
Abstract
Background
Obstructive sleep apnea syndrome, which is a risk factor for resistant hypertension, is characterized by chronic intermittent hypoxia (CIH) and is associated with many cardiovascular diseases. CIH elicits systemic oxidative stress and sympathetic hyperactivity, which lead to hypertension. Rho kinases (ROCKs) are considered to be major effectors of the small GTPase RhoA and have been extensively studied in the cardiovascular field. Upregulation of the RhoA/ROCK signaling cascade is observed in various cardiovascular disorders, such as atherosclerosis, pulmonary hypertension, and stroke. However, the exact molecular function of RhoA/ROCK in CIH remains unclear and requires further study.
Objective
This study aimed to investigate the role of the NADPH oxidase 4 (Nox4)-induced ROS/RhoA/ROCK pathway in CIH-induced hypertension in rats.
Methods
Male Sprague–Dawley rats were exposed to CIH for 21 days (intermittent hypoxia of 21% O2 for 60 s and 5% O2 for 30 s, cyclically repeated for 8 h/day). We randomly assigned 56 male rats to groups of normoxia (RA) or vertically implemented CIH together with vehicle (CIH-V), GKT137831 (CIH-G), N-acetyl cysteine (NAC) (CIH-N), or Y27632 (CIH-Y). The rats in the RA group were continuously exposed to room air, whereas the rats in the other groups were exposed to CIH. Systolic blood pressure (BP) was monitored at the beginning of each week. After the experiment, renal sympathetic nerve activity (RSNA) was recorded, and serum and renal tissues were subjected to molecular biological and biochemical analyses.
Results
Compared with the BP of RA rats, the BP of CIH-V rats started to increase 2 weeks after the beginning of the experiment, subsequently stabilizing at a high level at the end of the third week. CIH increased both RSNA and oxidative stress. This response was attenuated by treatment of the rats with GKT137831 or NAC. Inhibiting Nox4 activity or ROS production reduced RhoA/ROCK expression. Treatment with Y27632 reduced both BP and RSNA in rats exposed to CIH.
Conclusion
Hypertension can be induced by CIH in SD rats. The CIH-induced elevation of BP is at least partially mediated via the Nox4-induced ROS/RhoA/ROCK pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Obstructive sleep apnea (OSA) is a respiratory disease associated with sleep that is characterized by repeated upper airway occlusion. OSA causes cardiovascular diseases such as hypertension as a consequence of chronic intermittent hypoxia (CIH) [1]. Approximately, 50% of patients with OSA are hypertensive, and an estimated 30–40% of hypertensive patients have OSA [2].
It is generally agreed that several different mechanisms may be involved in CIH-induced hypertension. Several studies [3, 4] have concluded that sleep-related breathing disorders induce hypertension through oxidative stress, endothelial dysfunction, hyperactivity of the sympathetic nervous system, and effects on carotid body chemoreceptors and on the central nervous system, among other mechanisms. Recent studies [5, 6] have described the role of carotid bodies in CIH as well as the potent regulatory effect of carotid body-mediated chemoreflexes on blood pressure (BP). However, the potential mechanisms by which chemoreflexes lead to CIH are unclear. The kidneys are an important organ in the regulation of BP, as they receive nearly a quarter of the total resting cardiac output [7]. Studies [8, 9] have shown that the RhoA/Rho kinase (ROCK) pathway plays an important role in increased vascular resistance. ROCK is a downstream kinase of RhoA that plays an important role in local blood flow and BP regulation [10]. The Rho-kinase pathway is increased in hypertensive rats [11, 12] and hypertensive patients [13], and Rho-kinase inhibitor was reported to reduce arterial blood pressure in three experimental models of hypertension [14]. Enhanced production of reactive oxygen species (ROS) has been implicated in the development and progression of CIH and hypertension [15–19]. NADPH oxidase Nox1 and especially Nox4 (Renox) are major sources of ROS in the kidney and are critical mediators of redox signaling in vivo in the environment of CIH. Research [20–22] has shown that RhoA/ROCKs may be activated by ROS in vascular smooth muscle. Liming Jin et al. [23] reported that ROS increase membrane-bound Rho in aortic rings during maximum contraction. The aim of the present study was to investigate the role of the Nox4-derived ROS-mediated RhoA/ROCK pathway and the effect of antioxidants on the RhoA/ROCK pathway in CIH-induced hypertensive rats. We demonstrate that the elevation of BP in CIH may be at least partially mediated by Nox4-derived ROS/RhoA/ROCK pathway activity.
Materials and methods
Experimental protocols
Forty male Sprague–Dawley (SD) rats (body weight 190–210 g) were purchased from the Experimental Animal Center of Wuhan University (Wuhan, China). Eight rats (exposed to room air and injected i.p. with saline at 5 ml/kg·d; RA group) were randomly selected for experiments establishing the baseline levels of all biomarkers. The remaining 32 rats were randomly divided into four groups, denoted as follows: CIH-G [CIH + GKT137831 (Nox1 and Nox4 inhibitor) (40 mg/(kg·d), i.p., Selleck Chemicals, Shanghai, China)]; CIH-N [CIH + N-acetyl cysteine (NAC, ROS scavenger) (300 mg/(kg·d), i.p., Hangzhou Minsheng Pharmaceutical Co., Ltd., China)]; CIH-Y [CIH + Y27632 (RhoA inhibitor) (10 mg/(kg·d), i.p., Selleck Chemicals, Shanghai, China)]; and CIH-V [(CIH + saline or 0.1% DMSO in saline (5 ml/(kg·d), i.p.)]. This study was approved by the Ethics Committee of Wuhan University and was conducted in accordance with the Declaration of Helsinki and the Guide for the Care and Use of Laboratory Animals, as adopted and promulgated by the United States National Institutes of Health. The rats were housed in departmental animal chambers and maintained on a 12:12 h light-dark cycle under standard laboratory conditions (temperature, 25 ± 2 °C; humidity, 60 ± 5%). The rats were provided with standard rodent chow and allowed free access to water. Renal sympathetic nerve activity (RSNA) was measured at the end of the experiment, before all of the rats were sacrificed. Every effort was made to minimize the number of rats used and their suffering during the experiments.
The model of CIH was established according to previously published methods [24]. Sealed chambers were used to generate a hypoxic environment. Pure nitrogen and compressed air were distributed into each chamber through timed solenoid valves. Using 90 s cycles, pure nitrogen was infused into each chamber for the first 30 s until the minimum oxygen concentration reached 5%. Compressed air was infused for the remaining 60 s to allow the oxygen concentration in the chambers to gradually return to 20.9%. For the RA group, air was infused into the chamber. For all groups, exposure experiments were performed between 8 am and 4 pm.
Measurement of systolic BP (SBP)
SBP was measured every week using tail cuff plethysmography (RBP-1 non-invasive BP analyzer, Chengdu Technology, China). SBP was monitored in conscious animals, and three measurements were averaged.
RSNA recording
These experiments were performed under deep isoflurane anesthesia. RSNA was recorded using fine wire bipolar platinum electrodes placed on the left renal nerve under direct visualization using a surgical microscope. A left subcostal incision was made, and the kidney was approached from the retroperitoneal space under an anatomical microscope. A bundle of renal nerves was identified and gently freed from the surrounding tissue. One of the renal nerves was dissected and hooked by a pair of platinum electrodes. To insulate the electrodes and the nerve from the surrounding tissue and to prevent desiccation of the nerves, we covered the electrodes and the nerve preparation with liquid paraffin. Electrical changes in RSNA were amplified, filtered, and monitored on an oscilloscope with a low-frequency cutoff of 100 Hz and a high-frequency cutoff of 3000 Hz. RSNA was integrated at a time constant of 10 ms with a sampling frequency of 10 kHz. The rats were allowed to stabilize for 30–60 min after surgery before initiating the acute experimental protocols. The bipolar platinum electrode was connected to a biological polygraph (RM6240BD, Chengdu Technology, China) to record RSNA simultaneously. After stabilization of the signal, the rats were i.p. administered a daily dose of GKT137831 (40 mg/(kg·d)) to CIH-G, NAC (300 mg/(kg·d)) to CIH-N, Y27632 (10 mg/(kg·d)) to CIH-Y, and DMSO/saline (5 ml/(kg·d )), or saline (5 ml/(kg·d)) to CIH-V through single intraperitoneal injection, and changes in RSNA were observed. Integrated RSNA was simultaneously recorded. The postmortem background signal was determined, and the experimental data were corrected for the background signal. The difference in RSNA between before and after the intervention was expressed as the percent change from baseline.
Collection and storage of blood and tissues
Rats from each experimental group were sacrificed via blood collection through intracardiac puncture. Blood samples were subjected to centrifugation, and the resulting serum was stored at −80 °C. The kidneys were harvested and fixed in 4% phosphate-buffered formaldehyde for histopathological observation. The remaining portions of renal tissue were immediately frozen in liquid nitrogen and stored at −80 °C.
Measurement of oxidative stress biomarkers in the kidney
Blood samples from the rats were homogenized in tissue lysis buffer (Beyotime, China). After lysis for 15 min on ice, the homogenates were centrifuged at 3000 rpm for 15 min. The MDA contents in the supernatant were measured using commercially available kits (Jiancheng Bioengineering, Nanjing, China). Briefly, the MDA contents of homogenates were determined spectrophotometrically by measuring the levels of thiobarbituric acid-reactive substances. Three milliliters of 1% phosphoric acid and 1 ml of 0.6% thiobarbituric acid solution were added to 0.5 ml of plasma that had been pipetted into a tube. The mixture was then heated in boiling water for 45 min. After the mixture had cooled, the colored fraction was extracted into 4 ml of n-butanol. The absorbance at 532 nm was measured using a microplate spectrophotometer (Multiskan MK3, Thermo Scientific). The amount of lipid peroxide was calculated as the concentration of thiobarbituric acid-reactive substances of lipid peroxidation in nanomoles per milligram of protein (nmol/mg protein) according to a standard graph prepared from measurements of standard solutions (1,1,3,3-tetramethoxypropane). ROS levels were assessed in serum extracts using the DCFH-DA fluorescence assay (Molecular Probes). After preincubation with the above-mentioned chemicals, tissue was exposed to DCFH-DA (2 mM, in DMSO) for an additional 60 min to detect ROS production. DCF fluorescence of the samples was measured at room temperature with excitation and emission wavelengths of 488 and 525 nm, respectively. The activity of superoxide dismutase (SOD) in renal tissue was measured using a commercial assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions. Briefly, this assay kit employs a thiazole salt for the detection of superoxide anions to produce a colored product. The absorbance was measured at a wavelength of 450 nm. One unit (U) of SOD was defined as the total enzyme needed to produce 50% dismutation of superoxide radicals. The total tissue protein concentration was measured with a commercial kit (Beyotime Institute of Biotechnology, Shanghai, China), and SOD activity was expressed as U of SOD per milligram of protein (U/mg protein).
Serum norepinephrine (NE) assays
Blood samples were centrifuged at 3000 rpm for 10 min and stored at −20 °C until use. Serum NE levels were determined using a rat NE detection kit (Elabscience Biotechnology Co., Ltd., Wuhan, China) according to the manufacturer’s instructions.
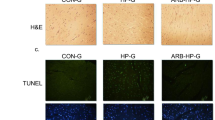
Terminal deoxynucleotidyl transferase-mediated dUTP digoxigenin nick-end labeling (TUNEL) assay
Apoptosis was evaluated via the in situ TUNEL assay according to the manufacturer’s instructions (Roche, USA). Serial sagittal kidney sections were digested with 20 μg/ml proteinase K (Dako, Glostrup, Denmark) for 15 min, immersed in 3% hydrogen peroxide for 5 min, and incubated with terminal deoxynucleotidyl transferase at 37 °C for 1 h. The sections were subsequently incubated with a peroxidase-labeled anti-digoxigenin antibody at 37 °C for 30 min, visualized with diaminobenzidine, and counterstained with hematoxylin. Apoptotic cells were counted manually under a light microscope at a magnification of 200×. The apoptotic index was calculated as the percentage of cells showing positive TUNEL staining. TUNEL(+) cells were counted from five random fields for each rat.
Western blotting
Renal tissues were used for western blot analysis. Protein concentrations were measured using the bicinchoninic acid (BCA) assay (Thermo Scientific, USA). Equal amounts of boiled protein in loading buffer were separated via SDS-PAGE using 10% Bis-Tris NuPAGE gels (Life Technologies, USA) and then electrophoretically transferred to polyvinylidene difluoride membranes (Millipore, USA). The membranes were subsequently incubated with primary antibodies (rabbit anti-ROCK, 1:30,000, Abcam, UK; rabbit anti-Nox4, 1:300, Elabscience, China; rabbit anti-Nox1, 1:200, BOSTER, Wuhan, China; and anti-GAPDH, Cell Signaling Technology, USA) overnight at 4 °C, followed by incubation with a secondary antibody (HRP-conjugated goat anti-rabbit IgG, 1:50,000, BOSTER, Wuhan, China) for 1 h at room temperature. The membranes were washed three additional times with TBST; positive antibody reactions were detected using an enhanced chemiluminescence system and Hyperfilm X-ray film. The resultant protein bands were quantified via densitometry (Quantity One 4.5.0 software; Bio-Rad Laboratories, Richmond, CA, USA).
RhoA activity assay
RhoA activation was assessed using a RhoA pull-down assay kit (NewEast Bioscience, Cat. No. 80601, USA) according to the manufacturer’s protocol. Briefly, renal tissue was lysed with lysis buffer from the RhoA pull-down assay kit, and the total protein concentration was measured using a protein assay kit (Beyotime, Cat. No. P0010S, China). Each sample was adjusted to 1 ml with 1× assay/lysis buffer from the RhoA pull-down assay kit. Then, 1 μl of a monoclonal anti-active RhoA antibody was added to the tube. Next, 20 μl of protein A/G agarose beads was quickly added to each tube, and the mixtures were incubated at 4 °C for 1 h with gentle agitation. The tubes were centrifuged for 1 min at 5000×g. The beads were washed three times with 0.5 ml of 1× assay/lysis buffer. The bead pellet was resuspended in 20 μl of 2× reducing SDS-PAGE sample buffer. Samples were boiled for 5 min, and the proteins in the supernatants were subjected to 10% SDS-PAGE and transferred to a PVDF membrane. The proteins were then analyzed via western blot using an anti-RhoA antibody from the RhoA pull-down assay kit at a 1:800 dilution.
Statistical analysis
All data are expressed as means ± S.E.M. Statistical analysis was performed using SPSS v17.0. Statistical comparisons between groups were conducted using one-way ANOVA, and the LSD test was employed for multiple comparisons. A value of P < 0.05 indicated a significant difference.
Results
CIH induced increased hypertension and hyperactivity of RSNA in rats
As shown in Fig. 1, compared with the RA group, of CIH-V group of SD rats showed a significant SBP elevation beginning in the second week, and SBP gradually increased throughout the experimental period in response to CIH. Moreover, CIH resulted in a significant increase in RSNA compared with room air exposure, which was (0.77 ± 0.09) μv/s and (0.26 ± 0.05) μv/s, respectively, P < 0.0001 (Fig. 2 a, e, f, g). RSNA was not altered by administration of DMSO or saline. In addition, CIH resulted in a marked increase in the NE level in serum (Fig. 3). TUNEL assays showed that CIH-V group exhibited a significant elevation of apoptosis relative to RA group (Fig. 4).

SBP in SD rats exposed to CIH (with and without drug intervention; n = 8) vs. room-air conditions (RA; n = 8).*p < 0.05, significant difference. n.s. not significant

Original recording and integrated RSNA (renal sympathetic nerve activity) showing in each group. a CIH-V. b CIH-G. c CIH-N. d CIH-Y. e RA. f △RSNA of baseline *P < 0.05 vs. CIH-V. g Baseline of RSNA *P < 0.001 vs. RA, # P < 0.0001 vs. RA. Data are presented as mean ± SEM, n = 8 rats per group; ANOVA

NE levels in each group. *p < 0.05, significant difference

a Representative microscopy images of tubular epithelial cell apoptosis (TUNEL assay, original magnification ×200). b Percentage (%) of TUNEL (+) cells in kidney tissues. *p < 0.05, significant difference
CIH induced increased expression of RhoA/ROCK
Western blotting was conducted to measure RhoA-GTP and ROCK protein expression in renal tissues from the CIH-V and RA groups. The expression of RhoA-GTP and ROCK proteins in renal tissues was increased by 1.76- and 1.02-fold, respectively, in rats exposed to CIH compared with room air-exposed rats (Fig. 7).
Y27632 suppressed CIH-induced hypertension and hyperactivity of RSNA
As shown in Fig. 1, treatment with Y27632 was associated with a significant decrease in SBP compared with vehicle treatment from the second week of CIH exposure until the end of the experiment, although there was also a significant difference between the CIH-Y and RA groups. In addition, RSNA was inhibited by Y27632 treatment, in spite of exposure to CIH (Fig. 2 d, f, g). Y27632 supplementation significantly reduced the CIH-induced increase in NE levels in serum and apoptosis in renal tissue compared with vehicle treatment (Fig. 3 and 4). Y-27632 partially inhibited the increases in SBP and NE levels induced by CIH. Based on these results, we concluded that the RhoA/ROCK pathway was involved in the mechanism of CIH-induced hypertension.
CIH increased oxidative stress and Nox4 expression in the kidney
Nox4 expression was increased by 1.85-fold in the CIH-V group relative to the RA group (Fig. 5). However, there was no significant difference in the expression of Nox1 between the CIH-V and RA groups (P > 0.05). The SOD level in the CIH-V group was decreased by 0.45-fold relative to that in the RA group. The MDA and ROS levels were increased by 0.82- and 1.71-fold, respectively, in the CIH-V group relative to the RA group. Treatment with GKT137831 or NAC resulted in reductions in MDA and ROS formation as well as recovery of SOD activity in the CIH-V group (Fig. 6).

a, c Expression of NOX1 and NOX4 in both CIH-V and RA groups. b, d NOX1 and NOX4 changes in renal tissue of rats in both CIH-V and RA groups. *p < 0.05 versus RA. n.s. not significant

SOD activity (a), MDA content (b), and ROS level (c). Results are expressed as mean ± SEM, n = 8 rats per group. *p < 0.05, significant difference
GKT137831 and NAC inhibited the elevation of SBP and enhancement of RSNA in rats exposed to CIH
Figure 1 showed that SBP was significantly lower in the CIH-G and CIH-N groups than in the CIH-V group. Acute intervention with an antioxidant (GKT137831) or an active oxygen scavenger (NAC) significantly decreased RSNA during CIH compared with vehicle treatment (Fig. 2 b, c, f, g). In addition, the NE levels of both the CIH-G and CIH-N groups were lower than those of the CIH-V group. Administration of GKT137831 and NAC dramatically attenuated CIH induced apoptosis (Fig. 4). This evidence demonstrates that GKT137831 and NAC can partially suppress the increases in SBP and RSNA induced by CIH.
GKT137831 and NAC inhibited the CIH-induced increases in RhoA/ROCK expression and oxidative stress
The RhoA-GTP and ROCK expression levels in the CIH-G group were decreased by 44 and 35%, respectively, and those in the CIH-N group were decreased by 45 and 37%, respectively, relative to those in the CIH-V group. Neither DMSO nor saline altered RhoA-GTP or ROCK expression (P > 0.05, Fig. 7). Although increased oxidative stress was observed in rats exposed to CIH, as demonstrated by elevated renal tissue MDA and ROS levels and decreased SOD levels in the CIH-V group, oxidative stress was significantly reduced by GKT137831 or NAC treatment (P < 0.05, Fig. 6). Based on the results, it revealed that GKT137831 and NAC can partially suppress the increases in SBP and RSNA induced by CIH through inhibition of RhoA/ROCK pathway.

a Expression of RhoA-GTP, total RhoA, and b ROCK in the kidney of different groups. RhoA-GTP/total RhoA (c) and ROCK (d) results are expressed as mean ± SEM. n = 8 rats per group. *p < 0.05, significant difference
Discussion
OSA syndrome is usually associated with daytime sleepiness, fatigue, and deficits in attention and executive function [25, 26]. In fact, OSA has been considered to represent an independent risk factor for systemic hypertension and resistant hypertension [27]. The sleeper is spared asphyxia by brief arousal, but the adverse consequences of this event include sleep fragmentation, further adrenergic activation coupled with vagal inhibition, surges in BP and heart rate, and a burst of oxidative stress [28]. Although various mechanisms have been proposed to underlie the development of hypertension in OSA, the detailed mechanism linking these two conditions is still unclear. Hypoxia-reoxygenation is used as a representative model of OSA [29]. The rat CIH model, in which hypoxia-reoxygenation episodes are simulated and several cardiovascular pathologic features of OSA are reproduced, is the gold-standard model for studying the mechanisms involved in OSA [16, 26, 30, 31]. The present study used an intermittent hypoxia model under normal pressure conditions of hypoxia (30 s) and reoxygenation (60 s) in SD rats, and these conditions were equivalent to severe sleep apnea [32].
Silva et al. [33] have suggested that sympathetic hyperactivity is associated with OSA-related hypertension. Renal sympathetic innervation may contribute to BP fluctuations. Denton et al. [34] showed in animal experiments that RSNA and renal postglomerular vascular resistance were increased in response to hypoxia. Linz et al. [35, 36] have demonstrated that repetitive obstructive respiratory events in pigs result in a postapneic BP increase. These changes are mainly sympathetically driven, as they are attenuated by renal sympathetic denervation. Bao et al. [37] reported that renal artery denervation eliminated the chronic diurnal changes in mean BP and plasma and renal tissue catecholamine levels in response to episodic hypoxia. Coincidentally, Fletcher et al. [38] demonstrated that animals that underwent renal artery denervation showed unchanged or reduced BP in response to episodic hypoxia, but that sham-operated animals showed a progressive, sustained increase in resting BP in response to compressed air. The nature of sympathetic hyperactivity in CIH-exposed animals was further elucidated by Huang et al. [39]. In the present study, we found that BP and RSNA were significantly higher after 21 days of CIH than before CIH. In addition, the NE contents in serum were higher in the CIH-V than in the RA group. Our findings are consistent with those of previous studies. This evidence indicates that CIH causes hypertension and hyperactivity of renal sympathetic nerves.
RhoA is a member of the Rho subfamily within the RAS superfamily of monomeric GTPases. RhoA, which exhibits 30% amino acid homology with Ras, has been identified as a key signaling molecule and plays an important role in various physiological processes. Rho-family proteins can possess lipid modifications that target them to cell membranes and can cycle between GTP- and GDP-bound states [40]. RhoA affects cell migration and cytokinesis through its downstream effector ROCK. The RhoA/ROCK pathway plays an important role in mediating cell functions and is related to the regulation of vascular tone and oxidative stress [9]. Activation of RhoA, which involves its migration to the plasma membrane, increases ROCK activity. The RhoA/ROCK pathway can change the sensitivity of Ca2+ to the systolic system to influence BP [41]. Specifically, cytosolic Ca2+ not only exerts direct control over myosin light chain phosphorylation but also influences vascular smooth muscle contraction by enhancing the sensitivity of the contractile apparatus, a process referred to as “Ca2+ sensitization”, which appears to be mediated by the small monomeric G protein RhoA and its target ROCK [42]. Increased RhoA expression and/or activity could augment smooth muscle tone and thus contribute to the pathogenesis of highly prevalent disorders such as hypertension [14]. Firth et al. [43] and Lee et al. [44] have detected increased expression of RhoA/ROCK in spontaneously hypertensive rats. Uehata et al. [14] demonstrated that Y-27632 dramatically reduces BP in rat models of hypertension. Furthermore, de Frutos et al. revealed that activation of ROCK in the vasculature could be induced by CIH [45]. Kyan J. Allahdadi et al. [46] demonstrated that the augmented constrictor response in CIH-induced hypertension is dependent on ROCK. Furthermore, de Frutos et al. revealed that activation of ROCK could be induced by CIH in the vasculature [45]. Moreover, Ito et al. found that activation of the Rho/Rho-kinase pathway in the nucleus tractus solitarius (NTS) contributed to the maintenance of basal BP via the sympathetic nervous system in spontaneously hypertensive rats [47–49]. In the current study, we found that the rats in the CIH-Y group exhibited a non-significant tendency towards increased B compared with the CIH-V group. RhoA and ROCK expression was significantly higher in the CIH-V group than in the RA group. In addition, the RSNA of CIH-exposed rats was attenuated by Y27632 treatment. These findings suggest that the RhoA/ROCK pathway may be a pathological mechanism involved in CIH-induced hypertension. Several studies [50–54] have demonstrated that excess RhoA protein signaling is involved in the pathogenesis of ischemia-reperfusion-induced target organ damage and that the pattern of hypoxia-reoxygenation is similar to that observed for the pathogenesis of ischemia-reperfusion.
Noxs are membrane-associated enzymes that catalyze the single-electron reduction of molecular oxygen using either NADH or NADPH as an electron donor. To date, seven members of the Nox family have been identified—Nox1–5 and the dual oxidases Duox1 and Duox2 [55, 56]. Nox4 is a 578-amino-acid protein that exhibits 39% sequence identity to Nox2, with specific conservation of the six membrane-spanning regions and binding sites for NADPH, flavin adenine dinucleotide (FAD), and heme [57–60]. Renal cortical Nox4, which is required for high ROS production and subsequent cell injury in mesangial cells, podocytes, and tubular epithelial cells, is the most important enzyme in CIH [61]. Cuevas et al. [62] investigated renal Nox4 expression and activity as well as ROS production during the development of hypertension in mice. Jha et al. [63] demonstrated that deletion of Nox4, but not Nox1, reduced renal ROS production. The deleterious effects of ROS on the cardiovascular system are primarily exerted via modulation of specific signaling pathways [16]. Chi et al. and Knock et al. [64, 65] found that ROS caused RhoA activation in vascular smooth muscle cells and endothelial cells through an unknown mechanism. Jackson et al. [66] demonstrated that intrarenal tempol (superoxide dismutase mimetic) or Y27632 (Rho kinase inhibitor) administration abolished the interaction between UK14,304 (alpha(2)-adrenoceptor agonist) and angiotensin II both in vivo and in vitro; the investigators concluded that Nox/superoxide and RhoA/ROCK are involved in the interaction between alpha(2)-adrenoceptors and angiotensin II in renal vascular resistance. In the present study, RhoA and ROCK expression in renal tissue was significantly decreased in both the CIH-G and CIH-N groups compared with the CIH-V group. Although GKT137831 is a dual Nox1 and Nox4 inhibitor, there is some indication that its actions in our experiments are due to its targeting of Nox4 in the kidney [67]. Although we cannot exclude the possibility that other enzymes are also involved in CIH, Nox4 may be particularly important in the kidney, where its expression was increased by CIH and decreased by treatment with specific inhibitors of Nox1/4, especially in association with renal oxidative stress and damage.
Based on these findings, we propose the following mechanisms of CIH-induced hypertension: (1) CIH induced hypertension and overexpression of RhoA and ROCK in rats and inhibition of the RhoA/ROCK pathway resulted in a decrease in hypertension; thus, we infer that the RhoA/ROCK pathway plays an important role in CIH-induced hypertension. (2) Administration of GKT137831 inhibited CIH-induced overexpression of RhoA and ROCK and reduction in BP. Based on this evidence, we conclude that GKT137831 reduces SBP by inhibiting the RhoA/ROCK pathway; that is, hypertension induced by CIH was at least partially mediated through the Nox4-derived ROS/RhoA/ROCK pathway.
Conclusion
In conclusion, we demonstrated a potential role of NOX4 in the pathogenesis of CIH-induced hypertension. Our results indicate that the elevation of BP induced by CIH is at least partially mediated via the Nox4-derived ROS/RhoA/ROCK pathway. Inhibition of this pathway may have clinical significance to the treatment of OSA patients.
References
Mooe T, Franklin KA, Wiklund U, Rabben T, Holmstrom K (2000) Sleep-disordered breathing and myocardial ischemia in patients with coronary artery disease. Chest 117:1597–1602
Calhoun DA (2010) Obstructive sleep apnea and hypertension. Curr Hypertens Rep 12:189–195
Prabhakar NR, Kumar GK (2010) Mechanisms of sympathetic activation and blood pressure elevation by intermittent hypoxia. Respir Physiol Neurobiol 174:156–161
Xing T, Pilowsky PM, Fong AY (2014) Mechanism of sympathetic activation and blood pressure elevation in humans and animals following acute intermittent hypoxia. Prog Brain Res 209:131–146
Peng YJ, Yuan G, Khan S, Nanduri J, Makarenko VV, Reddy VD et al (2014) Regulation of hypoxia-inducible factor-alpha isoforms and redox state by carotid body neural activity in rats. J Physiol 592:3841–3858
Prabhakar NR, Peng YJ, Kumar GK, Nanduri J (2015) Peripheral chemoreception and arterial pressure responses to intermittent hypoxia. Compr Physiol 5:561–577
Eckardt KU, Bernhardt WM, Weidemann A, Warnecke C, Rosenberger C, Wiesener MS, et al. (2005) Role of hypoxia in the pathogenesis of renal disease. Kidney Int SupplS46-S51
Loirand G, Pacaud P (2010) The role of Rho protein signaling in hypertension. Nat Rev Cardiol 7:637–647
Nguyen DCA, Touyz RM (2011) Cell signaling of angiotensin II on vascular tone: novel mechanisms. Curr Hypertens Rep 13:122–128
Nunes KP, Rigsby CS, Webb RC (2010) RhoA/Rho-kinase and vascular diseases: what is the link? Cell Mol Life Sci 67:3823–3836
Mukai Y, Shimokawa H, Matoba T, Kandabashi T, Satoh S, Hiroki J et al (2001) Involvement of Rho-kinase in hypertensive vascular disease: a novel therapeutic target in hypertension. FASEB J 15:1062–1064
Moriki N, Ito M, Seko T, Kureishi Y, Okamoto R, Nakakuki T et al (2004) RhoA activation in vascular smooth muscle cells from stroke-prone spontaneously hypertensive rats. Hypertens Res 27:263–270
Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A (2001) Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension 38:1307–1310
Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T et al (1997) Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389:990–994
Dikalov SI, Ungvari Z (2013) Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol 305:H1417–H1427
Dumitrascu R, Heitmann J, Seeger W, Weissmann N, Schulz R (2013) Obstructive sleep apnea, oxidative stress and cardiovascular disease: lessons from animal studies. Oxidative Med Cell Longev 2013:234631
Harrison DG, Gongora MC (2009) Oxidative stress and hypertension. Med Clin North Am 93:621–635
O’Halloran KD (2015) Chronic intermittent hypoxia creates the perfect storm with calamitous consequences for respiratory control. Respir Physiol Neurobiol
Rubattu S, Pagliaro B, Pierelli G, Santolamazza C, Castro SD, Mennuni S et al (2015) Pathogenesis of target organ damage in hypertension: role of mitochondrial oxidative stress. Int J Mol Sci 16:823–839
Denniss SG, Jeffery AJ, Rush JW (2010) RhoA-Rho kinase signaling mediates endothelium- and endoperoxide-dependent contractile activities characteristic of hypertensive vascular dysfunction. Am J Physiol Heart Circ Physiol 298:H1391–H1405
Schluter T, Steinbach AC, Steffen A, Rettig R, Grisk O (2008) Apocynin-induced vasodilation involves Rho kinase inhibition but not NADPH oxidase inhibition. Cardiovasc Res 80:271–279
Sun Q, Yue P, Ying Z, Cardounel AJ, Brook RD, Devlin R et al (2008) Air pollution exposure potentiates hypertension through reactive oxygen species-mediated activation of Rho/ROCK. Arterioscler Thromb Vasc Biol 28:1760–1766
Jin L, Ying Z, Webb RC (2004) Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am J Physiol Heart Circ Physiol 287:H1495–H1500
Feng SZ, Tian JL, Zhang Q, Wang H, Sun N, Zhang Y et al (2011) An experimental research on chronic intermittent hypoxia leading to liver injury. Sleep Breath 15:493–502
Idiaquez J, Santos I, Santin J, Del RR, Iturriaga R (2014) Neurobehavioral and autonomic alterations in adults with obstructive sleep apnea. Sleep Med 15:1319–1323
Iturriaga R, Andrade DC, Del RR (2014) Enhanced carotid body chemosensory activity and the cardiovascular alterations induced by intermittent hypoxia. Front Physiol 5:468
Marcus JA, Pothineni A, Marcus CZ, Bisognano JD (2014) The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep 16:411
Floras JS (2015) Hypertension and sleep apnea. Can J Cardiol 31:889–897
Levy P, Tamisier R, Arnaud C, Monneret D, Baguet JP, Stanke-Labesque F et al (2012) Sleep deprivation, sleep apnea and cardiovascular diseases. Front Biosci (Elite Ed) 4:2007–2021
Del RR, Moya EA, Parga MJ, Madrid C, Iturriaga R (2012) Carotid body inflammation and cardiorespiratory alterations in intermittent hypoxia. Eur Respir J 39:1492–1500
Peng YJ, Raghuraman G, Khan SA, Kumar GK (1985) Prabhakar NR (2011) angiotensin II evokes sensory long-term facilitation of the carotid body via NADPH oxidase. J Appl Physiol 111:964–970
Parati G, Lombardi C, Hedner J, Bonsignore MR, Grote L, Tkacova R et al (2013) Recommendations for the management of patients with obstructive sleep apnoea and hypertension. Eur Respir J 41:523–538
Silva AQ, Schreihofer AM (2011) Altered sympathetic reflexes and vascular reactivity in rats after exposure to chronic intermittent hypoxia. J Physiol 589:1463–1476
Denton KM, Shweta A, Anderson WP (2002) Preglomerular and postglomerular resistance responses to different levels of sympathetic activation by hypoxia. J Am Soc Nephrol 13:27–34
Linz D, Mahfoud F, Linz B, Hohl M, Schirmer SH, Wirth KJ et al (2014) Effect of obstructive respiratory events on blood pressure and renal perfusion in a pig model for sleep apnea. Am J Hypertens 27:1293–1300
Linz D, Mahfoud F, Schotten U, Ukena C, Neuberger HR, Wirth K et al (2012) Renal sympathetic denervation suppresses postapneic blood pressure rises and atrial fibrillation in a model for sleep apnea. Hypertension 60:172–178
Bao G, Metreveli N, Li R, Taylor A (1985) Fletcher EC (1997) blood pressure response to chronic episodic hypoxia: role of the sympathetic nervous system. J Appl Physiol 83:95–101
Fletcher EC, Bao G, Li R (1999) Renin activity and blood pressure in response to chronic episodic hypoxia. Hypertension 34:309–314
Huang J, Lusina S, Xie T, Ji E, Xiang S, Liu Y et al (2009) Sympathetic response to chemostimulation in conscious rats exposed to chronic intermittent hypoxia. Respir Physiol Neurobiol 166:102–106
Somlyo AP, Somlyo AV (2004) Signal transduction through the RhoA/Rho-kinase pathway in smooth muscle. J Muscle Res Cell Motil 25:613–615
Berridge MJ (2008) Smooth muscle cell calcium activation mechanisms. J Physiol 586:5047–5061
Urena J, Lopez-Barneo J (2012) Metabotropic regulation of RhoA/Rho-associated kinase by L-type Ca2+ channels. Trends Cardiovasc Med 22:155–160
Firth AL, Choi IW, Park WS (2012) Animal models of pulmonary hypertension: Rho kinase inhibition. Prog Biophys Mol Biol 109:67–75
Lee TM, Chung TH, Lin SZ, Chang NC (2014) Endothelin receptor blockade ameliorates renal injury by inhibition of RhoA/Rho-kinase signalling in deoxycorticosterone acetate-salt hypertensive rats. J Hypertens 32:795–805
de Frutos S, Caldwell E, Nitta CH, Kanagy NL, Wang J, Wang W et al (2010) NFATc3 contributes to intermittent hypoxia-induced arterial remodeling in mice. Am J Physiol Heart Circ Physiol 299:H356–H363
Allahdadi KJ, Walker BR, Kanagy NL (2007) ROK contribution to endothelin-mediated contraction in aorta and mesenteric arteries following intermittent hypoxia/hypercapnia in rats. Am J Physiol Heart Circ Physiol 293:H2911–H2918
Ito K, Hirooka Y, Kimura Y, Shimokawa H, Takeshita A (2005) Effects of hydroxyfasudil administered to the nucleus tractus solitarii on blood pressure and heart rate in spontaneously hypertensive rats. Clin Exp Hypertens 27:269–277
Ito K, Hirooka Y, Kishi T, Kimura Y, Kaibuchi K, Shimokawa H et al (2004) Rho/Rho-kinase pathway in the brainstem contributes to hypertension caused by chronic nitric oxide synthase inhibition. Hypertension 43:156–162
Ito K, Hirooka Y, Sakai K, Kishi T, Kaibuchi K, Shimokawa H et al (2003) Rho/Rho-kinase pathway in brain stem contributes to blood pressure regulation via sympathetic nervous system: possible involvement in neural mechanisms of hypertension. Circ Res 92:1337–1343
Cui Q, Zhang Y, Chen H, Li J (2013) Rho kinase: a new target for treatment of cerebral ischemia/reperfusion injury. Neural Regen Res 8:1180–1189
Noma K, Kihara Y, Higashi Y (2012) Striking crosstalk of ROCK signaling with endothelial function. J Cardiol 60:1–6
Sari AN, Kacan M, Unsal D, Sahan FS, Kemal BC, Vezir O et al (2014) Contribution of RhoA/Rho-kinase/MEK1/ERK1/2/iNOS pathway to ischemia/reperfusion-induced oxidative/nitrosative stress and inflammation leading to distant and target organ injury in rats. Eur J Pharmacol 723:234–245
Satoh K, Fukumoto Y, Shimokawa H (2011) Rho-kinase: important new therapeutic target in cardiovascular diseases. Am J Physiol Heart Circ Physiol 301:H287–H296
Shi J, Wei L (2013) Rho kinases in cardiovascular physiology and pathophysiology: the effect of fasudil. J Cardiovasc Pharmacol 62:341–354
Lassegue B, San MA, Griendling KK (2012) Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110:1364–1390
Selemidis S, Sobey CG, Wingler K, Schmidt HH, Drummond GR (2008) NADPH oxidases in the vasculature: molecular features, roles in disease and pharmacological inhibition. Pharmacol Ther 120:254–291
Gorin Y, Block K (2013) Nox4 and diabetic nephropathy: with a friend like this, who needs enemies? Free Radic Biol Med 61:130–142
Gorin Y, Wauquier F (2015) Upstream regulators and downstream effectors of NADPH oxidases as novel therapeutic targets for diabetic kidney disease. Mol Cells 38:285–296
Lassegue B, Griendling KK (2010) NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30:653–661
Nistala R, Whaley-Connell A, Sowers JR (2008) Redox control of renal function and hypertension. Antioxid Redox Signal 10:2047–2089
Eid AA, Lee DY, Roman LJ, Khazim K, Gorin Y (2013) Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol Cell Biol 33:3439–3460
Cuevas S, Zhang Y, Yang Y, Escano C, Asico L, Jones JE et al (2012) Role of renal DJ-1 in the pathogenesis of hypertension associated with increased reactive oxygen species production. Hypertension 59:446–452
Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T et al (2014) Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 25:1237–1254
Chi AY, Waypa GB, Mungai PT, Schumacker PT (2010) Prolonged hypoxia increases ROS signaling and RhoA activation in pulmonary artery smooth muscle and endothelial cells. Antioxid Redox Signal 12:603–610
Knock GA, Snetkov VA, Shaifta Y, Connolly M, Drndarski S, Noah A et al (2009) Superoxide constricts rat pulmonary arteries via rho-kinase-mediated Ca(2+) sensitization. Free Radic Biol Med 46:633–642
Jackson EK, Gillespie DG, Zhu C, Ren J, Zacharia LC, Mi Z (2008) Alpha2-adrenoceptors enhance angiotensin II-induced renal vasoconstriction: role for NADPH oxidase and RhoA. Hypertension 51:719–726
Gorin Y, Cavaglieri RC, Khazim K, Lee DY, Bruno F, Thakur S et al (2015) Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am J Physiol Renal Physiol 308:F1276–F1287
Acknowledgements
We thank Dr. Jing Feng and Prof. Baoyuan Chen, Respiratory Department, Tianjin Medical University General Hospital, China, for their support with the intermittent hypoxia chamber and the gas control delivery system used in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The National Natural Science Foundation of China provided financial support in the form of national natural science funding (No.81070065, 81370181). The sponsor had no role in the design or conduct of this research.
Conflict of interest
The authors declare that they have no conflict of interest.
Animal experiments
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Author contributions
WL and JK: Directly participated in the execution of the study and molecular biology experiment
KH: Study planning, analysis of the study and writing of the manuscript
ST, XZ, LX, YL, and SY: CIH exposure of rats and gas control delivery
Rights and permissions
About this article

Cite this article
Lu, W., Kang, J., Hu, K. et al. The role of the Nox4-derived ROS-mediated RhoA/Rho kinase pathway in rat hypertension induced by chronic intermittent hypoxia. Sleep Breath 21, 667–677 (2017). https://doi.org/10.1007/s11325-016-1449-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11325-016-1449-2