10.1016/j.ecoenv.2021.112525
Traas T, Van de Meent D, Posthuma L, Hamers T, Kater BJ, De Zwart D, Aldenberg T (2002) The potentially affected fraction as a measure of ecological risk. In Species Sensitivity Distributions in Ecotoxicology pp 315– 344
Tropsha A (2010) Best practices for QSAR model development, validation, and exploitation. Mol Inform 29(6–7):476–488. https://doi.org/10.1002/minf.201000061
USEPA (1985) Guidelines for deriving numerical national water quality criteria for the protection of aquatic organisms and their uses. Office of Research and Development, Washington DC
USEPA (1992) Framework for ecological risk assessment. Risk Assessment Forum, Office of Research and Development, Washington DC
USEPA (1993) Technical basis for deriving sediment quality criteria for nonionic organic contaminants for the protection of benthic organisms by using equilibrium partitioning. Office of Water, U.S. Environmental Protection Agency, Washington DC
Valavanidis A, Vlahogianni T, Dassenakis M, Scoullos M (2006) Molecular biomarkers of oxidative stress in aquatic organisms in relation to toxic environmental pollutants. Ecotox Environ Safe 64(2):178–189. https://doi.org/10.1016/j.ecoenv.2005.03.013
Vighi M, Finizio A, Villa S (2006) The evolution of the environmental quality concept: from the US EPA Red Book to the European Water Framework Directive. Environ Sci Pollut Res 13(1):9–14. https://doi.org/10.1065/espr2006.01.003
Wang X, Yan Z, Liu Z, Zhang C, Wang W, Li H (2014) Comparison of species sensitivity distributions for species from China and the USA. Environ Sci Pollut Res 21(1):168–176. https://doi.org/10.1007/s11356-013-2110-2
Wang ZJ, Liu SS, Huang P, Xu YQ (2021) Mixture predicted no-effect concentrations derived by independent action model vs concentration addition model based on different species sensitivity distribution models. Ecotox Environ Safe 227:112898. https://doi.org/10.1016/j.ecoenv.2021.112898
Wang S, Zhang X, Gui B, Xu X, Su L, Zhao YH, Martyniuk CJ (2022) Comparison of modes of action between fish, cell and mitochondrial toxicity based on toxicity correlation, excess toxicity and QSAR for class-based compounds. Toxicology 470:153155. https://doi.org/10.1016/j.tox.2022.153155
Wang K, Lv Y, He M, Tian L, Nie F, Shao Z, Wang Z (2023) A quantitative structure–activity relationship approach to determine biotoxicity of amide herbicides for ecotoxicological risk assessment. Arch Environ Contam Toxicol 84(2):214–226. https://doi.org/10.1007/s00244-023-00980-9
Wu J, Liu Z, Yan Z, Yi X (2015) Derivation of water quality criteria of phenanthrene using interspecies correlation estimation models for aquatic life in China. Environ Sci Pollut Res 22(12):9457–9463. https://doi.org/10.1007/s11356-015-4091-9
Xu L, Zhang WJ (2001) Comparison of different methods for variable selection. Anal Chim Acta 446(1):475–481. https://doi.org/10.1016/S0003-2670(01)01271-5
Xu J, Wang L, Wang L, Shen X, Xu W (2011) QSPR study of Setschenow constants of organic compounds using MLR, ANN, and SVM analyses. J Comput Chem 32(15):3241–3252. https://doi.org/10.1002/jcc.21907
Yang L, Wang Y, Chang J, Pan Y, Wei R, Li J, Wang H (2020) QSAR modeling the toxicity of pesticides against Americamysis bahia. Chemosphere 258:127217. https://doi.org/10.1016/j.chemosphere.2020.127217
Yang L, Sang C, Wang Y, Liu W, Hao W, Chang J, Li J (2021) Development of QSAR models for evaluating pesticide toxicity against Skeletonema costatum. Chemosphere 285:131456. https://doi.org/10.1016/j.chemosphere.2021.131456
Yap C W (2011) Padel-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comp Chem 32(7):1466–1474. https://doi.org/10.1002/jcc.21707
Zhang L, Zhou P, Yang F, Wang Z (2007) Computer-based QSARs for predicting mixture toxicity of benzene and its derivatives. Chemosphere 67(2):396–401. https://doi.org/10.1016/j.chemosphere.2006.09.018
Zhang S, Wang L, Wang Z, Fan D, Shi L, Liu J (2017) Derivation of freshwater water quality criteria for dibutyltin dilaurate from measured data and data predicted using interspecies correlation estimate models. Chemosphere 171:142–148. https://doi.org/10.1016/j.chemosphere.2016.12.046
Zhao B, Huang F, Zhang C, Huang G, Xue Q, Liu F (2020) Pollution characteristics of aromatic hydrocarbons in the groundwater of China. J Contam Hydrol 233:103676. https://doi.org/10.1016/j.jconhyd.2020.103676
Zhu M, Ge F, Zhu R, Wang X, Zheng X (2010) A DFT-based QSAR study of the toxicity of quaternary ammonium compounds on Chlorella vulgaris. Chemosphere 80(1):46–52. https://doi.org/10.1016/j.chemosphere.2010.03.044
Zvinavashe E, van den Berg H, Soffers AEMF, Vervoort J, Freidig A, Murk AJ, Rietjens IMCM (2008) QSAR models for predicting in vivo aquatic toxicity of chlorinated alkanes to fish. Chem Res Toxicol 21(3):739–745. https://doi.org/10.1021/tx700367c
Acknowledgements
Many thanks to the editor and reviewers for their recommendations and advice to make the manuscript better.
Funding
This work was supported by the CNPC Scientific Research and Technology Development Programme (2021DJ6605), PetroChina Innovation Foundation (Grant No. 2016D-5007–0702), and the Yangtze Talents Fund (2020–2023).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Formal analysis, investigation, methodology, validation, and writing original draft preparation were performed by Xiudi Lv. Formal analysis, investigation, validation, writing original draft preparation, supervision, project administration, and funding acquisition were performed by Mei He. Formal analysis, investigation, and validation were performed by Jiajia Wei and Qiang Li. Methodology, formal analysis, validation, and project administration were performed by Fan Nie. Methodology, validation, supervision, and project administration were performed by Zhiguo Shao. Validation, supervision, and project administration were performed by Zhansheng Wang. Formal analysis, investigation, methodology, validation, writing reviewing and editing, project administration, and funding acquisition were performed by Lei Tian. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
There was no animal experiment in this manuscript.
Consent to participate and publish
The manuscript is approved by all authors and has not been submitted to more than one journal for simultaneous consideration. The manuscript described has not been published before. All authors have agreed to its submissions for publication.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Marcus Schulz
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
•The effective hazardous concentrations (HC5) of 40 AHCs were derived by SSD models.
• Three reliable QSAR models were developed to obtain the hazard thresholds of AHCs.
• The derived hazard thresholds of AHCs were approximate to the published limits.
• Electrophilicity and topological descriptors mostly impacted the risks of AHCs.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lv, X., He, M., Wei, J. et al. Development of an effective QSAR-based hazard threshold prediction model for the ecological risk assessment of aromatic hydrocarbon compounds. Environ Sci Pollut Res 31, 47220–47236 (2024). https://doi.org/10.1007/s11356-024-34016-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-024-34016-z