
Abstract
The change in environmental conditions during the transportation of contaminated soil and sediment was expected to affect the transformation of heavy metal fractionation. This study disclosed the serious contamination of copper (Cu), lead (Pb), and zinc (Zn) in the sewer sediment of an e-waste dismantling community in Thailand which may be caused by flushed contaminated soil and e-waste fragments. Two environmental conditions were simulated to observe the transformation of heavy metal fractionation. The anoxic sewer condition was induced using high substrate and sulfate in a closed container. The aeration of anoxic contaminated sediment was applied to simulate the transformation to an oxidative environment. The BCR sequential extraction was applied for heavy metal fractionation in this study. The study results exhibited that when heavy metal contaminated soil was transferred into this induced anoxic condition, fractionation was redistributed based on the chemical change of system that tends to be associated into F3 (oxidizable fraction) > F2 (reducible fraction) > F1 (acid soluble/exchangeable fraction). Cu exhibited the outstanding capability association to F3. The iron sulfide was not observed as usual due to its lower capability than Cu, Pb, and Zn. When contaminated sediment was transported to a more oxidative environment, the heavy metals fractionation would be redistributed again among those new environment media. It is noteworthy that F3 of Cu was stable even in oxic conditions. F2 of Fe was not developed by this oxic condition, possibly because its dehydration process was limited. The redistribution under an oxic environment became F1 > F2 > F3 indicating their more available form. This transformation was imperative and should be taken into account in heavy metal contaminated site management and control.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Heavy metal is a hazardous inorganic pollutant group commonly found at contaminated sites as a result of metal mining, milling processes, industrial wastes, and electronic waste (e-waste). Heavy metals are accumulated in soil and sediment by various mechanisms. It could precipitate as its own compound such as a metal hydroxide, metal carbonate, and well-known metal sulfides formed under anoxic conditions. Sulfide is well-known to principally associate with heavy metals rather than other soil materials via its covalent bond. Another major process in accumulating heavy metals in soil is adsorption by various mechanisms on the surface of many soil materials, especially, on ferric oxide and organic matter (Bradl 2004). Amorphous ferric oxides in soil are natural materials having great capability to sorb heavy metals through isomorphic substitution and ion exchange mechanisms (Hooda 2010).
The heavy metal fractionation in soil is a concept to classify heavy metals in soil into some form related to the environmental states or chemical forms (e.g., exchangeable form, reducible form, oxidizable form, and stable form) (Filgueiras et al. 2002). Changing of soil chemicals due to the shifting of the surrounding environment leads to the change of soil chemicals, thus transforming heavy metal fractionation in soil.
Oxidation-reduction potential (ORP) is a measure of electron pressure in a solution. It is often used to access the degree of electrochemical reduction of an environmental system. The chemical and biological transformations take place as coupled oxidation in which the organic substrate acts as the electron donor and as a reduction reaction in which various chemicals act as electron accepters. For the anoxic respiration, nitrate is one of the first electron accepters at an ORP of about + 250 mV followed by manganese at an ORP of about + 225 mV. The reduction of iron (Fe) from ferric (Fe3+) to ferrous (Fe2+) occurred at an ORP of about + 100 to − 100 mV. Sulfide (S2−) is developed from the reduction of sulfate (SO42−) occurring at an ORP of − 100 to − 200 mV. These reduction reaction was rather occurred sequentially from higher ORP to lower ORP (e.g., if available ferric in the system was still plenty or sufficient for biological activity, sulfate will not be used).
Some studies demonstrated that the change of ORP of the surrounding environment affects the form of heavy metals in soil and sediment. The study of Kelderman and Osman (2007) showed that when anoxic sediment was exposed to the air, some heavy metals were released due to the oxidation of metal sulfide, and then redistributed to exchangeable and carbonate fractions that were labile binding phase. Hartley and Dickinson (2010) demonstrated the effect of the ORP of water that slowly flows through the sediment column on the mobility of some metals. They found that iron and soluble organic carbon were released into the flowing water during negative ORP (− 157 mV) while some heavy metals (Cu, Pb, Zn) were released when the ORP was manipulated to more than + 300 mV in contrast to iron and soluble organic carbon which recoil to limited concentrations. Vink et al. (2010) simulated floodplain soil inundated by water which was a reductive environment. The result demonstrated that the heavy metals were released or detained kinetically depending on its local condition relating to microbial activity. The heavy metals in soil were released due to the loss of the reactive ferric-oxyhydroxide sorption phase, and then dissolved in water by association with dissolved organic matter or precipitated as metal sulfide via bonding with sulfides. This was dependent on metal species, organic matter in soil, and available sulfate in the system.
The soil in the e-waste dismantling area was usually polluted seriously by copper (Cu), lead (Pb), and zinc (Zn) while nickel (Ni), cadmium (Cd), and mercury (Hg) were found at elevated levels (Damrongsiri et al. 2016; Leung et al. 2006; Jun-hui and Hang 2009; Luo et al. 2011; Tang et al. 2010). Metal contamination through wind and water transportation affected adjacent areas and accumulated in many food plants (Fu et al. 2008; Jun-hui and Hang 2009; Luo et al. 2011; Olafisoye et al. 2013; Pradhan and Kumar 2014).
The serious heavy metal contamination in soil was disclosed in a dismantling community—Sue Yai Utit—located in Bangkok, the capital city of Thailand (Damrongsiri et al. 2016). The contaminated soil and small e-waste fragments were expected to be transported to the drainage collection system by runoff which accumulates in sewers and may be further transported to the river. The drainage system in this study area is a combined system that receives both surface runoff and untreated household wastewater. Thus, the biological degradation of organic substrate occurs naturally. Due to the confined environment in the sewer, the oxygen is normally depleted by microbial activity resulting in the occurrence of anaerobic respiration transforming various chemicals to a reduced state. Subsequently, this anoxic sediment would be finally transported to the river, which has a more oxidative environment, therefore transforming many chemicals to a higher oxidative state.
This study aims to investigate the transformation of heavy metal fractionation by simulating the change in environmental conditions. Heavy metal contamination in the sewer system in the Sue Yai Utit e-waste dismantling area was observed. Soil and sewer sediment in this area was examined in this study. Copper (Cu), lead (Pb), and zinc (Zn) exhibited very high concentrations in soil in this area (Damrongsiri et al. 2016) and so were selected as the representatives of heavy metal pollution in the soil and sewer sediment.
Materials and method
Sampling sites, sample collection, and preparation
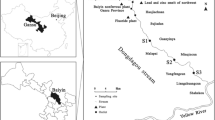
The soil and sediment sampling activities at Sue Yai Utit were conducted during April and June 2016, respectively. The details of this dismantling area and its heavy metal concentrations in the soil were demonstrated in the study of Damrongsiri et al. (2016). The wastewater collection system in this area was a combined sewer system. The direction of flow in the sewer is shown in Fig. 1a. The sediment samples from the sewer were collected from six manholes as shown in Fig. 1a. The manholes #1 and #2 were located in the upstream area, #3 and #4 were among the hotspots of the contaminated area, and #5 and #6 were downstream of the contaminated area before the discharge to a canal. Soil samples were collected from eight sampling points in the hotspot area (Damrongsiri et al. 2016) as shown in Fig. 1b. All samples were collected to a depth of 10 cm using a stainless steel trowel. The sediment samples were stored in HDPE bottles and were kept moist at 4 °C in a refrigerator. The polyethylene bags (Ziploc) were employed to store the soil samples.

Map of the Sue Yai Utit study area: a position of manhole and wastewater direction and b soil sampling position
Observation for heavy metals in sewer sediment
The sediment samples were dried in an oven at 40 °C. The vegetation, stones, and other coarse materials were removed before grinding and sieving (< 2 mm). The samples were then homogenized and stored in a desiccator prior to digestion and analysis for Cu, Pb, and Zn concentration.
Environmental transformation experiment
There were two transformation simulations in this study: (1) transformation from a surface soil environment to anoxic sewer sediment and (2) transformation from anoxic sewer sediment to aerobic suspended solid in the waterway.
Surface soil to anoxic sewer sediment
This experiment was designed for the case of surface soil (oxidative environment) being moved to the sewer and submerged with wastewater (anoxic environment). The sample was prepared as a dried soil sample by the method described earlier and applied in this experiment. The equal weights of those eight soil samples were mixed together to be a composite sample. The prepared soil samples were sieved to separate the fine soil particles (Fsoil) and coarse soil particles (Csoil). Fine soil particles (< 0.063 mm) were aligned with silt and clay, while coarse soil particles (0.063–2 mm) were comparable to the size of sand according to the grain size classification for soil by Wentworth (1922). The soil samples were quantified for pH, sulfide, organic matter (OM), Fe, Cu, Pb, and Zn.
To induce anoxic environment, 30 g of soil sample was mixed with 150 mL synthetic wastewater in a 200 mL glass bottle. The bottle was closed tightly and kept in the dark at room temperature (OECD 2002) for 2 months. The synthetic wastewater was prepared from Lauryl Tryptose Broth (Difco tm) and Na2SO4 (99%, MERCK) to derive 3000 mg L−1 of chemical oxygen demand (COD) and 3000 mg SO42− L−1. This 3000 mg SO42− L−1 was equivalent to 5000 mg S2− kg−1 in soil while 3000 mg COD L−1 was ten times the average COD in domestic wastewater in Thailand (Department of Industrial Works 2002). After 2 months, the sample was measured for the pH and ORP of the system. Then, soil and water were separated by centrifugal approach. The sulfate, COD, total concentration of Fe, Cu, Pb, and Zn in water; and sulfide, OM, total concentration and fractionation of Fe, Cu, Pb, and Zn in soil were quantified. This experiment was performed in triplicate for both Fsoil and Csoil.
Anoxic sewer sediment to aerobic suspended solid
This experiment was set to simulate the contaminated sediment being released to the river and suspended in an aerobic water column. A selected contaminated sediment sample which had high concentrations of heavy metals was used in this study. The wet sediment sample was used to minimize the loss of sulfide due to the reaction with oxygen and dehydration of some hydrated compounds (Koopmans and Groenenberg 2011; Qi et al. 2014). This sediment sample was separated into fine sediment particles (< 0.063 mm, Fsed) and coarse sediment particles (0.063–2 mm, Csed) by a wet sieving technique. The sieving water was prepared using 18-MΩ DI water purged with N2 to degas any dissolved oxygen. The separated sediment samples were centrifuged to dispel excess water. Then, both coarse and fine sediment samples were measured for pH, ORP, sulfide, OM, total concentration and fractionation of Fe, Cu, Pb, and Zn.
Then, the 20 g of wet sediment sample was mixed with 200 mL of 18-MΩ DI water in a 250-mL glass bottle and aerated continuously. The attached sediment on the glass wall above the water surface was moved back to the water every day. The DI water was filled to the control level every day to maintain sediment sample to water ratio. This experiment was performed in triplicate for both coarse and fine sediment. After 2 months of aeration, the sample was measured for the pH and ORP of the system, and then water and sediment were separated by centrifuge before analysis for sulfate, COD, total concentration of Fe, Cu, Pb, and Zn in water; and sulfide, OM, total concentration and fractionation of Fe, Cu, Pb, and Zn in sediment.
Measurement
The sample was dried as described earlier. The digestion was performed following US-EPA 3050b guidelines described here briefly. One gram of dried sample was transferred to a 100-mL beaker and placed on a hotplate. The sample was repeatedly refluxed using HNO3 (65%, EMSURE®, MERCK), then oxidized by 30% H2O2 (30%, MERCK) until the general appearance of the sample was unchanged. Then, it was refluxed with HCl (37%, EMSURE®, MERCK) for a while. The sample was then filtered through Whatman no. 41 filters, transferred to a 50-mL volumetric flask, and diluted by 18-MΩ DI water. The water sample was digested following US-EPA 3005a using HNO3 and HCl which were heated by a hotplate. The digested samples were analyzed for Fe, Cu, Pb, and Zn by inductively coupled plasma optical emission spectroscopy (ICP-OES, Plasma Quant® PQ 9000 Elite, Analytik Jena). Reagent blanks and the analytical triplicates were used to control the accuracy of the analysis.
A sequential extraction procedure proposed by the Commission of the European Community Bureau of Reference (BCR) was adapted for use in this study (Ure et al. 1993); however, the US-EPA digestion procedure was applied for the last extraction step because only the total environmental availability was considered in this study. The wet sample was used to minimize the alteration of phase of the heavy metals in the sample (Koopmans and Groenenberg 2011; Qi et al. 2014). The different extracting agents were applied in each extraction step which is summarized here briefly. First step is 0.11 M acetic acid (100%, EMSURE®, MERCK). Second step is 0.1 M hydroxylammonium chloride (96%, CARLO ERBA) adjusted with HNO3 to pH 2. Third step is 30% H2O2 followed by ammonium acetate (98%, QRëc) adjusted with nitric acid to pH 2. Fourth step is hot acid digestion following US-EPA method 3050b. Following this procedure, the heavy metals in the soil can be divided into four fractions (Ure et al. 1993): (1) acid soluble/exchangeable fraction or easily mobile fraction (F1), (2) reducible fraction or metal bound to oxide of iron and manganese (F2), (3) oxidizable fraction or metal bound to organic matter (F3), and (4) residual fraction (F4) that is not released under normal natural conditions. The extractant from each step was digested using US-EPA method 3005a and measured by ICP-OES for Fe, Cu, Pb, and Zn. The pseudo-total concentrations of metals were the sum of F1, F2, F3, and F4.
Some important parameters influencing heavy metal fractionation which quantified in this study were organic matter which represent by OM, sulfide which represent by acid volatile sulfide (AVS) and amorphous ferric oxide which was the extracted iron during step 2 of BCR extraction. The OM was measured by the Walkley–Black modified acid-dichromate digestion-FeSO4 titration method. The AVS in the sample was quantified by the method modified from Yekta et al. (2012). An evaporating dish with plastic stand was set up in a 250-mL wide mouth glass bottle. One gram of wet sample and a magnetic bar were then placed into the bottle. The 10 mL of sulfide antioxidant buffer (SAOB) was filled up to the evaporating dish. A sulfide antioxidant buffer (SAOB) containing 2 M NaOH (100% EMPLURA®, MERCK), 0.1 M EDTA (100%, CARLO ERBA), and 0.1 M ascorbic acid (99.96%, Fisher Scientific) was used to trap the evaporated sulfide and prevent its evaporation and its reaction with oxygen. Then, 20 mL of 1 M HCl (37%, EMSURE®, MERCK) was added into the bottle and the lid was closed instantly. The sample was stirred for 2 h, and then the SAOB sample was taken for AVS measurement using a sulfide sensor (Ag/S ion selective electrode HI4015 HANNA instruments). The sulfide solution was prepared from Na2S·xH2O (30% as Na2S, PanReac AopliChem) in SAOB solution and used for sensor calibration.
The aqueous COD, sulfate, and sulfide were quantified following the method in standard methods for the Examination of Water and Wastewater (Eaton et al. 2005), which were the open reflux method, turbidimetric method, and iodometric method, respectively. The pH meter (inoLab® 740 connected with pH probe, WTW) and ORP meter (pH 3210 with ORP electrode, WTW) were used to measure the pH and ORP of the system, respectively.
Statistics
The statistical analysis was conducted using the SPSS program (ver. 22). The t test was applied to examine the heavy metal concentration in some interesting issues. The p value of less than 0.05 was used to identify the significance level.
Results
Heavy metal contamination in sewer sediment
The sediment samples from manholes upstream (# 1 and #2) and downstream (#5 and #6) were black in appearance with general mud smells. The sediment from manholes #3 and #4 were different, black in color with many e-waste fragments (e.g., copper wire, circuit board, metal nut, etc.). During sieving, both samples were quite viscous—especially for #4—and bad smell of organic solvent were obvious, and thus it can be surmised that both sediment samples #3 and #4 were contaminated by organic solvent. The heavy metal concentrations in these sediment samples are shown in Table 1.
The concentrations of Fe, Cu, Pb, and Zn in sediments #1 #2, #5, and #6 were considered in the range of background concentrations. The Zn concentrations in #1 and #5 were quite high. Heavy metal concentrations in sediments #3 and #4 were far greater than in other samples. These results indicated the serious contamination from e-waste dismantling activity to sewer sediment in the dismantling zone. The contamination may be caused by polluted surface soil and fragments of e-waste which were transported to the sewer via surface runoff. It should be noted that Pb concentration in sediment #3 was incredibly high. However, the contamination downstream was not as obvious as had been expected. The field survey found that sewers in this e-waste dismantling area faced a clogging problem for a long time, making this contaminated sediment difficult to transport. Besides, the sewer lines from manhole #2 to #6 and from #1 to #5 were the main sewer lines to the river, thus, they were dredged every 1–2 years. Thus, the anticipated elevated concentration of heavy metals was not found in sediments #5 and #6.
Effect of environmental transformation on the fractionation of heavy metals in soil and sediment
Surface soil to anoxic sewer sediment
The composite soil samples were prepared as Fsoil and Csoil as described in “Environmental transformation experiment.. The color of both samples was brown and similar to general surface soil. The soil sample characteristics were shown in Table 2 and the fractionation of heavy metals can be seen in Fig. 2. Total concentrations of most heavy metals in the samples before and after the simulation were not significantly different (p > 0.05). Almost all the total concentrations of each heavy metal and their pseudo-total concentrations derived from BCR extraction were not significantly different (p > 0.05), except Pb in Fsoil after the simulation which pseudo-total concentrations was 91.6% recovery comparing to its total concentration.

Fractionation of Cu, Pb, Zn, and Fe in surface soil before and after the anoxic sewer simulation
Surface soil
The OM of Fsoil was much higher than Csoil which was greater than general soil in Bangkok (0.7–8.9% OM, Wilcke et al. 1998). The high OM value was presumed to be caused by contamination of organic solvent and oil released from some e-waste dismantling process. Sulfide was not detected, indicating the oxidative condition of the prepared soil sample.
Cu was distributed among all four fractions. The greatest Cu component in the soil samples was in F4, followed by F3, while some was in F2 and F1. The major Pb and Fe components were in F4 (> 80%). Zn was fractionated among all four fractions. However, F4 of Zn was least, while, F1 was greatest among these four studied metals. The F4 of those metals were quite high and their fractionation in Fsoil and Csoil was similar. A distribution pathway of heavy metal contamination in e-waste dismantling sites was proposed by Damrongsiri et al. (2016), beginning with the deposition of e-waste scrap on to the soil surface. It acts as a source of heavy metals which then corroded and were released via natural processes and redistributed among the soil material.
Soil after the anoxic sewer simulation
After 2 months of anoxic sewer simulation, the soil was completely black in color with a bad smell. The soil and water characteristics are shown in Table 2 and the heavy metal fractionation is depicted in Fig. 2. The COD and sulfate of synthetic wastewater were markedly reduced (86–88% for COD, 33–42% for sulfate) indicating the biological degradation activity in the simulated system. The ORP of the Fsoil and Csoil systems were lower than − 200 mV indicating that anoxic conditions were in place (Mitsch and Gosselink, 2000) in the simulation. Under these very low ORPs, the available ferric, sulfate, and some organic compounds or carbon dioxide could be reduced. Obviously, sulfide in soil was developed conform to the ORP value (less than − 100 mV; Mitsch and Gosselink 2000). The OM in both Fsoil and Csoil was increased, possibly due to growth in microorganisms.
In comparison between situations before and after the simulation, the F4 (residual fraction) of every studied metal of every sample was reduced significantly (p < 0.05), thus the non-residual fraction was increased. For the non-residual fraction, the change of Cu was obvious in that all Cu was in F3. The non-residual fraction of Pb was not much changed, predominantly in F3 and F2. Zn was still mainly in F1 and F2 but the F1 was increased while the F2 was decreased. Generally, after the anoxic simulation, the residual fraction was decreased. The F3 of Cu, Pb, and Zn were increased clearly, and the F2 of Pb, Fe, and especially Zn was also increased. The F1 of Cu, Pb, and Zn seems reduced while the F1 of Fe was emerged.
Anoxic sewer sediment to aerobic suspended solid
A high concentration of heavy metals was found in the sediment samples from manholes #3 and #4. However, the wet sieving could not be carried out for sediment #4 due to the viscosity of the sample that was possibly caused by the contamination with oil. Thus, sediment #3 was used in this experiment. After the preparation, the sample was still totally black with the bad smell of sulfide and organic solvent. The characteristics of the prepared sediment samples—Fsed and Csed—are shown in Table 3 and the fractionation of heavy metals is shown in Fig. 3. Total concentrations of heavy metals in each sample before and after aeration were not significantly different (p > 0.05). All the total concentrations of each heavy metal and their pseudo-total concentrations from BCR extraction in all samples were not significantly different (p > 0.05).

Fractionation of Cu, Pb, Zn, and Fe in sewer sediment before and after aeration
Sewer sediment
The ORP of the sieved sediment samples was less than − 100 mV which was in the range that indicates sulfate reduction. The sulfide was quite high (about 10,000 mg kg−1) compared to other observations (500–16,000: Larner et al. 2008; Peng et al. 2004; Yu et al. 2001). The measured OM was irregularly high (naturally OM is less than 5%: Hou et al. 2013; Peng et al. 2004; Yu et al. 2001), especially for Fsed. This high OM was anticipated to be the result of contaminated organic liquid and some plastic fragments in this sediment sample. The concentration of all studied heavy metals in Fsed was higher than in Csed. Fine-grained soil or sediment generally contains higher heavy metal concentrations due to its large surface area and large surface activity which enhance adsorption properties (Bradl 2004; Houhou et al. 2009). The stable form of Cu was small while most of the non-residual fraction was in F3, similar to other studies on anoxic sediment from rivers and lakes (Hou et al. 2013; Kelderman and Osman 2007; Larner et al. 2008; Peng et al. 2004; Sobczynski and Siepak 2001; Yu et al. 2001). The concentration of lead in Fsed was enormous. Most of the non-residual fraction of lead in Fsed was in F3 while for Csed it was distributed in F3 and F2. Various distribution patterns of the non-residual fraction of lead in anoxic sediments were disclosed in the literature (Hou et al. 2013; Kelderman and Osman 2007; Larner et al. 2008; Peng et al. 2004; Sobczynski and Siepak 2001; Yu et al. 2001), which related to various sediment parameters and heavy metal contents in soil. Most of the Zn was in non-residual fractions which were distributed in F3 and F2 for Fsed and extended to F1 for Csed. Similar to lead, various distribution patterns of Zn in sediment samples have been reported (Hou et al. 2013; Kelderman and Osman 2007; Larner et al. 2008; Peng et al. 2004; Sobczynski and Siepak 2001; Yu et al. 2001). Iron in this contaminated sewer sediment was mainly in F4 and F1, which differed from other studies (Sobczynski and Siepak 2001; Yu et al. 2001) in that the non-residual fraction of iron was mostly associated with sulfide (F3) and iron oxide (F2) and could be found as derived from the oxic surface of the sediment (Yu et al. 2001).
Sediment after aeration
After aeration, the OM and sulfide in the sediment were reduced largely. The COD of water appeared, indicating the presence of dissolved oil or organic compounds from the sediment or suspended microorganisms. The sulfate was emerged, indicating the oxidation of sulfide compounds in the soil. The release of heavy metals into water was very limited. The fractionation of Cu in soil was still in F3. Pb in F3 was decreased, especially for Fsed, while F1 was emerged. The F3 and F2 of Zn were decreased while the F1 was increased. The F4 of Fe was increased while the F1 was decreased in contrast with Pb and Zn.
Discussion
Considering the experimental results, the fractionation of Cu, Pb, Zn, and Fe in the same environment exhibited similar tendencies which could be divided into different issues for consideration.
The pH was not changed markedly
The anoxic simulation used highly biodegradable organic substrate and high sulfate resulting in small increase of pH (6.9–8.0). The pH was elevated a little due to the high concentration of organic substances and high Fe concentration which consumes protons during the ferric reduction process (Mitsch and Gosselink 2000). In contrast, the aeration of sewer sediment resulted in a small decrease in average pH but this was not significant (p < 0.05) for the Fsed system. There are many aerobic reactions that conduce to opposite consequences; the oxidation of sulfide produces protons while the oxidation of Fe2+ consumes protons (Mitsch and Gosselink 2000). The changing of pH is very complex and is related to the various chemical reactions. However, the natural soil materials have great buffering capacity and thus the generally occurring biological reactions would not alter the pH markedly (Mitsch and Gosselink 2000).
Residual and non-residual fraction of metals
Based on these study results, the high or greater proportion of F4 tends to be related to oxidative environments. The aeration of sewer sediment resulting in the increase of F4 of almost all samples (p < 0.05, except Cu in Fsed), especially for Fe, conformed to the study of Larner et al. (2008). In contrast, the low or reduced residual fraction seems to be related to reductive environments. The decrease of F4 was found in soil samples from the anoxic sewer simulation which conforms to the study of El Samrani et al. (2004) and Houhou et al. (2009) who found that the surface of metal fragments was sulfurized when submerged in anoxic sediment.
Form of Fe
At basis, the major non-residual fraction of soil Fe in the oxidative environment was ferric oxide (Fe2O3) which was present in F2 (amorphous ferric oxide) and F4 (crystallized ferric oxide), while for the reductive environment it was ferrous sulfide (FeS) which was present in F3 of our applied sequential extraction.
The aeration of sediment represented a change from a reductive environment to an oxidative one. However, the result was not as expected whereby F2 of Fe was not extended. It may be described that when the sediment was altered to an oxidative condition, Fe2+ was oxidized to Fe3+, then Fe3+ would be adsorbed or precipitated as Fe(OH)3(s). However, the Fe2O3 (F2) would be formed by the dehydration of Fe(OH)3(s), thus could not occur in a submerged system. Nevertheless, the extended form of F4 was found and it was still curious. Similar findings arose in the study of Larner et al. (2008) in which the sediment was exposed to the air.
Under the reductive environment of the anoxic sewer sediment simulation, the ORP of system was lower than − 100 mV indicating that the ferric reduction and sulfate reduction took place. The resulting Fe2+ may dissolve into flowing aqueous solution (Hartley and Dickinson 2010) or would be redistributed among chemicals and materials in the soil. The developed Fe2+ generally binds with sulfide and precipitates as FeS that is a general form of metal sulfide in anoxic sediments (Guo et al. 1997; Sobczynski and Siepak 2001; Yu et al. 2001). However, FeS, which is represented by F3 of Fe, was not formed in our experiment—not as expected—but presented in weaker bonds as F1 and F2 indicating that Fe2+ is rather adsorbed to soil materials (this will be discussed later).
Distribution of Cu, Pb, Zn, and Fe in an anoxic environment
There were similar distribution patterns in the heavy metal fractionation of sewer sediment and soil simulating anoxic conditions: (1) most of the non-residual fractions of Cu were F3, (2) Pb was distributed primarily in F3 and F2, (3) Zn was distributed in F1 to greater extent than Pb, and (4) Fe was absent or very slightly associated to F3 while present in F1.
The development and breakdown of soil sulfide during the experiment and the transformation of its heavy metal fractionation indicate its important role in this study. Generally, the metal sulfides in soil are iron-sulfide compounds. It is the primary natural heavy metal in reductive soil (De Jonge et al. 2011). The Fe concentration in soil was generally greater than 10,000 mg kg−1, while the others were generally less than 1000 mg kg−1; for example, the background metal concentrations in the study area were 17,000 mg kg−1 for Fe, 370 mg kg−1 for Mn, 230 mg kg−1 for Zn, 90 mg kg−1 for Cu, and 70 mg kg−1 for Pb (Damrongsiri et al. 2016). However, the iron-sulfide compound (represent by F3 of Fe) was mostly not observed in contaminated soil or sediment in this study. This unusual finding was probably related to the presence of other heavy metals rather than Fe (Cu, Pb, and Zn) that were present in much greater than normal quantities in the soil (Tables 2 and 3).
In general, as the reaction between sulfide and metal was very fast and very strong, if the sulfide/heavy metals mole ratio was greater than 1, the heavy metals were assumed to be bonded with sulfide and not available to organisms, or considered non-toxic to the environment (Yu et al. 2001; Zhang et al. 2014). The concentrations of heavy metals and sulfide in mole units of the study soil and sediment are shown in Table 4.
The values in Table 4 showed that the contaminated soil and sediment in this study had less sulfide concentration than heavy metals. Thus, the association of sulfide with these heavy metals should be related to the formation and precipitation competency of these solid metal sulfides. The solubility product constant (Ksp) is the equilibrium constant for a solid substance in an aqueous solution which represents the maximum extent to which a solid can be dissolved in solution. As the forms of metal sulfide related to this study were similar (FeS, CuS, PbS, and ZnS), the reaction quotient of metal sulfides could be expressed by similar equations: reaction quotienti = metali concentration × sulfide concentration, where “i” represents certain metal species. If its reaction quotient was greater than its Ksp, its precipitate would be formed. Thus, a metal sulfide which has a lower Ksp would more easily form a solid precipitate. The Ksp values for the studied metals from low to high were CuS = 10–35.96, PbS = 10–28.05, ZnS = 10–21.97, and FeS = 10–16.84 (Benjamin 2002). The Ksp of CuS was far lower than the other metal sulfides (almost 20 times lower than FeS). Thus, based on these Ksp, sulfides in the system would chemically form principally with Cu, then followed by Pb, Zn, and Fe.
However, for the soil samples, moles of sulfide were less than Cu, and thus it could not associate with all the non-residual Cu and certainly not with Pb and Zn which have a higher Ksp. Thus, the soil fractionation result would become irrational. As a matter of fact, Cooper and Morse (1998) found that FeS, PbS, and ZnS could be dissolved by 1 M of HCl but it was low for Cu-sulfide due to its very strong bond. Therefore, the measured sulfide in this study was not total sulfide in soil since it was highly contaminated by Cu, but presumed to be derived from PbS, ZnS, and a part of Cu-sulfide.
In addition to sulfide, the metals associated with organic matter were also extracted in F3. Organic matter in soil could complex and sorb heavy metals in soil via its negative functional group resulting in various immobilization mechanisms. The sequence of heavy metal capability to associate with organic matter could be estimated by the stability constant of metal-fulvic acid (FA): Cu-FA = 105.8, Pb-FA = 103.1, Zn-FA = 101.7 (Stevenson and Ardakani 2010). A higher value of stability constant indicated a greater capability to form metal-FA compounds that was in the same order with metal sulfides.
Iron oxide is a major sink of heavy metals among soil materials. The study of Gadde and Laitinen (1974) demonstrated that oxides of iron could sorb Pb to a greater extent than Zn which conforms to this study result that the F3 fraction of Pb was larger than that of Zn. However, differences in the fractionation of Pb and Zn were found in many studies (Hartley and Dickinson 2010; Kelderman and Osman 2007; Peng et al. 2004; Sobczynski and Siepak 2001) dependent on the concentration of Pb and Zn in soil. Carbonate is another compound affecting the form of metals in soil, especially for Pb which particularly bonds and precipitates with carbonate (Hou et al. 2013). However, the carbonate content was not quantified in this study.
The sequence of metal association described earlier was quite consistent with the metal fractionation in these samples. The non-residual fractions of metals were primarily bonded to sulfide. Most non-residual fractions of Cu were in F3 due to the very low Ksp of CuS, high stability constant of Cu-FA, and very high Cu concentration in these samples. The remaining sulfide and organic matter were then associated by Pb and Zn. The reason why there was no Fe in F3 was that there was not enough sulfide and organic matter to make any association with Fe due to it having much less bonding competence than Cu, Pb, and Zn; thus, it was redistributed to F1. The latter Pb and Zn were then fractionated to F2 that was associated with ferric oxide, and F1 which was carbonate bonded and involved general ion adsorption. The proportion of F2 to F1 of Pb was greater than Zn due to the stronger association of Pb (Gadde and Laitinen, 1974). This was one reason why F1 of Zn was usually found in greater proportions than other metals.
Distribution of Cu, Pb, Zn, and Fe in aerated sediment
The distribution patterns of heavy metal fractionation in soil and aerated sediment which was an oxidative condition were described earlier. Their F3 fractions in oxidative conditions were obviously less than that in reductive conditions. There were two interesting alterations—not as expected—during the aeration of sewer sediment: (1) the non-residual fraction of Cu was still mostly in F3 and (2) the F3 of Pb and Zn decreased and redistributed to F1.
The fraction of Cu before and after aeration was still the same and mostly in F3. The study of Zhou et al. (1999) found that Cu-sulfide was not oxidized after exposure to aerated water for 35 days. Thus, the stable form of Cu in this study was believed to be a Cu-sulfide compound.
The decreasing tendency of Pb and Zn in F3 after exposure to oxidizing conditions was found to conform to the study of Kelderman and Osman (2007) due to the oxidization of sulfide and degradation of organic matter. The Pb and Zn were released into water (Hartley & Dickinson, 2010) and redistributed to soil material. Normally, the heavy metal in an oxidative environment would be mainly distributed in F2 associating to ferric oxide. However, F2 of Pb and Zn in this study was decreased. This F2 decreasing trend was also observed by Kelderman and Osman (2007). Nevertheless, due to the ferric oxide probably not forming in the submerged condition as described earlier, the released Pb and Zn were then adsorbed to other soil materials having weaker bonds that redistributed to F1. Thus, the F1 of Pb and Zn of the sediment samples was expanded after aeration. However, the increase of residual Fe remains a question.
Conclusion
This study discloses the contamination problem in sewer sediment located in an e-waste dismantling area. The sediment was contaminated with high concentrations of Cu, Pb, Zn, and unknown organic oil. The Fe in soil was greatly elevated as well. Many e-waste fragments were also found mingled in the sediment samples.
This study revealed that when heavy metal contaminated soil was transferred to a reductive environment, the metal fractionation was redistributed based on the chemicals of the sediment that tend to associate into F3 > F2 > F1. The redistribution was also related to the capability of heavy metals to associate with each compound. Sulfide played an important role in this anoxic condition. The F4 of heavy metals tends to be decreased via sulfurization.
If these contaminated anoxic sediments were transported to a more oxidative environment, these heavy metals would be released due to the metal sulfide being oxidized—except Cu-sulfide—and redistributed again among those new environment media. The ferric oxide may not be developed under this submerged condition. The redistribution under an oxic environment became F1 > F2 > F3 indicating the greater availability of these forms.
This study indicated the effects of environmental condition on the fractionation of heavy metals in soil and sediment which could occur at any source of heavy metal contamination. The alteration of fractionation results in changes in its mobility and environmental availability and this should be considered for the management and control of heavy metal contamination.
References
Benjamin MM (2002) Water chemistry, 1st edn. New York, McGraw-Hill Science
Bradl HB (2004) Adsorption of heavy metal ions on soils and soils constituents. J Colloid Interf Sci 277:1–18
Cooper DC, Morse JW (1998) Extractability of metal sulfide minerals in acidic solutions: application to environmental studies of trace metal contamination within anoxic sediments. Environ Sci Technol 32:1076–1078
Damrongsiri S, Vassanadumrongdee S, Tanwattana P (2016) Heavy metal contamination characteristic of soil in WEEE (waste electrical and electronic equipment) dismantling community: a case study of Bangkok, Thailand. Environ Sci Pollut Res 23:17026–17034
De Jonge M, Eyckmans M, Blust R, Bervoets L (2011) Are accumulated sulfide-bound metals metabolically available in the benthic oligochaete Tubifex tubifex? Environ Sci Technol 45:3131–3137
Department of industrial works (2002) Water pollution treatment system, 2nd edn. Water technology and industrial pollution management bureau, Bangkok
Eaton AD, Clesceri LS, Rice EW, Greenberg AE (2005) Standard methods for the examination of water and wastewater, 21st edn. Port City Press, Maryland
El Samrani AG, Lartiges BS, Ghanbaja J, Yvon J, Kohler A (2004) Trace element carriers in combined sewer during dry and wet weather: an electron microscope investigation. Water Res 38:2063–2076
Filgueiras AV, Lavilla I, Bendicho C (2002) Chemical sequential extraction for metal partitioning in environmental solid samples. J Environ Monit 4:823–857
Fu J, Zhou Q, Liu J, Liu W, Wang T, Zhang Q, Jiang G (2008) High levels of heavy metals in rice (Oryzasativa L.) from a typical E-waste recycling area in southeast China and its potential risk to human health. Chemosphere 71(7):1269–1275
Gadde RR, Laitinen HA (1974) Studies of heavy metal adsorption by hydrous iron and manganese oxides. Anal Chem 46(13):2022–2026
Guo T, Delaune RD, Patrick Jr WH (1997) The influence of sediment redox chemistry active forms of arsenic, cadmium, and zinc in estuarine sediment. Environ Int 23(3):305–316
Hartley W, Dickinson NM (2010) Exposure of an anoxic and contaminated canal sediment: mobility of metal(loid)s. Environ Pollut 158:649–657
Hooda PS (2010) Trace elements in soils. Blackwell Publiching Ltd, United Kingdom
Hou D, He J, Lu C, Ren L, Fan Q, Wang J, Xie Z (2013) Distribution characteristics and potential ecological risk assessment of heavy metals (Cu,Pb,Zn,Cd) in water and sediments from Lake Dalinouer, China. Ecotoxicol Environ Saf 93:135–144
Houhou J, Lartiges BS, Montarges-Pelletier E, Sieliechi J, Ghanbaja J, Kohler A (2009) Sources, nature, and fate of heavy metal-bearing particles in the sewer system. Sci Total Environ 407:6052–6062
Jun-hui Z, Hang M (2009) Eco-toxicity and metal contamination of paddy soil in an e-wastes recycling area. J Hazard Mater 165(1–3):744–750
Kelderman P, Osman AA (2007) Effect of redox potential on heavy metal binding forms in polluted canal sediments in Delf (The Netherlands). Water Res 41:4251–4261
Koopmans GF, Groenenberg JE (2011) Effects of soil oven-drying on concentrations and speciation of trace metals and dissolved organic matter in soil solution extracts of sandy soils. Geoderma 161:147–158
Larner BL, Palmer AS, Seen AJ, Townsend AT (2008) A comparison of an optimised sequential extraction pro metal partitioning. Anal Chim Acta 608:147–157
Leung A, Cai ZW, Wong MH (2006) Environmental contamination from electronic waste recycling at Guiyu, Southeast China. J Mater Cycles Waste Manage 8:21–33
Luo C, Liu C, Wang Y, Liu X, Li F, Zhang G, Li X (2011) Heavy metal contamination in soils and vegetables near an e-waste processing site, South China. J Hazard Mater 186:481–490
Mitsch WJ, Gosselink JG (2000) Wetlands, 3rd edn. New York, John Wiley & Sons
OECD (Organisation for Economic Co-operation and Development) (2002) Aerobic and anaerobic transformation in aquatic sediment systems. OECD Guideline Test Chem 308
Olafisoye OB, Adefioye T, Osibote OA (2013) Heavy metals contamination of water, soil, and plants around an electronic waste dumpsite. Pol J Environ Stud 22(5):1431–1439
Peng SH, Wang WX, Li X, Yen YF (2004) Metal partitioning in river sediments measured by sequential extraction and biomimetic approaches. Chemosphere 57:839–851
Pradhan JK, Kumar S (2014) Informal e-waste recycling: environmental risk assessment of heavy metal contamination in Mandoli industrial area, Delhi. India. Environ Sci Pollut Res 21(13):7913–7928
Qi Y, Huang B, Darilek JL (2014) Effect of drying on heavy metal fraction distribution in rice paddy soil. PLoS One 9(5):e97327
Sobczynski T, Siepak J (2001) Speciation of heavy metals in bottom sediments of lakes in the area of Wielkopolski National Park. Pol J Environ Stud 10(6):463–474
Stevenson FJ, Ardakani MS (2010) Organic matter reactions involving micronutrients in soils. In: Hooda PS (ed) Trace elements in soils. John Wiley & Sons Ltd, United Kingdom
Tang X, Shen C, Shi D, Cheema SA, Khan MI, Zhang C, Chen Y (2010) Heavy metal and persistent organic compound contamination in soil from Wenling: an emerging e-waste recycling city in Taizhou area, China. J Hazard Mater 173:653–660
Ure AM, Quevauviller Ph MH, Griepink B (1993) Speciation of heavy metals in soils and sediments. An account of the improvement and harmonization of extraction techniques undertaken under the auspices of the BCR of the commission of the european communities. Int J Environ Anal Chem 51:135–151
Vink JPM, Harmsen J, Rijnaarts H (2010) Delayed immobilization of heavy metals in soils and sediments under reducing and anaerobic conditions; consequences for flooding and storage. J Soils Sediments 10:1633–1645
Wentworth CK (1922) A scale of grade and class terms for clastic sediments. J Geol 30:377–392
Wilcke W, Muller S, Kanchanakool N, Zech W (1998) Urban soil contamination in Bangkok: heavy metal and aluminium partitioning in topsoils. Geoderma 86:211–228
Yekta SS, Gustavsson J, Svensson BH, Skyllberg U (2012) Sulfur K-edge XANES and acid volatile sulfide analyses of changes in chemical speciation of S and Fe during sequential extraction of trace metals in anoxic sludge from biogas reactors. Talanta 89:470–477
Yu KC, Tsai LJ, Chen SH, Ho ST (2001) Chemical binding of heavy metals in anoxic river sediments. Water Res 35(17):4086–4094
Zhang Q, Ye J, Chen J, Xu H, Wang C, Zhao M (2014) Risk assessment of polychlorinated biphenyls and heavy metals in soils of an abandoned e-waste site in China. Environ Pollut 185:258–265
Zhou W, Hesterberg D, Hansen PD, Hutchison KJ, Sayers DE (1999) Stability of copper sulfide in a contaminated soil. J Synchrotron Rad 6:630–632
Funding
The authors thank the Chulalongkorn Academic Advancement in its second century project and the S&T Postgraduate Education and Research Development Office (PERDO) for the financial support of the Research Program and thanks the Ratchadaphiseksomphot Endowment Fund, Chulalongkorn University for the Research Unit.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Philippe Garrigues
Rights and permissions
About this article

Cite this article
Damrongsiri, S. Transformation of heavy metal fractionation under changing environments: a case study of a drainage system in an e-waste dismantling community. Environ Sci Pollut Res 25, 11800–11811 (2018). https://doi.org/10.1007/s11356-018-1495-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-018-1495-3