
Abstract
Sulfate-reducing bacteria (SRB) are an attractive option for treating acid mine drainage (AMD) and are considered to be of great significance in the natural attenuation of AMD, but the available information regarding the highly diverse SRB community in AMD sites is not comprehensive. The Hengshi River, which is continually contaminated by AMD from upstream mining areas, was selected as a study site for investigation of the distribution, diversity, and abundance of SRB. Overall, high-throughput sequencing of the 16S rRNA and dsrB genes revealed the high diversity, richness, and OTU numbers of SRB communities, suggesting the existence of active sulfate reduction in the study area. Further analysis demonstrated that AMD contamination decreased the richness and diversity of the microbial community and SRB community, and led to spatiotemporal shifts in the overall composition and structure of sediment microbial and SRB communities along the Hengshi watershed. However, the sulfate reduction activity was high in the midstream, even though AMD pollution remained heavy in this area. Spatial distributions of SRB community indicated that species of Clostridia may be more tolerant of AMD contamination than other species, because of their predominance in the SRB communities. In addition, the results of CCA revealed that environmental parameters, such as pH, TS content, and Fe content, can significantly influence total microbial and SRB community structure, and dissolved organic carbon was another important factor structuring the SRB community. This study extends our knowledge of the distribution of indigenous SRB communities and their potential roles in natural AMD attenuation.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Acid mine drainage (AMD) is typically characterized by low pH and high concentrations of sulfate and heavy metals (Bao et al. 2017; Gao et al. 2019). AMD severely impacts aquatic ecosystems, degrading the quality of water used for drinking, crop irrigation, or aquaculture and rendering water unsafe for human consumption (Mohapatra et al. 2011; Xu et al. 2020). Due to the extreme toxicity of AMD, numerous technologies involving physical, chemical, and biological approaches have been developed to remove toxic substances, mainly soluble metals and sulfates. Biological treatment strategies take advantage of the ability of microorganisms occurring in AMD to metabolize iron and sulfate (Klein et al. 2014), and biological treatment applying sulfate-reducing bacteria (SRB) is an attractive option for treating AMD and recovering metals (Sánchez-Andrea et al. 2014). Furthermore, SRB also have been demonstrated to play a role in the natural attenuation of AMD (Coggon et al. 2012).
Sulfate reduction by SRB has been successfully applied to decrease the acidity of and remove metals and sulfate from AMD (Coggon et al. 2012; Sánchez-Andrea et al. 2014; Sun et al. 2020), due to their ability to induce alkalinity and produce sulfides (Eq. (1)), which neutralize AMD and precipitate metals (M2+) simultaneously (Eq. (2)).
Sulfate-reducing bacteria mainly belong to Deltaproteobacteria, Clostridia, Thermodesulfobacteria, Thermodesulfobiaceae, Nitrospirae, Euryarchaeota, and Crenarchaeota (Dev et al. 2016). Generally, most of the identified SRB grow optimally at neutral pH (Sánchez-Andrea et al. 2013; Mardanov et al. 2017). Although acidophilic and acid-tolerant species have been found in acidic mine waters and sediments (Sen and Johnson 1999; Giloteaux et al. 2013; Mardanov et al. 2017; Bao et al. 2018), such organisms are not common among SRB due to the low pH and low availability of organic carbon in AMD sites (Koschorreck 2008; Bao et al. 2018). To date, only very few SRB species growing at low pH have been isolated and characterized (Bertel et al. 2012; Petzsch et al. 2015). Most of them belong to the genera Desulfosporosinus (Clostridia) and Desulfovibrio (Deltaproteobacteria) (Mardanov et al. 2017; Bao et al. 2018).
Located in South China, the Hengshi watershed is heavily contaminated by long-term AMD from the upstream Dabaoshan Mine. Previous studies have shown that the AMD in the Dabaoshan Mine is characterized by low pH (< 3.0) and a high concentration of SO42− (> 2000 mg/L), and various kinds of dissolved metals such as Fe, Pb, Cd, and Cu (Chen et al. 2015, 2018; Bao et al. 2017). AMD impacts over 40 km of streams within the Hengshi watershed basin, creating sulfate gradients along the water flow (Bao et al. 2017). Compared with the heavily contaminated mud impoundments, the downstream Hengshi River is moderately to lightly contaminated overall. Therefore, this watershed allows the investigation of the spatiotemporal distribution of the SRB community along an AMD gradient. Importantly, little is known about the acidophilic and acid-tolerant indigenous SRB in AMD sites.
In the present study, we investigated the following: (i) the physicochemical details of AMD-contaminated watershed sediment in the Dabaoshan Mining District, (ii) the composition of the microbial community and SRB community within this watershed and the relationship between community composition and physiochemical parameters, and (iii) the abundance of the SRB community based on qPCR, providing insight into SRB community characteristics.
Materials and methods
Sampling and physicochemical analyses
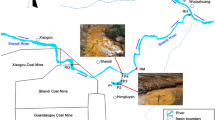
The Dabaoshan Mine (South China (24° 31′ 37″ N; 113° 42′ 49″ E)) is a large polymetallic sulfide deposit and a range of metal ores (mainly iron and copper) have been extensively mined since the 1970s. The Hengshi watershed is continually contaminated by AMD produced from the upstream Dabaoshan Mine (Fig. 1). Two impoundments (S0 and S1) were built to dam the AMD. However, a large amount of AMD overflow continues to flow to the downstream Hengshi River. The Hengshi River, which is joined by a few tributaries and is surrounded by scattered villages, flows through the Wengcheng city area and then into the Wengjiang River. AMD is attenuated as the river flows. The significant confluence points S6–S8 and S10–S11 (41 km from the dam S1) were chosen in this study to represent the moderately and lightly contaminated regions, respectively. In addition, the Taiping River, which is one of the most important tributaries with no AMD pollution, was selected as the control stream (C4).

Location of the Hengshi River watershed and the locations of 7 sampling sites in the Hengshi River watershed
Field measurements and sampling were carried out in January and November 2016 at 7 sites (Fig. 1). Approximately 500 g of surface sediment (up to 5 cm deep) was collected per site. Samples from each site were pooled, homogenized, and then subpacked in sterile 50-ml Falcon tubes. The sediments for chemical analyses and molecular analyses were stored at − 20 °C prior to any treatment. Genomic DNA extraction was finished within 1 week, and sulfur species determination was finished within 2 weeks. For total sulfur (TS) and heavy metal analyses, sediments were processed by vacuum freeze drying and passed through a 100-mesh sieve.
Measurements of sediment pH and temperature (T) were carried out in situ using a multi-parameter tester (SG2-T SevenGopro™ MTD, Switzerland). The sediment pH was expressed by the pH of the water. The sediment dissolved organic carbon (DOC) was extracted following the method described by Jones and Willett (2006), then measured by a total organic carbon analyzer (TOC-V CPN, Shimadzu). Measurements of heavy metals (including Fe, Pb, Zn, Cu, Cr, and Cd) were performed by atomic absorption spectrophotometry (Z-2000, Hitachi, Japan). Sulfur species (including acid volatile sulfide (AVS), chromium reducible sulfide (CRS), elemental sulfur (ES), exchangeable sulfate (ExS), and TS) of the sediments were extracted and analyzed using the method described by Chen et al. (2015).
DNA extraction
Genomic DNA was extracted from each sediment sample using a MOBIO™ PowerSoil® DNA Isolation Kit (MOBIO Laboratories Inc., Carlsbad, CA, USA) following the manufacturer’s protocol. The extracted DNA was stored at − 80 °C prior to further analyses.
High-throughput sequencing and data processing
Microbial and SRB community structure, richness, and diversity were studied using Illumina MiSeq PE300 based on 16S rRNA and dsrB gene sequencing, respectively. Amplification of the 16S rRNA gene (∼ 468 bp, V3-V4 region) and dsrB gene (∼ 350 bp) was performed using the primer pairs 341F/806R and DSRp2060F/DSR4R, respectively (Supplementary Table S1); these primer pairs were widely employed in previous studies (Zhang et al. 2017). PCR amplification was performed in a S1000 thermal cycler (Bio-Rad, USA). Amplifications were carried out in a 50-μl reaction mixture containing 25 μl of 2× SYBR® Premix Ex Taq (TaKaRa), 3 μl of template cDNA (∼ 20 ng/μl), 1 μl of each primer at 10 mM, and 20 μl of sterile distilled water. The amplification conditions were as follows: initial denaturation at 94 °C for 5 min, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 52 °C for 30 s, and elongation at 72 °C for 30 s, and a final step at 72 °C for 10 min. The yield and quality of the PCR products were examined on a 1% agarose gel stained with ethidium bromide. The PCR products were then mixed at equidense ratios and purified using an EZNA Gel Extraction Kit (Omega, USA). 16S rRNA and dsrB gene libraries were subsequently prepared using an NEBNext® Ultra™ DNA Library Prep Kit (New England Biolabs, USA), and the libraries were finally sequenced on the Illumina MiSeq (PE300) platform.
Sequences were analyzed with QIIME software and the UPARSE pipeline (Caporaso et al. 2010). After assignment based on each sample and merging (Magoè and Salzberg 2011), low-quality reads (16S rRNA and dsrB) were filtered with QIIME (Caporaso et al. 2010). Subsequently, UPARSE (Edgar 2013) was used to cluster operational taxonomic units (OTUs) at 97% and 90% similarity cutoffs for the 16S rRNA gene and dsrB gene, respectively (Zhang et al. 2017). In addition, the most abundant sequence from each OTU was selected as the representative sequence. Phylogenetic analysis was performed with 16S rRNA and dsrB gene sequences obtained from the Greengenes and NCBI (National Center for Biotechnology Information) databases, respectively. Reads that did not match any sequences in the database were clustered into the unclassified group. Raw sequences of 16S rRNA and SRB genes have been submitted to NCBI Sequence Read Archive database with no. PRJNA642338 and no. PRJNA642374, respectively.
Quantification of gene copies in sediments
The microbial 16S rRNA gene and the dsrB gene in the sediment samples were amplified using the primers 341F/518R and DSRp2060F/DSR4R, respectively (Supplementary Table S1). Quantification of the 16S rRNA and dsrB genes was performed with the methods described by Zhang et al. (2017) and Wang et al. (2016), respectively. Real-time PCR was conducted using a SYBR® Premix Ex Taq™ II kit (Takara). Then, the PCR products were cloned into pMD19-T Easy Vector (Takara) to draw standard curves.
Statistical analyses
Gene richness and diversity were assessed by the Chao1 richness estimator (Chao 1984) and by the Simpson evenness index (Simpson 1949) and nonparametric Shannon diversity index (Shannon 1948), respectively, as implemented in QIIME (Schloss et al. 2009). The similarity of microbial communities among different sediment samples was determined by the unweighted pair group method with arithmetic mean (UPGMA) clustering based on a weighted UniFrac distance matrix calculated by QIIME. Nonmetric multidimensional scaling (NMDS) based on weighted UniFrac distances was performed to provide a visual comparison of the pairwise UniFrac distances among samples.
The interrelationships among the sampling sites, the microbial community structures, and the environmental variables were analyzed using canonical correspondence analysis (CCA) with CANOCO 4.5 multivariate data analysis software (Microcomputer Power, Ithaca, NY, USA). The significant physiochemical parameter-community interactions were tested using Monte Carlo permutation tests. Finally, the CANODRAW 4.0 program (in the Canoco package) was used to graphically present the ordination results.
Results
Physicochemical characterization and sulfur distributions in sediments
The physicochemical parameters measured in the sediment samples are summarized in Fig. S1. In general, temporal variations of the physicochemical parameters between the two sampling campaigns were more marked in the downstream of Shangba (S8–S11), compared with the two mud impoundments (S0 and S1). In terms of spatial variations, natural attenuation processes result in a strong spatial pollution gradient along the water flow. The sediment pH and DOC increased along the Hengshi River. The two mud impoundments (S0 and S1) were the headwaters of the Hengshi River and were the most heavily contaminated areas. Sediments in the two mud impoundments had a pH < 3.0 and a large number of precipitated heavy metals. AMD overflowed from the S1 mud impoundment into the Hengshi River, and the pH increased along the Hengshi River. S6 and S8 are located in the AMD-attenuating zone, which is mildly acidic and represents a moderately AMD-contaminated area. The pH values in these regions (S6–S8) ranged from 3.3 to 5.2 in January 2016, whereas in November 2016, they increased to 4.6 and 6.4 in S6 and S8, respectively. Downstream (S10 and S11), the sediment pH was close to neutral, which was similar to the pH in the control site (C4), indicating slight AMD contamination in this area. Among the heavy metals (Fig. S1), the Fe and Pb concentrations in sediment declined rapidly along the river. Studies have demonstrated that Fe and Pb have a distance-metal concentration decay pattern (Taylor 2007; Chamani et al. 2016). However, no regular changes in Zn, Cu, Cr, and Cd concentrations in the sediment were found throughout the Hengshi watershed.
Sulfur species in sediments along the Hengshi River had different characteristics. In general, the sediment TS and ExS decreased along the river (Fig. 2). TS values at S0 and S1 (ranging from 25.69 to 40.10 g/kg) were markedly higher than those at other sites. ExS was the main sulfur species in the upstream and midstream sediments, especially at S6 in January 2016, where ExS accounted for 63.5% of the TS. Chen et al. (2015) suggested that the high TS and ExS contents in acidic sediments result from the precipitation of sulfate-rich minerals and sulfate adsorption by minerals. In addition, several reduced sulfur species (ES, AVS, and CRS) were detected, indicating the existence of active sulfate reduction in this watershed. However, compared with ExS, the ratio of reduced sulfur species was low (< 5.6% of TS). The total reduced sulfur species at S6 and S8 was often higher than that at the other sites, and CRS was present at the highest percentage in S6 in January 2016 (~ 5.4% of TS) and S8 in November 2016 (3.7%), suggesting that the sulfate reduction activity was high at these two sites.

Sulfur species in sediments along the Hengshi River (TS: total sulfur, ExS: exchangeable sulfate, ES: element sulfur, AVS: acid volatile sulfide, CRS: chromium reducible sulfide). a and b are samples collected in January 2016; c and d are samples collected in November 2016
16S rRNA gene analysis
A total of 566,152 valid 16S rRNA gene sequences were obtained from the 14 sediment samples through Illumina MiSeq PE300 sequencing analysis (Table S2). The valid sequences were clustered into OTUs with 97% sequence similarity. Accordingly, microbial richness and diversity estimates were calculated for each sample and are shown in Table S2. A total of 342–4113 OTUs with 97% similarity were identified in the 14 sediment samples. The OTU numbers increased along the watershed, and the sediment samples from the two mud impoundments (S0 and S1) presented the smallest number of OTUs in each sampling period. The species richness index (Chao 1) and diversity indices (Shannon indices) showed trends similar to that of the number of OTUs across the watershed. The lowest and highest richness and diversity indices were obtained for the headwater (S0 and S1) and the downstream sections (S10 and S11), respectively. These results revealed shifts in microbial richness and diversity linked to AMD pollution gradients.
More than 15 prokaryotic phyla were detected across all sediment samples, and the dominant phyla (> 1.0%), which accounted for more than 93.7% of the total sequences in each sample, are shown in Fig. 3a. Proteobacteria, Bacteroidetes, and Firmicutes were the core phyla, accounting for nearly 68.5% of the total. For the majority of the sediment samples, Proteobacteria was the most abundant phylum and accounted for 25.9–65.9% of the total sequences. Within Proteobacteria, the majority of sequences were assigned to Gammaproteobacteria (13.2–99.3%), Betaproteobacteria (0.3–72.1%), and Alphaproteobacteria (0.3–47.2%) (Fig. 3b). The relative abundance of Gammaproteobacteria decreased along the watershed, whereas the relative abundance of Betaproteobacteria increased along the watershed (Fig. 3b). Bacteroidetes displayed a high relative abundance in all sediment samples, except for the two samples from the two mud impoundments collected in November 2016. Firmicutes displayed a high relative abundance in all sediment samples, especially at the S0 site in November 2016, accounting for 53.2% of the total sequences.

16S rRNA gene-based relative abundance of dominant phyla (a) and different classes of Proteobacteria (b) in sediment samples collected from the Hengshi watershed
As shown in Table S4, a total of 18 families of potential SRB were inferred for the microbial community by 16S rRNA gene sequencing, with relative abundances ranging from 4.45 to 35.05% (12.46% on average).
dsrB gene analysis
The total number of high-quality sequences of the dsrB gene obtained from all samples was 2,072,813. Using a similarity cutoff of 90%, these sequences were classified into 53,098 OTUs. The corresponding species richness (Chao 1 estimator) and diversity estimates (Shannon indices) were calculated and are listed in Table S3. In general, the richness and diversity indices of the dsrB gene increased along the watershed with the spatial gradient of AMD from the first impoundment (S0) to the confluence of the Hengshi River and Wengjiang River (S11).
Our results showed that SRB community composition varied with geographical zone. At the class level, Clostridia and Deltaproteobacteria were the predominant SRB groups in all the sediment samples, accounting for 30.0–91.0% and 8.8–69.7% of the total dsrB sequences, respectively (Fig. 4a). Generally, the relative abundances of Clostridia increased along the river. Clostridia were the most abundant class in most sediments, except for the sediments from downstream of Shangba (S8–S11) in November 2016 and from the control river (C4).

Community structure of the dsrB gene in sediment samples at the class level (a), the family level (b), and the genus level (c)
Significant differences at the family level along the AMD contamination gradient were also observed (Fig. 4b). Peptococcaceae represented a significant fraction in all 14 libraries, accounting for 25.2–98.2% of the total dsrB sequences, and tended to decrease along the watershed. Syntrophobacteraceae and Desulfobacteraceae were also abundant in all 14 libraries, representing 1.4–32.5% and 1.6–45.9% of the total dsrB sequences, respectively. The relative abundances of Syntrophobacteraceae decreased along the watershed, while those of Desulfobacteraceae increased along the watershed. Desulfobulbaceae was found at relatively high abundances in sediment samples from downstream of Liangqiao (S6–S11) and from the control river (C4).
As shown in Fig. 4c and Table S5, the majority of sequences (ranging from 56.3 to 93.9%) could not be assigned at the genus level, especially in the heavily contaminated areas (S0 and S1). Among the detected genera, Desulfotomaculum, Desulfovibrio, and Desulfomicrobium were dominant, while Desulfosporosinus, Desulfobacter, and Desulfotignum were rare in these sediments. Desulfotomaculum was abundant in all 14 libraries, accounting for 5.8–43.5% of the total dsrB sequences, with the highest relative abundances in the heavily contaminated areas (S0 and S1). Desulfovibrio and Desulfomicrobium were found in only some sediment samples. Desulfovibrio showed relatively high abundances in less indices areas (S8–S11, and C4).
Linking microbial and SRB community structures to AMD pollution gradients
The similarities and dissimilarities of the microbial communities in different sediments were quantified through NMDS analysis (Fig. 5a) and UPGMA clustering (Fig. S2a) based on weighted UniFrac distances. Overall, the microbial community structures in the sediment samples from the two impoundments (S0 and S1) and Liangqiao (S6) collected in January 2016 were similar, and those in the samples collected in November 2016 from the three sites also clustered. The microbial community structures in the sediment samples from downstream of Liangqiao (S8-S11) clustered together and were closer to those of the control stream (C4) in January 2016 and November 2016, respectively. This finding that the sequences tended to group into clusters was consistent with major physicochemical changes along the watershed, as well as along the pollution gradients. However, analysis of the cluster revealed that the samples collected in January 2016 formed a separate group from the samples collected in November 2016, revealing differences in microbial community structure between the two sampling campaigns.

Nonmetric multidimensional scaling (NMDS) plots derived from weighted UniFrac distances for the 16S rRNA (a) and dsrB (b) genes in sediments. Blue squares represent the sediment samples collected in January 2016; red circles represent the sediment samples collected in November 2016
Much more significant similarities and dissimilarities in the SRB communities were observed through NMDS analysis (Fig. 5b). UPGMA clustering (Fig. S2b) further confirmed the patterns revealed by NMDS. The three major clusters shown in Fig. 5b, i.e., the most heavily contaminated sediments (S0 and S1), the lightly contaminated sediments (S8, S10 and S11) and the control collected in January 2016, and the lightly contaminated sediments and the control collected in November 2016, suggested that the phylogenetic composition of SRB varied with AMD pollution gradients and sampling times.
CCA was conducted to discern the possible correlations between environmental variables and microbial community structure, as represented by the major taxonomic groups (genera for 16S rRNA genes and families for dsrB genes) (Fig. 6). CCA revealed that the environmental factors most strongly correlated with the total microbial and SRB communities were pH and the TS, ExS, and Fe contents. In addition, DOC appeared to play an important role in SRB community structure. As indicated by the length of the environmental variable arrows in the CCA biplot, the strongest determinant of the overall microbial and the SRB communities was pH. As shown in Fig. 6a, the relative abundances of Leptospirillum, Acidocella, and Acidiphilium were negatively correlated with pH. Leptospirillum, Acidocella, and Acidiphilium were the commonly detected acidophilic bacteria in AMD (Méndez-García et al. 2015; Xu et al. 2020). As shown in Fig. 6b, the relative abundances of Peptococcaceae and Syntrophobacteraceae were negatively correlated with pH and positively correlated with the TS and ExS contents. In contrast, the relative abundances of Desulfobulbaceae and Desulfomicrobiaceae were positively correlated with reduced sulfur species (ES, AVS, and CRS).

Ordination diagrams from canonical correspondence analysis (CCA) of environmental parameters and the 16S rRNA (a) and dsrB genes (b). Blue samples represent the sediment samples collected in January 2016; black samples represent the sediment samples collected in November 2016
Abundance of total bacterial 16S rRNA and dsrB genes
The abundance of total bacterial 16S rRNA and dsrB genes in the sediment samples from the Hengshi River was determined via qPCR (Fig. 7). The results showed that the abundances of the 16S rRNA and dsrB genes gradually increased with increasing pH along the river, which was consistent with the prediction of an increase in microbial abundances in the river flow direction since a decrease in AMD pollution would be related to a decrease in environmental harshness.

16S rRNA (filled) and dsrB (hollow) gene copy numbers in the sediments
Discussion
Sulfate-reducing bacteria (SRB) are an attractive option for treating AMD and are considered to be of great significance for the natural attenuation of AMD. This study aimed to characterize the microbial and SRB communities in Hengshi watershed sediment to provide new insights into the diversity of and shift in SRB communities in response to AMD contamination gradients. The diversity of microbial and SRB communities was determined using an approach combining 16S rRNA and dsrB gene high-throughput sequencing with qPCR analyses. This information will enable us to learn more about the functional groups involved in the natural attenuation of AMD and to exploit SRB capacities for AMD bioremediation.
Physicochemical parameters shape microbial and SRB abundance, community diversity, and structure
Several statistical analyses of the 16S rRNA and dsrB sequences suggested that AMD contamination decreased the richness and diversity of microbial and SRB communities and led to spatial shifts in the overall composition and structure of sediment microbial and SRB communities along the Hengshi watershed. The lowest microbial and SRB abundances and diversities were found in the upstream sediments (S0 and S1), which had been heavily contaminated by AMD, indicating that AMD does reduce the microbial and SRB communities diversity. Previous studies demonstrated that the extreme toxicity and acidity of AMD destroy aquatic ecosystems and render water resources less habitable for various biota; thus, AMD sites often have very low biodiversity (Baker and Banfield 2003; Simmons et al. 2005; Delavat et al. 2013; Bao et al. 2017). Meanwhile, the abundance and activities of SRB are usually limited by the small amount of organic substrate in AMD sites (Kolmert and Johnson 2001; Bao et al. 2018) because of the low biomass and net primary productivity in these environments (Hamsher et al. 2002; Simmons et al. 2005). In this study, the low DOC and the relatively low pH (< 3.0) eventually resulted in a low abundance and diversity of SRB in the upstream sediments. In the midstream (S6 and S8), increasing richness and diversity of microbial and SRB communities were detected due to the decrease in AMD pollution. Along with the increased DOC and abundant available sulfate in this area, the activity of SRB was expected to result in a high level of reduced sulfur species (Fig. 2). In the downstream sites (S10 and S11), the richness and diversity of the microbial and SRB communities were similar to those in the control site (C4) due to the similar physicochemical characteristics among these sites.
Clustering analysis of the detected 16S rRNA and dsrB sequences separated all samples into several major clusters (Fig. 5 and Fig. S2), and the sample groupings were largely consistent with the differences in the AMD pollution gradient and sampling time, indicating spatiotemporal variation in microbial and SRB community structures. Similar spatial or spatiotemporal shifts in the phylogenetic composition and structure of overall microbial communities have been observed in other AMD-contaminated watersheds, such as the Rio Tinto in southwestern Spain (García-Moyano et al. 2012), the Amous River in southern France (Volant et al. 2014), and the Aha watershed in Southwest China (Sun et al. 2015). Similarly, spatiotemporal variations in SRB community structure have also been found in Carnoulès AMD in France (Giloteaux et al. 2013). In this study, the effects of temporal fluctuations on SRB community structure were more notable at the lightly contaminated sites (S8, S10, and S11) and the control site (C4) (Fig. 5b) than at the other sites, probably because of the temporal fluctuation of physicochemical parameters in these areas. A previous study by Giloteaux et al. (2013) suggested that fluctuating physicochemical parameters was the decisive factor for seasonal fluctuations of SRB diversity. In this study, a clear spatial pattern of microbial and SRB community composition was observed (Fig. 5 and Fig. S2). One of the notable findings was that pH may be the primary environmental parameter structuring the differences in microbial and SRB communities between upstream and downstream samples (Fig. 6a). In addition, as indicated by CCA, the TS, ExS, and Fe contents were three additional major factors that shaped the microbial and SRB community composition. As the major contaminants in AMD sites, iron and sulfur compounds might have influenced the overall microbial communities primarily by controlling the distribution of Fe- and S-metabolizing microorganisms (Sun et al. 2015; Bao et al. 2017; Sun et al. 2020). Similarly, available sulfate, which was represented by ExS in this study, could be a decisive factor that affects SRB community composition. In addition, many SRB species can not only reduce sulfate, but also have their ability to reduce Fe(III) (Bao et al. 2018). This could explain why Fe content was also important for SRB communities distribution in this study. Moreover, organic substrate, which was represented by DOC in this study, was also demonstrated as a major factor for the SRB community, as organic substrate is used as the electron donor by SRB during sulfate reduction.
Composition of the SRB communities
Our high-throughput sequencing analyses revealed high SRB diversity in Hengshi watershed sediments, even in the heavily AMD-contaminated sections. The 16S rRNA sequencing analysis identified many potential SRB. 18 potential SRB families were detected, accounting for 4.45–35.05% (12.46% on average) of the total 16S rRNA gene sequences (Table S4), even in the heavily AMD-contaminated sediments (S0 and S1) with a pH lower than 3.0. This finding is consistent with the results of a previous study in this region that found numerous sequences affiliated with potential SRB (Bao et al. 2017). This result was not surprising, as SRB have been found in many AMD environments, such as mine tailings near Timmins in Canada (Fortin et al. 2000), the Carnoulès AMD in France (Giloteaux et al. 2013), and the Aha watershed in Southwest China (Sun et al. 2015). Overall, the results in this study confirm the high abundance of potential SRB groups in sediments of the Hengshi watershed and suggest the existence of active biological sulfate reduction in this area, which could therefore play a role in the natural attenuation of AMD.
The dsrB gene sequencing analysis performed in this study identified two phyla (Firmicutes and Proteobacteria), and Clostridia (within Firmicutes) and Deltaproteobacteria (within Proteobacteria) represented the dominant SRB. Our data were consistent with previous observations obtained from various environments, such as lake hypolimnion and sediments (Hamilton et al. 2016), marine crustal fluids (Robador et al. 2015), marine and estuary sediments (Jiang et al. 2009; Zouch et al. 2017), and petroleum reservoirs (Tian et al. 2017), suggesting that Clostridia and Deltaproteobacteria have a wide adaptation range. Notably, previous studies found that Deltaproteobacteria was the most abundant class of SRB communities in sea sediment cores, salt marshes (Zhang et al. 2017; Cui et al. 2017), and even sediment from AMD sites (Mardanov et al. 2017). Surprisingly, Clostridia were the most abundant class in most sediments in this study, especially the heavily contaminated upstream regions, suggesting that species in this class are more tolerant than other species to AMD contamination in the study area. Within Clostridia, Peptococcaceae was the most abundant family and was negatively correlated with pH, indicating that Peptococcaceae is well adapted and tolerant of AMD contamination in the study area. Most of the identified SRB grow optimally at a pH of 6–8 (Widdel 1988), and only very few acidophilic/acid-tolerant SRB have been isolated and characterized up to now (Bertel et al. 2012; Petzsch et al. 2015). Desulfosporosinus is the most commonly identified acidophilic/acid-tolerant bacterium among the SRB, and several species belonging to this genus have been isolated (Bertel et al. 2012; Petzsch et al. 2015). Interestingly, this genus happens to belong to the family Peptococcaceae. Desulfosporosinus was detected by both 16S rRNA gene and dsrB gene sequencing, but it was not the dominant genus in the dsrB gene sequencing analysis. Desulfosporosinus is generally enriched and isolated with low molecular weight organic carbon (glycerol, glucose, and lactate) as the electron donor (Alazard et al. 2010; Bertel et al. 2012); this could explain why it was not the dominant species in natural environment where low molecular weight organic carbon was not the main organic substance available. Moreover, the most abundant genus was Desulfotomaculum (within Peptococcaceae) in this study, which showed a relatively high abundance in the heavily contaminated regions. Previous studies suggested that most of the isolated Desulfotomaculum grow optimally at neutral pH (Jabari et al. 2013; Krishnamurthi et al. 2013), but members of Desulfotomaculum are somewhat acid tolerant and have been found in some moderately acidic environments (Dugan 1975; Sánchez-Andrea et al. 2013). Although an acid-tolerant species belonging to Desulfovibrio has been isolated (Mardanov et al. 2017), Desulfovibrio showed relatively high abundances in less contaminated areas in this study.
Notably, a large fraction of the dsrB sequences could not be identified at the genus level in this study, and only a relative abundance ranging from 6.1 to 43.7% of the dsrB sequences could be assigned at the genus level (Fig. 4c and Table S5). These findings suggest that the AMD-contaminated environment contains a large proportion of currently unknown SRB lineages, which need to be studied in more detail in future studies. Indeed, similar results were widely found in many studies of various environments, mainly due to the limited number of reference sequences. For example, approximately 62% of dsrB sequences in surface sediments from the adjacent area of the Changjiang estuary had no clear phylogenetic affiliation with previously cultured SRB (Zhang et al. 2016); 40.91 to 73.36% of dsrB sequences in sediment cores from the East China Sea could not be assigned at the family or genus level (Zhang et al. 2017); and 60% of dsrA sequences in heavy metal-contaminated sediments from a salt marsh in the Medway estuary (UK) showed no clear phylogenetic affiliation with known SRB lineages (Quillet et al. 2012).
Ecological implications
Sulfate-reducing bacteria have been investigated and found to be important functional bacteria in diverse ecological habitats. These bacteria are known to be of great significance in the carbon and sulfur cycles of ecosystems and have a wide variety of applications in wastewater treatment, remediation of pollutants, and other processes (Zhang et al. 2017; Tian et al. 2017). Many studies have demonstrated that sulfate reduction by SRB can decrease the acidity of and remove metals and sulfate from AMD (Coggon et al. 2012; Bao et al. 2018). By quantifying and deep sequencing the dsrB genes involved in dissimilatory sulfate reduction, we found that diverse SRB were widely distributed in the AMD-contaminated Hengshi watershed, and it might be expected that SRB would play important roles in AMD attenuation in the Hengshi watershed.
Specifically, the high abundance and diversity of SRB would very likely fuel sulfate reduction and accelerate the natural attenuation of AMD. This is because SRB reduce sulfate to sulfide which can precipitate metals, and this process promotes alkalinity and then neutralizes AMD (Coggon et al. 2012; Sun et al. 2020). AMD attenuation by SRB has been demonstrated in many studies. Coggon et al. (2012) observed that the biological reduction of iron-sulfate minerals by SRB has the potential to naturally attenuate AMD. Ergas et al. (2006) suggested that the bacteria that play roles in the natural attenuation of AMD are acidophilic and acid-tolerant Fe(III)- and sulfate-reducing bacteria due to their tolerance of the acidity of AMD.
However, a question remains: what is the relationship between the reduced sulfur species contents in sediment and the detected SRB? The findings of the present study suggested widely distributed SRB and the existence of active sulfate reduction in the AMD-contaminated watershed. However, compared with pH and TS, reduced sulfur species (ES, AVS, and CRS) appeared to have no significant correlation with the SRB community based on CCA (Fig. 6b). The diversity of SRB and the potential sulfate reduction activity might still be overestimated or underestimated. Studies have shown that certain named SRB might actually lack the ability to reduce sulfate in anaerobic environments, such as Pelotomaculum and Sporotomaculum, which are not able to respire anaerobically with sulfite/sulfate/organosulfonates, even though they are closely affiliated with SRB (Brauman et al. 1998; Imachi et al. 2006). Moreover, the functional dsr gene sued by SRB is sometimes present in certain sulfide-oxidizing bacteria (SOB), which have the reverse function of sulfide oxidation (Hipp et al. 1997; Loy et al. 2009; Fan et al. 2012). In addition, physicochemical factors, such as pH, heavy metals, sulfate, and carbon availability, might exhibit moderate inhibition or stimulation of the activity of certain strains of SRB (Coggon et al. 2012; Giloteaux et al. 2013; Robador et al. 2015). Or perhaps it resulted from the fact that SRB are active upstream but their produced sulfides are not in the solid phase because they are dissolved by acidity. Therefore, further studies should focus on this issue.
References
Alazard D, Joseph M, Battaglia-Brunet F, Cayol J, Ollivier B (2010) Desulfosporosinus acidiphilus sp. nov.: a moderately acidophilic sulfate-reducing bacterium isolated from acid mining drainage sediments. Extremophiles 14:305–312
Baker BJ, Banfield JF (2003) Microbial communities in acid mine drainage. FEMS Microbiol Ecol 44:139–152
Bao Y, Guo C, Wang H, Lu G, Yang C, Chen M, Dang Z (2017) Fe- and S-metabolizing microbial communities dominate an AMD-contaminated river ecosystem and play important roles in Fe and S cycling. Geomicrobiol J 34:695–705
Bao Y, Guo C, Lu G, Yi X, Wang H, Dang Z (2018) Role of microbial activity in Fe(III) hydroxysulfate mineral transformations in an acid mine drainage-impacted site from the Dabaoshan Mine. Sci Total Environ 616-617:647–657
Bertel D, Peck J, Quick TJ, Senko JM (2012) Iron transformations induced by an acid-tolerant Desulfosporosinus species. Appl Environ Microbiol 78:81–88
Brauman A, Müller JA, Garcia JL, Brune A, Schink B (1998) Fermentative degradation of 3-hydroxybenzoate in pure culture by a novel strictly anaerobic bacterium, Sporotomaculum hydroxybenzoicum gen. nov. sp. nov. Int J Syst Bacteriol 48:215–221
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Chamani PM, Marasinghe W, Anne MT, Frank K, William AM (2016) Sediment metal concentration survey along the mine-affected Molonglo River, NSW, Australia. Arch Environ Contam Toxicol 70:572–582
Chao A (1984) Nonparametric estimation of the number of classes in a population. Scand J Stat 11:265–270
Chen M, Lu G, Guo C, Yang C, Wu J, Huang W, Yee N, Dang Z (2015) Sulfate migration in a river affected by acid mine drainage from the Dabaoshan mining area, South China. Chemosphere 119:734–743
Chen M, Lu G, Wu J, Yang C, Niu X, Tao X, Shi Z, Yi X, Dang Z (2018) Migration and fate of metallic elements in a waste mud impoundment and affected river downstream: a case study in Dabaoshan Mine, South China. Ecotoxicol Environ Saf 164:474–483
Coggon M, Becerra CA, Nüsslein K, Miller K, Yuretich R, Ergas SJ (2012) Bioavailability of jarosite for stimulating acid mine drainage attenuation. Geochim Cosmochim Acta 78:65–76
Cui J, Chen X, Nie M, Fang S, Tang B, Quan Z, Li B, Fang C (2017) Effects of Spartina alterniflora invasion on the abundance, diversity, and community structure of sulfate reducing bacteria along a successional gradient of coastal salt marshes in China. Wetlands 37:221–232
Delavat F, Lett MC, Lièvremont D (2013) Yeast and bacterial diversity along a transect in an acidic, As-Fe rich environment revealed by cultural approaches. Sci Total Environ 463-464:823–828
Dev S, Roy S, Bhattacharya J (2016) Understanding the performance of sulfate reducing bacteria based packed bed reactor by growth kinetics study and microbial profiling. J Environ Manag 177:101–110
Dugan PR (1975) Bacterial ecology of strip mine areas and its relationship to the production of acidic mine drainage. Ohio J Sci 75:266–279
Edgar RC (2013) UPARSE: highly accurate OTU sequencesfrom microbial amplicon reads. Nat Methods 10:996–998
Ergas SJ, Harrison J, Bloom J, Forloney K, Ahlfeld DP, Nüsslein K, Yuretich RF (2006) Natural attenuation of acid mine drainage by acidophilic and acidotolerant Fe(III)- and sulfate-reducing bacteria. ACS Symp Ser 940:105–127
Fan L, Tang S, Chen C, Hsieh H (2012) Diversity and composition of sulfate- and sulfite-reducing prokaryotes as affected by marine-freshwater gradient and sulfate availability. Microb Ecol 63:224–237
Fortin D, Roy M, Rioux JP, Thibault PJ (2000) Occurrence of sulfate-reducing bacteria under a wide range of physico-chemical conditions in Au and Cu-Zn mine tailings. FEMS Microbiol Ecol 33:197–208
Gao P, Sun X, Xiao E, Xu Z, Li B, Sun W (2019) Characterization of iron-metabolizing communities in soils contaminated by acid mine drainage from an abandoned coal mine in Southwest China. Environ Sci Pollut Res 26(10):9585–9598
García-Moyano A, González-Toril E, Aquilera Á, Amils R (2012) Comparative microbial ecology study of the sediments and the water column of the Río Tinto, an extreme acidic environment. FEMS Microbiol Ecol 81:303–314
Giloteaux L, Duran R, Casiot C, Bruneel O, Elbaz-Poulichet F, Goñi-Urriza M (2013) Three-year survey of sulfate-reducing bacteria community structure in Carnoule’s acid mine drainage (France), highly contaminated by arsenic. FEMS Microbiol Ecol 83:724–737
Hamilton TL, Bovee RJ, Sattin SR, Mohr W, Gilhooly I, William P, Lyons TW, Pearson A, Macalady JL, Spear JR, Loy A (2016) Carbon and sulfur cycling below the chemocline in a meromictic lake and the identification of a novel taxonomic lineage in the FCB Superphylum, Candidatus Aegiribacteria. Front Microbiol 7:598
Hamsher SE, Casamatta DA, Filkin NR, McClintic AS, Chiasson WB, Verb GR, Vis ML (2002) A new method for studying nutrient limitation of Periphyton: a case study from acid mine drainage streams. J Phycol 38(s1):15
Hipp W, Pott A, Thum-Schmitz N, Faath I, Dahl C, Trüper HG (1997) Towards the phylogeny of APS reductases and sirohaem sulfite reductases in sulfate-reducing and sulfur-oxidizing prokaryotes. Microbiology 143:2891–2902
Imachi H, Sekiguchi Y, Kamagata Y, Loy A, Qiu YL, Hugenholtz P, Kimura N, Wagner M, Ohashi A, Harada H (2006) Non-sulfate-reducing, syntrophic bacteria affiliated with Desulfotomaculum cluster I are widely distributed in methanogenic environments. Appl Environ Microbiol 72:2080–2091
Jabari L, Gannoun H, Cayol JL, Hamdi M, Ollivier B, Fauque G, Fardeau ML (2013) Desulfotomaculum peckii sp nov., a moderately thermophilic member of the genus Desulfotomaculum, isolated from an upflow anaerobic filter treating abattoir wastewaters. Int J Syst Evol Microbiol 63:2082–2087
Jiang LJ, Zheng YP, Peng XT, Zhou HY, Zhang CL, Xiao X, Wang F (2009) Vertical distribution and diversity of sulfate-reducing prokaryotes in the Pearl River estuarine sediments, Southern China. FEMS Microbiol Ecol 70:249–262
Jones DL, Willett VB (2006) Experimental evaluation of methods to quantify dissolved organic nitrogen (DON) and dissolved organic carbon (DOC) in soil. Soil Biol Biochem 38:991–999
Klein R, Tischler JS, Mühling M, Schlömann M (2014) Bioremediation of mine water. Adv Biochem Eng Biotechnol 141:109–172
Kolmert A, Johnson DB (2001) Remediation of acidic waste waters using immobilised, acidophilic sulfate-reducing bacteria. J Chem Technol Biotechnol 76:836–843
Koschorreck M (2008) Microbial sulphate reduction at a low pH. FEMS Microbiol Ecol 64:329–342
Krishnamurthi S, Spring S, Kumar PA, Mayilraj S, Klenk HP, Suresh K (2013) Desulfotomaculum defluvii sp. nov., a sulfate-reducing bacterium isolated from the subsurface environment of a landfill. Int J Syst Evol Microbiol 63:2290–2295
Loy A, Duller S, Baranyi C, Mußmann M, Ott J, Sharon I, Béjà O, Paslier DL, Dahl C, Wagner M (2009) Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur-oxidizing prokaryotes. Environ Microbiol 11:289–299
Magoè T, Salzberg SL (2011) FLASH: fast lengthadjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963
Mardanov AV, Beletskii AV, Ivasenko DA, Pimenov NV, Karnachuk OV, Ravin NV (2017) Sulfate-reducing bacteria in the microbial community of acidic drainage from a gold deposit tailing storage. Microbiology 86(2):286–288
Méndez-García C, Peláez AI, Mesa V, Sánchez J, Golyshina OV, Ferrer M (2015) Microbial diversity and metabolic networks in acid mine drainage habitats. Front Microbiol 6:475
Mohapatra BR, Gould WD, Dinardo O, Koren DW (2011) Tracking the prokaryotic diversity in acid mine drainage-contaminated environments: a review of molecular methods. Miner Eng 24:709–718
Petzsch P, Poehlein A, Johnso BD, Daniel R, Schlömann M, Mühling M (2015) Genome sequence of the moderately acidophilic sulfate-reducing Firmicute desulfosporosinus acididurans (strain M1T). Genome Announc 3(4):e00881–e00815
Quillet L, Besaury L, Popova M, Paissé S, Deloffre J, Ouddane B (2012) Abundance, diversity and activity of sulfate-reducing prokaryotes in heavy metal-contaminated sediment from a salt marsh in the Medway estuary (UK). Mar Biotechnol 14:363–381
Robador A, Jungbluth SP, LaRowe DE, Bowers RM, Rappé MS, Cowen JP (2015) Activity and phylogenetic diversity of sulfate-reducing microorganisms in low-temperature subsurface fluids within the upper oceanic crust. Front Microbiol 5:748
Sánchez-Andrea I, Stams AJM, Amils R, Sanz JL (2013) Enrichment and isolation of acidophilic sulfate-reducing bacteria from Tinto River sediments. Environ Microbiol Rep 5(5):672–678
Sánchez-Andrea I, Sanz JL, Bijmans MFM, Stams AJM (2014) Sulfate reduction at low pH to remediate acid mine drainage. J Hazard Mater 269:98–109
Schloss PD, Westcott SL, Ryabin T, Hall JR,Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, SahlJW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source,platform-independent, community-supported software for describing and comparingmicrobial communities. Appl Environ Microbiol 75:7537–7541
Sen AM, Johnson B (1999) Acidophilic sulphate-reducing bacteria: candidates for bioremediation of acid mine drainage. Process Metall 9:709–718
Shannon CE (1948) A mathematical theory of communication. Bell Syst Tech J 27:379–423
Simmons JA, Lawrence ER, Jones TG (2005) Treated and untreated acid mine drainage effects on stream periphyton biomass, leaf decomposition, and macroinvertebrate diversity. J Freshw Ecol 20(3):413–424
Simpson EH (1949) Measurement of diversity. Nature 163:688–688
Sun W, Xiao T, Sun M, Dong Y, Ning Z, Xiao E, Tang S, Li J (2015) Diversity of the sediment microbial community in the Aha watershed (Southwest China) in response to acid mine drainage pollution gradients. Appl Environ Microbiol 81:4874–4884
Sun W, Sun X, Li B, Xu R, Young LY, Dong Y, Zhang M, Kong T, Xiao E, Wang Q (2020) Bacterial response to sharp geochemical gradients caused by acid mine drainage intrusion in a terrace: relevance of C, N, and S cycling and metal resistance. Environ Int 138:105601
Taylor MP (2007) Distribution and storage of sediment-associated heavy metals downstream of the remediated Rum Jungle Mine on the East Branch of the Finniss River, Northern Territory, Australia. J Geochem Explor 92:55–72
Tian H, Gao P, Chen Z, Li Y, Li Y, Wang Y, Zhou J, Li G, Ma T (2017) Compositions and abundances of sulfate-reducing and sulfur-oxidizing microorganisms in water-flooded petroleum reservoirs with different temperatures in China. Front Microbiol 8:143
Volant A, Bruneel O, Desoeuvre A, Héry M, Casiot C, Bru N, Delpoux S, Fahy A, Javerliat F, Bouchez O, Duran R, Bertin PN, Elbaz-Poulichet F, Lauga B (2014) Diversity and spatiotemporal dynamics of bacterial communities: physicochemical and other drivers along an acid mine drainage. FEMS Microbiol Ecol 90:247–263
Wang H, Guo CL, Yang CF, Lu GN, Chen MQ, Dang Z (2016) Distribution and diversity of bacterial communities and sulphate-reducing bacteria in a paddy soil irrigated with acid mine drainage. J Appl Microbiol 121(1):196–206
Widdel F (1988) Microbiology and ecology of sulfate- and sulfur-reducing bacteria. In: Zehnder AJB (ed) Biology of anaerobic microorganisms. Wiley, New York, pp 469–585
Xu R, Li B, Xiao E, Young LY, Sun X, Kong T, Dong Y, Wang Q, Yang Z, Chen L, Sun W (2020) Uncovering microbial responses to sharp geochemical gradients in a terrace contaminated by acid mine drainage. Environ Pollut 261:114226
Zhang Y, Zhen Y, Mi TZ, He H, Yu ZG (2016) Molecular characterization of sulfate-reducing bacteria community in surface sediments from the adjacent area of Changjiang estuary. J Ocean Univ China 15:107–116
Zhang Y, Wang X, Zhen Y, Mi T, He H, Yu Z (2017) Microbial diversity and community structure of sulfate-reducing and sulfur-oxidizing bacteria in sediment cores from the East China Sea. Front Microbiol 8:2133
Zouch H, Karray F, Armougom F, Chifflet S, Hirschler-Réa A, Kharrat H, Kamoun L, Hania WB, Ollivier B, Sayadi S, Quéméneur M (2017) Microbial diversity in sulfate-reducing marine sediment enrichment cultures associated with anaerobic biotransformation of coastal stockpiled phosphogypsum (Sfax, Tunisia). Front Microbiol 8:1583
Funding
This study was financially supported by the National Natural Science Foundation of China (nos. 41720104004 and 41931288), the National Key Research and Development Program of China (no. 2017YFD0801000), the Fund of Science and Technology Bureau of Shaoguan City (no. 2017SGTYFZ201), and the Guangdong Basic and Applied Basic Research Foundation (no. 2019A1515110811).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible Editor: Diane Purchase
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 136 kb)
Rights and permissions
About this article

Cite this article
Bao, Y., Jin, X., Guo, C. et al. Sulfate-reducing bacterial community shifts in response to acid mine drainage in the sediment of the Hengshi watershed, South China. Environ Sci Pollut Res 28, 2822–2834 (2021). https://doi.org/10.1007/s11356-020-10248-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-020-10248-7