
Abstract
Introduction
Globally, breast cancer (BC) is leading at the top of women's diseases and, as a multifactorial disease, there is the need for the development of new approaches to aid clinicians on monitoring BC treatments. In this sense, metabolomic studies have become an essential tool allowing the establishment of interdependency among metabolites in biological samples.
Objective
The combination of nuclear magnetic resonance (NMR) and gas chromatography–quadrupole mass spectrometry (GC–qMS) based metabolomic analyses of urine and breast tissue samples from BC patients and cancer-free individuals was used.
Methods
Multivariate statistical tools were used in order to obtain a panel of metabolites that could discriminate malignant from healthy status assisting in the diagnostic field. Urine samples (n = 30), cancer tissues (n = 30) were collected from BC patients, cancer-free tissues were resected outside the tumor margin from the same donors (n = 30) while cancer-free urine samples (n = 40) where obtained from healthy subjects and analysed by NMR and GC–qMS methodologies.
Results
The orthogonal partial least square discriminant analysis model showed a clear separation between BC patients and cancer-free subjects for both classes of samples. Specifically, for urine samples, the goodness of fit (R2Y) and predictive ability (Q2) was 0.946 and 0.910, respectively, whereas for tissue was 0.888 and 0.813, revealing a good predictable accuracy. The discrimination efficiency and accuracy of tissue and urine metabolites was ascertained by receiver operating characteristic curve analysis that allowed the identification of metabolites with high sensitivity and specificity. The metabolomic pathway analysis identified several dysregulated pathways in BC, including those related with lactate, valine, aspartate and glutamine metabolism. Additionally, correlations between urine and tissue metabolites were investigated and five metabolites (e.g. acetone, 3-hexanone, 4-heptanone, 2-methyl-5-(methylthio)-furan and acetate) were found to be significant using a dual platform approach.
Conclusion
Overall, this study suggests that an improved metabolic profile combining NMR and GC–qMS may be useful to achieve more insights regarding the mechanisms underlying cancer.
Graphic abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Up to date, breast cancer (BC) is leading at the top of women’s illnesses after coronary diseases, accounting with around 2.1 million cases diagnosed in 2018 and expected to increase 1.1 million by 2040, according to GLOBOCAN series of the International Agency for Research on Cancer (IARC) (Bray et al., 2018). Being a multifactorial disease, with highly variable clinical behavior and response to therapy, it can be curable when detected in early stages. Although the extensive investigation on new therapy targets and diagnosis, there is the need for the development of new approaches to aid clinicians to monitor BC treatments and follow-up together with the current diagnostic tools, namely mammography, ultrasound or tumor markers (Lavra et al., 2015). Nowadays, the available diagnostic tools have supported in BC detection leading to the improved of survival rates. In this regard, metabolomics (metabolomic profiling) studies have emerged as a powerful approach to study the metabolic changes in several diseases including cancer (Chen et al., 2015; Dona et al., 2016; Gu et al., 2011; Trifonova et al., 2013; Zhang et al., 2013). Moreover, metabolomics plays an important role in disease profiling being a promising approach for the pursuit of new biomarkers in biological matrices, such as cell extracts, tissues or biological fluids (Johnson et al., 2016). As a fast growing field that focus on the investigation of metabolites present in biological systems, it reflects the altered metabolism and the physiological status (Koek et al., 2011). Normally, these studies are combined with analytical techniques being the most popular nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS) that have gained attention in this field as strong tools for the identification of potential biomarkers in a variety of clinical fields (Bingol, 2018; Chen et al., 2015). Usually, MS includes a separation stage using liquid chromatography (LC) or gas chromatography (GC) and can discriminate between compounds based on mass-to-charge (m/z) ratio in charged particles. Regarding NMR, it is an appealing technique that allows the investigation of metabolism due to its advantages as non-destructive, non-invasive, highly reproducible, giving information about biological samples environment offering both qualitative and quantitative measure. When compared with NMR, MS exhibits a greater sensitivity, although sample preparation is laborious and dependent on metabolite chemical properties (Oakman et al., 2011). On the other hand, MS lacks accuracy and precision producing an enhanced resolution profile with several peaks. In addition, these platforms can be used together, often applied to complex samples and coupled with advanced chemometric tools integrating the datasets obtained by the analytical techniques. If used in combination, these techniques enable the identification of a more comprehensive panel of metabolites involved in metabolic alterations and help unraveling the possible correlations and the underlying mechanisms induced by a disease (Chen et al., 2015; Marshall & Powers, 2017; Robertson et al., 2011). A variety of studies have been conducted by NMR and direct analysis in real time (DART)-MS using this approach to find serum biomarkers for BC (Gu et al., 2011). Another study was developed by Chen et al. (2015) that used the dual platform of NMR and MS methods to establish the urinary metabolomic profile of bipolar disorder (BD) subjects with a diagnosis purpose. Marshall et al. (2015) combined direct infusion electrospray ionization mass spectrometry (DI-ESI–MS) and NMR to analyze the impact of neurotoxins involvement in dopaminergic cell death which is relevant to Parkinson’s disease. Wei et al. (2013) combined NMR, liquid chromatography–mass spectrometry (LC–MS) and statistical analysis to predict the response to chemotherapy in the neoadjuvant setting using serum samples from 28 patients with BC. In turn, Falegan et al. (2017) used urine and serum samples from renal cell carcinoma (RCC) patients with the aim of distinguishing between stages of the disease and also to make a distinction between benign renal tumors and RCC. In summary, multiplatform approaches are useful tools to achieve a comprehensive analysis of the broad variety of metabolic alterations in cancer progression and development.
The current work describes the combination of NMR and GC–qMS based metabolomic analyses of urine and breast tissue samples from BC patients and cancer-free individuals combined with multivariate statistical tools in order to obtain a panel of metabolites together with their metabolic profiles that can discriminate malignant from healthy status, thus assisting in the diagnostic field.
2 Methods
2.1 Reagents and materials
All solvents and chemicals used in the experimental work were analytical grade.
For the NMR analysis, 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt (TSP) and deuterium oxide (D2O) were supplied by Acros Organics (Geel, Belgium) while potassium dihydrogen phosphate (KH2PO4), sodium azide (NaN3), phosphate buffer solution (PBS), potassium deuteroxide solution (KOD) were purchased from Panreac (Barcelona, Spain) and Sigma Aldrich (St. Louis, MO), respectively. Methanol (MeOH) and chloroform (CHCl3) were purchased from Thermo Fisher Scientific (Waltham, Massachusetts).
For the GC–qMS analysis, sodium chloride (NaCl), hydrochloric acid (HCl) and 4-methyl-2-pentanol (internal standard, IS) were supplied by Panreac (Barcelona, Spain) and Sigma Aldrich (St. Louis, MO), respectively. The SPME holder for manual sampling together with 75 µm carboxen/polydimethylsiloxane (CAR/PDMS) fiber was purchased from Supelco (Bellefonte, PA).
2.2 Subjects and sample collection
2.2.1 Subjects
The healthy participants (for urine samples collection) for this study were recruited from Blood Transfusion Medicine Service at Funchal Hospital and had no history of breast malignancy, or metabolic diseases such as diabetes, or hypertension. In addition, they were non-smoking and were not using medical drugs. The research was approved by the Ethics Committee of Funchal Central Hospital Dr. Nélio Mendonça (Approval no. S.1708625/2017) and has been performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. All the participants were fully informed of the objectives of the study and signed the informed consent.
2.2.2 Urine samples
Urine samples (first morning urine) from BC patients (BC, n = 30, age range 44–85, average 67) were taken at the Haemato-Oncology Unit from Dr. Nélio Mendonça Hospital, while the urine collection from healthy individuals (CF, n = 40, age range 43–80, average 64) (Table 1) was provided by the Blood Transfusion Medicine Service from the same Hospital.
Participants were instructed to collect the first morning urine (after the rejection of the first urine stream) into a sterile cup. The collected urine from either patients or healthy volunteers (CF) were aliquoted into 4 mL glass vials and stored at − 80 °C for further analysis by GC–qMS and NMR. Prior to analysis, all urine samples were centrifuged at 4000 rpm (~ 2000×g) for 20 min at 4 °C, and the supernatant used for the investigation. All the analyses were performed in triplicate.
2.2.3 Tissue samples
Regarding the tissues, 30 samples from patients with breast cancer (BC, n = 30, age range 44–85, average 67), and 30 samples from cancer-free tissue (CF, n = 30, age range 44–85, average 67) without malignant infiltration were resected from the same donor. The resected samples were divided into the active carcinoma and cancer-free tissue outside the tumor margin and were immediately frozen in liquid nitrogen, in a total set of 60 samples. The tissues were stored at − 80 °C until extraction. Regarding tissue, the analysis of cancer and cancer-free tissue from the same subject can also aid to balance the effect of external interferents, such as diet or environmental exposure. These samples were obtained at the Pathologic Anatomy Unit of Hospital Dr. Nélio Mendonça (Funchal, Portugal) according to Table 1. The resected BC tissues were classified using the TNM (tumor, node, and metastasis) staging approach which included five cases of stage IA, ten cases of stage IIA, one case of stage IIIA B, seven cases of stage IIB, five cases of stage IIIB and two of stage IIIC. BC has four stages, I, II III and IV. Stage I and II have subcategories A and B, while stage III can have A, B or C. The letters describe the size of the tumor and if the tumor has spread to the axillary lymph nodes or the lymph nodes near the breastbone.
2.3 Extraction of metabolites
2.3.1 HS-SPME procedure for urine samples
The extraction procedure for urine samples was performed according to our previous study (Silva et al., 2019a, b, c). Prior to HS-SPME procedure, urines were thawed and then 4 mL of urine was placed into 8 mL vials together with 17% NaCl (w/v) and 100 µL of the, 4-methyl-2-pentanol (IS = 1.6 mg/L). The pH was adjusted to 2 with small amounts of HCl 5 M. Then, the vial was capped with a Teflon (PTFE) septum using a screw cap and the CAR/PDMS fiber was introduced and exposed into the headspace during 75 min at 50 °C at 800 rpm (0.5 mm × 0.1 mm bar) using a thermostatic magnetic stirrer plate mixer agitator. After this period, the fiber was removed from the vial and inserted into the GC injection port and the extracted metabolites were desorbed for 10 min at 250 °C. Each sample was analyzed in triplicate and blanks were performed before each analysis.
2.3.2 Urine samples by NMR
Urine samples were prepared according to our previous work (Silva et al., 2019a, b, c). Briefly, urine samples were thawed and centrifuged at 8000 rpm (~ 6000×g, for 5 min) to remove suspended cells and any proteins present in samples. Then, 540 µL of urine was mixed with 60 µL of a buffer solution (KH2PO4, 1.5 M in D2O) containing 0.1% of TSP-d4 (used as chemical shift reference) and 2 mM NaN3. The pH was adjusted to 7.00 ± 0.02 by adding small amounts of KOD.
2.3.3 HS-SPME for tissue samples
The extraction was performed based on Silva et al. (2019a, b, c) method. Tissue samples were thawed and then portions of 100 mg were weighted into 20 mL vials together with 17% NaCl (w/v), 1000 µL of ultrapure water and 100 µL of the 4-methyl-2-pentanol (Internal standard (IS) = 1.6 mg/L). The pH was adjusted to 2 with small amounts of HCl 5 M. Then, the vial was capped with a Teflon (PTFE) septum using a screw cap and the SPME fiber was introduced and exposed into the headspace during 75 min at 50 °C at 800 rpm (0.5 mm × 0.1 mm bar). After this period, the fiber was removed from the vial and inserted into the GC injection port and the analytes were desorbed for 10 min at 250 °C. Each sample was analyzed in triplicate and blanks were performed before each analysis. In addition, the GC peaks obtained from ultrapure water were removed from analysis. A SPME extraction using the same procedure was performed using ultrapure water and the data regarding the peaks was removed from tissue analysis.
2.3.4 Tissue samples by NMR
The intact frozen tissues were weighed, and portions of 100 mg were transferred into a glass vial. Then, 5 mL of PBS was added to remove any blood residues from the samples. After this, 5 mL of cold MeOH and CHCl3 were added prior to homogenization and vortex. The vials were placed at − 20 °C and the vials were vortexed three times every 10 min. After this procedure, the vials were centrifuged at 4000×g for 15 min at 4 °C. The upper phase (methanol) containing the polar metabolites was transferred into another vial and the MeOH was removed under a nitrogen stream before lyophilisation. At the time of the NMR analysis, 540 µL of D2O was added to the obtained extract and mixed with 60 µL of a buffer solution (KH2PO4, 1.5 M in D2O) containing 0.1% of TSP-d4 (used as chemical shift reference) and 2 mM NaN3. The pH was adjusted to 7.00 ± 0.02 by adding small amounts of KOD.
3 Gas chromatography quadrupole mass–spectrometry (GC–qMS) conditions
After the extraction procedure (for urine and tissue samples), the SPME fiber with the analytes was inserted into the injection port of an Agilent Technologies 6890 N Network gas chromatograph system (Palo Alto, CA), where the analytes were desorbed at 250 °C for 10 min. The GC was equipped with a 60 m × 0.25 mm I.D. × 0.25 µm film thickness, BP-20 (SGE, Dortmund, Germany) fused silica capillary column and interfaced with an Agilent 5975 quadrupole inert mass selective detector. The following oven temperature profile was set: (a) 5 min at 45 °C; (b) increase temperature until 150 °C, at a rate of 2 °C min−1 (hold for 10 min); (c) 150 °C for 10 min; (d) increase temperature until 220 °C, at a rate of 7 °C min−1; and (e) 220 °C for 10 min for a total GC run time of 87.5 min. The column flow was constant at 1.3 mL min−1 using Helium (He, N60, Air Liquide, Portugal) as carrier gas. The injection port was operated in the splitless mode and held at 250 °C. For the 5975 MS system, the operating temperatures of the transfer line, quadrupole and ionization source were 270, 150 and 230 °C, respectively, while electron impact mass spectra were recorded at 70 eV ionization voltage and the ionization current was 10 µA. Data acquisition was performed in the scan mode (30–200 m/z). The electron multiplier was set to the auto tune procedure. Metabolite’s identification was accomplished by manual interpretation through single ion monitorization (SIM) of spectra and matching against the Agilent MS ChemStation Software, equipped with a NIST05 mass spectral library with a similarity threshold higher than 80% and comparison with commercially available standards when available. The GC peak areas (relative peak areas) were obtained from the ratio between the area of each metabolite and the area of the IS. A series of C8–C20 n-alkanes were analyzed using the same extraction procedure to establish the Kóvat indices (KI), and to confirm the identity of the metabolites by comparison with the values reported in the literature for similar columns. In addition, the extraction of the standard solution (containing one standard of each chemical family) was performed each week and relative standard deviations (RSD) were lower than 5%. This approach was used to evaluate the reproducibility of the GC–MS analyses. The analyses were performed in triplicate and the results expressed by mean ± standard deviation.
3.1 NMR measurements
NMR spectral acquisition was performed using a Bruker Advance II Plus NMR spectrometer equipped with a 400 MHz magnet UltraShield™ 400 Plus at 300 K. All NMR spectra acquisition and pre-processing were performed under the control of a workstation with TopSpin 3.1 (Bruker BioSpin). Two different 1H-NMR spectra were acquired: a 1D 1H spectrum providing quantitative metabolite data for statistical analysis while 2D HSQC and 2D-Jres experiments supported in peak assignment and metabolite identification using the standard Bruker pulse programs. For each sample, a 1D nuclear overhauser enhancement spectroscopy (NOESY) pulse sequence (noesypr1d) was used, and solvent signal suppression was achieved by presaturation during relaxation and mixing time (SW 4807.692 Hz, TD 64 K data points, relaxation delay 5 s, 128 scans). The shimming was calibrated automatically. Also, all spectra were processed using a line broadening (1.0 Hz) and baseline automatically corrected. The NMR spectrum of each sample was calibrated with reference to the TSP signal at δ 0.00 ppm. Spectral regions within the range of 0.94 to 10 ppm were analysed after excluding the sub-region δ 4.55–6.05 to remove variability arising from water suppression and possible cross-relaxation effect on the urea signal (for the case of urine samples) via solvent exchanging protons. Each sample analysis was performed in triplicate and the relative standard deviation (RSD) was lower than 2%.
The analysis of NMR spectral data was performed using the Chenomx NMR Suite 8.2 (Chenomx Inc., Alberta, Canada) and relative concentrations were determined using the 400 MHz library from Chenomx NMR Suite 8.2, which compares the integral of a known reference signal (TSP) with signals derived from a library of compounds containing chemical shifts and peak multiplicities. In addition, the identification of metabolites was checked with the spectra library existing on Human Metabolome Database (Ellinger et al., 2013) and literature (Bouatra et al., 2013; Gramatyka et al., 2020; Jagannathan & Sharma, 2017; Li & Deng, 2017). Regarding to the metabolites that were not available in the library, their identification was accomplished by running a standard solution and the relative concentration was calculated manually.
4 Statistical analysis
Statistical analysis was performed using the web server Metaboanalyst 4.0 (Xia et al., 2016) using the mean values of the triplicate values obtained for each biological sample (urine and tissue) and analytical platform. First, data was transformed using cube root (e.g. cube root calculation of data values) and data autoscaling (e.g. mean-centered and divided by the standard deviation of each variable) procedure. In addition, the t test was used to compare the means of samples between the groups under study (t test, p < 0.05). Furthermore, the multivariate statistical analysis, namely the orthogonal projections to latent structures discriminant analysis (OPLS-DA) were applied on tissue and urine metabolomic dataset to provide insights into the groups under study for each analytical platform. The metabolites with variable importance in the projection (VIP) scores higher than 1.6 were considered significant and used for further analysis (Akarachantachote et al., 2014; Nguyen et al., 2019). The receiver operating characteristic curves (ROC) were also attained to verify which metabolites had the highest sensitivity/specificity for a potential BC diagnosis. The selected metabolites with significance level of 0.05, were used for the metabolite set enrichment analysis (MSEA) to identify significant patterns of metabolite concentration changes. MSEA uses a collection of predefined metabolite pathways and disease states obtained from the HMDB. Furthermore, to inspect the correlations between urine and tissue metabolites, samples from the same individuals were matched and the correlation matrices obtained for the results for each analytical platform (GC–qMS and NMR) separately. The matrices containing the mean values for each metabolite identified in samples and analyzed by each analytical platform were generated by calculating the Pearson´s correlation coefficient between each pair of variables from either NMR or GC–qMS. The results were generated and plotted using MATLAB R2018b Academic version (MathWorks, Natick, MA).
5 Results and discussion
5.1 Tissue and urine metabolomic pattern based on GC–qMS and 1H NMR spectroscopy
The analyses of urine and tissue samples from the same donors allowed the identification of 37 and 31 metabolites using NMR method, while for GC–qMS, 114 and 32 metabolites were identified, respectively (Supplementary Table S1 and S2). Prior to multivariate analysis, all datasets were scaled to unit variance so that each variable had the same weight and were used for further analysis (t test, p < 0.05). The combined datasets of tissue and urine samples composed of 30 metabolites (GC–qMS) towards 24 metabolites (NMR) in tissue samples and 52 metabolites (GC–qMS) towards 33 metabolites (NMR) in urine samples, were subjected to statistical analysis using the Metaboanalyst 4.0 (Xia et al., 2016) web server in order to obtain a preliminary information about data projection. OPLS-DA statistical analysis was generated to compare the metabolic profiles between BC patients and cancer-free individuals, maximizing the class discrimination. The quality of the model was evaluated by goodness of fit (R2Y) and predictive ability (Q2) values, which gave the variance explained and predicted ability for the model, respectively, thus confirming that the model was effective with a good predictable accuracy.
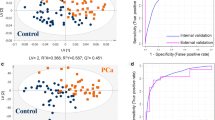
Figure 1a and b presents the results obtained for the multivariate analysis: OPLS-DA for tissue (R2Y = 0.888; Q2 = 0.813) and urine (R2Y = 0.946 and Q2 = 0.910) samples, respectively (Supplementary Fig. S1). As observed, the obtained OPLS-DA score plot showed a clear separation between BC patients and cancer-free subjects for both category of samples. In urine, α-hydroxyisobutyrate, glutamine, betaine and hypoxanthine, were the metabolites with highest impact in group discrimination, whereas for tissue were lactate, glutamate, valine and taurine. Additionally, the metabolites from each biological matrix with VIP values higher than 1.6 were used for the pathway analysis to verify which pathways were the relevant involved in the pathology under study.

Orthogonal partial least-squares discriminant analysis (OPLS-DA) score plots between BC patients and cancer-free (CF) individuals for tissue (a) (R2Y = 0.888; Q2 = 0.813) and urine (b) (R2Y = 0.946 and Q2 = 0.910) samples obtained by NMR and GC–qMS analysis
Figure 2a and b includes the top ten metabolites with the highest importance in the projection which comprised most of the metabolites arising from the tissue analysis by NMR when compared with the VIP values from urine analysis that derived from the GC–qMS methodology. Moreover, the ROC (Fig. 2c, d) curves for each type of biological sample (urine and tissue) were created using the metabolites with higher VIP values as described previously. Figure 2c and d demonstrate the values obtained using the ROC curves given by the area under the curve (AUC). Regarding the results obtained, we can observe that in the case of tissue (Fig. 2c) four metabolites, namely lactate, glutamate, o-phosphocholine and trimethylamine N-oxide (derived from NMR analysis) were enough to obtain an AUC of 0.864, whereas for urine an AUC of 0.931 was obtained using only three metabolites including glutamine, betaine and α-hydoxybutyrate (derived from NMR analysis) were sufficient to provide a good sensitivity and specificity to discriminate BC against CFs. These results are in accordance with the described in literature, namely Xia et al. (Xia et al., 2013) reported that an AUC between 0.9 and 1.0 is excellent and between 0.8 and 0.9 is good. Based on this classification, the results obtained were very good (AUC = 0.864 and 0.931, respectively).

Top ten significant features from tissue (a) and urine (b) samples based on variable importance in the projection (VIP) and receiver operating characteristic (ROC) curves for tissue (c) and urine (d) samples using the selected metabolites by VIP values
To go further, the metabolic pathway analysis was performed using a Metabolite Set Enrichment Analysis (MESA) in order to identify which pathways were affected in both cases.
This type of analysis is used as a mean to recognize biologically meaningful patterns that are enriched in metabolomic data (Marco-Ramell et al., 2018; Xia & Wishart, 2010). For the tissue analysis, the metabolic pathways mostly affected included those of lactate, valine, aspartate and glutamine whereas, for urine, only glutamine and hypoxanthine metabolisms presented variations (Fig. 3a, b).

Summary plots for Metabolite Set Enrichment Analysis (MESA) overview of tissue (a) and urine (b) samples and respective pathways associated. The color of the bar indicates the degree of positive association with the corresponding phenotype
Finally, in order to check the possible correlations between urine and tissue metabolites, samples from the same individuals were matched and the correlation matrices obtained for the results for each analytical platform (GC–qMS and NMR) are presented in Supplementary Fig. S2.
For this test, only the metabolites with correlation coefficient higher than 0.6 and p < 0.05 were used. Using this criterion, in this case, no correlations were observed for tissue vs urine in NMR or tissue NMR vs urine GC–qMS. By observing Supplementary Fig. S2, we can perceive that only five metabolites (acetone, 3-hexanone, 4-heptanone, 2-methyl-5-(methylthio)-furan and acetate) were found significant as indicated in Table 2, being that the correlation level increases with the intensity of colour.
Regarding the possible origin of these metabolites, 4-heptanone was already identified in urine samples from BC patients and cancer-free individuals being hypothesized that it arises from in vivo β-oxidation of 2-ethylhexanoic acid (EHA) from plasticizers, similar to formation of 3-heptanone from valproic acid (Silva et al., 2012; Statheropoulos et al., 2005). Acetate is a common metabolite found in most tissues and was also identified in biological specimens such as urine, saliva or faeces. The main pathways where acetate is involved includes pyruvate, aminoacid and aspartate metabolisms (Gray et al., 2014). Also, 2-methyl-5-(methylthio)-furan was identified in urine samples from cancer patients (Silva et al., 2012). However, to the best of our knowledge the possible origin of this metabolite is not yet reported in literature. Regarding acetone, their production is closely related to the higher oxidation rate of fatty acids derived from decarboxylation of acetoacetate and the dehydrogenation of isopropanol (Erhart et al., 2009). The main pathways associated and their origin arises from the synthesis and degradation of ketone bodies and propanoate metabolism through acetyl-CoA in the liver (Mediani et al., 2018). In addition, the results obtained with this preliminary research suggest the possibility to identify endogenous metabolites using a dual platform to discover potential BC biomarkers and provides a way to investigate the related metabolic pathways to improve the BC diagnostic tools.
6 Conclusions
In this work, two analytical platforms, namely NMR and GC–qMS were used to explore urine and tissue samples. In this sense, using GC–qMS methodology it was possible to identify 128 metabolites from urine and tissue samples, where 115 arose from urine and 32 from tissue samples. Also, it was observed that 17 metabolites were common to both matrices namely, toluene, hexanal, d-limonene, 2-pentylfuran, furfural, acetic acid, 2-ethyl-1-hexanol, benzaldehyde, 1-octanol, acetophenone, hexanoic acid, phenol, octanoic acid, 4-methyl-phenol, nonanoic acid, decanoic acid and p-tert-butyl-phenol. The metabolites with highest relative peak area identified in urine were 2-ethyl-1-hexanol and 4-methyl-phenol while in tissue were acetic acid and phenol. Concerning NMR analysis, a total of 49 metabolites were recognized from which 31 derived from tissue and 35 from urine, having 19 common metabolites including valine, 3-hydroxybutyrate, lactate, alanine, acetate, glutamine, acetone, succinate, dimethylamine, choline, betaine, trimethylamine N-oxide, taurine, 4-hydroxyphenylacetate, glycine, mannitol, guanidoacetate, creatine, and hypoxanthine. The metabolites with highest concentration included citrate, betaine and hippurate in urine while in tissue were lactate and taurine.
This research also allowed the combination of NMR and GC–qMS based metabolomic analyses of urine and breast tissue samples from BC patients and cancer-free individuals tandem with multivariate statistical tools in order to obtain a panel of metabolites that can discriminate malignant from healthy status thus assisting in the diagnostic field. Moreover, using a dual platform approach (NMR and GC–qMS), we could enlarge the panel of identified metabolites showing a promising BC diagnostic tool. Overall, this study suggests that an improved metabolic profile combining NMR and GC–qMS may be useful to achieve more insights regarding cancer mechanisms. Nevertheless, due to the challenge of identifying metabolites linked to BC, further studies are needed for a broader understanding of the mechanisms underlying BC.
References
Akarachantachote, N., Chadcham, S., Saithanu, K., Akarachantachote, N., Chadcham, S., & Saithanu, K. (2014). Cutoff threshold of variable importance in projection for variable selection. International Journal of Pure and Applied Mathematics, 94(3), 307–322.
Bingol, K. (2018). Recent advances in targeted and untargeted metabolomics by NMR and MS/NMR methods. High-Throughput, 7(2), 9.
Bouatra, S., Aziat, F., Mandal, R., Guo, A. C., Wilson, M. R., Knox, C., Bjorndahl, T. C., Krishnamurthy, R., Saleem, F., Liu, P., & Dame, Z. T. (2013). The human urine metabolome. PLoS ONE, 8(9), e73076.
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., & Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 68(6), 394–424.
Chen, J., Liu, Z., Fan, S., Yang, D., Zheng, P., Shao, W., Qi, Z. G., Xu, X. J., Li, Q., Mu, J., & Yang, Y. T. (2015). Combined application of NMR- and GC-MS-based metabonomics yields a superior urinary biomarker panel for bipolar disorder. Scientific Reports, 4(1), 5855.
Dona, A. C., Kyriakides, M., Scott, F., Shephard, E. A., Varshavi, D., Veselkov, K., & Everett, J. R. (2016). A guide to the identification of metabolites in NMR-based metabonomics/metabolomics experiments. Computational and Structural Biotechnology Journal, 14, 135–153.
Ellinger, J. J., Chylla, R. A., Ulrich, E. L., & Markley, J. L. (2013). Databases and software for NMR-based metabolomics. Current Metabolomics, 1(1), 28–40.
Erhart, S., Amann, A., Haberlandt, E., Edlinger, G., Schmid, A., Filipiak, W., Schwarz, K., Mochalski, P., Rostasy, K., Karall, D., & Scholl-Bürgi, S. (2009). 3-Heptanone as a potential new marker for valproic acid therapy. Journal of Breath Research, 3(1), 016004.
Falegan, O., Ball, M., Shaykhutdinov, R., Pieroraio, P., Farshidfar, F., Vogel, H., Allaf, M. E., & Hyndman, M. E. (2017). Urine and serum metabolomics analyses may distinguish between stages of renal cell carcinoma. Metabolites, 7(1), 6.
Gramatyka, M., Boguszewicz, L., Ciszek, M., Gabryś, D., Kulik, R., & Sokół, M. (2020). Metabolic changes in mice cardiac tissue after low-dose irradiation revealed by 1H NMR spectroscopy. Journal of Radiation Research, 61(1), 14–26.
Gray, L. R., Tompkins, S. C., & Taylor, E. B. (2014). Regulation of pyruvate metabolism and human disease. Cellular and Molecular Life Sciences, 71(14), 2577–2604.
Gu, H., Pan, Z., Xi, B., Asiago, V., Musselman, B., & Raftery, D. (2011). Principal component directed partial least squares analysis for combining nuclear magnetic resonance and mass spectrometry data in metabolomics: Application to the detection of breast cancer. Analytica Chimica Acta, 686(1), 57–63.
Jagannathan, N. R., & Sharma, U. (2017). Breast tissue metabolism by magnetic resonance spectroscopy. Metabolites, 7(2), 25.
Johnson, C. H., Ivanisevic, J., & Siuzdak, G. (2016). Metabolomics: Beyond biomarkers and towards mechanisms. Nature Publishing Group.
Koek, M. M., Jellema, R. H., van der Greef, J., Tas, A. C., & Hankemeier, T. (2011). Quantitative metabolomics based on gas chromatography mass spectrometry: Status and perspectives. Metabolomics: Official Journal of the Metabolomic Society, 7(3), 307–328.
Lavra, L., Catini, A., Ulivieri, A., Capuano, R., Baghernajad Salehi, L., Sciacchitano, S., Bartolazzi, A., Nardis, S., Paolesse, R., Martinelli, E., & Di Natale, C. (2015). Investigation of VOCs associated with different characteristics of breast cancer cells. Scientific Reports, 5, 13246.
Li, T., & Deng, P. (2017). Nuclear magnetic resonance technique in tumor metabolism. Genes and Diseases, 4(1), 28–36.
Marco-Ramell, A., Palau-Rodriguez, M., Alay, A., Tulipani, S., Urpi-Sarda, M., Sanchez-Pla, A., & Andres-Lacueva, C. (2018). Evaluation and comparison of bioinformatic tools for the enrichment analysis of metabolomics data. BMC Bioinformatics, 19(1), 1.
Marshall, D. D., Lei, S., Worley, B., Huang, Y., Garcia-Garcia, A., Franco, R., Dodds, E. D., & Powers, R. (2015). Combining DI-ESI–MS and NMR datasets for metabolic profiling. Metabolomics, 11(2), 391–402.
Marshall, D. D., & Powers, R. (2017). Beyond the paradigm: Combining mass spectrometry and nuclear magnetic resonance for metabolomics. Progress in Nuclear Magnetic Resonance Spectroscopy, 100, 1–16.
Mediani, A., Abas, F., Maulidiani, M., Sajak, A. A. B., Khatib, A., Tan, C. P., Ismail, I. S., Shaari, K., Ismail, A., & Lajis, N. H. (2018). Metabolomic analysis and biochemical changes in the urine and serum of streptozotocin-induced normal- and obese-diabetic rats. Journal of Physiology and Biochemistry, 74(3), 403–416.
Nguyen, T. M., Shafi, A., Nguyen, T., & Draghici, S. (2019). Identifying significantly impacted pathways: A comprehensive review and assessment. Genome Biology, 20(1), 203.
Oakman, C., Tenori, L., Biganzoli, L., Santarpia, L., Cappadona, S., Luchinat, C., & Di Leo, A. (2011). Uncovering the metabolomic fingerprint of breast cancer. The International Journal of Biochemistry & Cell Biology, 43(7), 1010–1020.
Robertson, D. G., Watkins, P. B., & Reily, M. D. (2011). Metabolomics in toxicology: Preclinical and clinical applications. Toxicological Sciences, 120(Supplement 1), S146–S170.
Silva, C., Perestrelo, R., Silva, P., Capelinha, F., Tomás, H., & Câmara, J. S. (2019a). Volatomic pattern of breast cancer and cancer-free tissues as a powerful strategy to identify potential biomarkers. The Analyst, 144(14), 4153–4161.
Silva, C. L., Olival, A., Perestrelo, R., Silva, P., Tomás, H., & Câmara, J. S. (2019b). Untargeted urinary1H NMR-based metabolomic pattern as a potential platform in breast cancer detection. Metabolites, 9(11), 269.
Silva, C. L., Perestrelo, R., Silva, P., Tomás, H., & Câmara, J. S. (2019c). Implementing a central composite design for the optimization of solid phase microextraction to establish the urinary volatomic expression: A first approach for breast cancer. Metabolomics, 15(4), 64.
Silva, C. L., Passos, M., & Câmara, J. S. (2012). Solid phase microextraction, mass spectrometry and metabolomic approaches for detection of potential urinary cancer biomarkers—a powerful strategy for breast cancer diagnosis. Talanta, 89, 360–368.
Statheropoulos, M., Sianos, E., Agapiou, A., Georgiadou, A., Pappa, A., Tzamtzis, N., Giotaki, H., Papageorgiou, C., & Kolostoumbis, D. (2005). Preliminary investigation of using volatile organic compounds from human expired air, blood and urine for locating entrapped people in earthquakes. Journal of Chromatography B, 822(1–2), 112–117.
Trifonova, O., Lokhov, P., & Archakov, A. (2013). Postgenomics diagnostics: Metabolomics approaches to human blood profiling. Omics: A Journal of Integrative Biology, 17(11), 550–559.
Wei, S., Liu, L., Zhang, J., Bowers, J., Gowda, G. A. N., Seeger, H., Fehm, T., Neubauer, H. J., Vogel, U., Clare, S. E., & Raftery, D. (2013). Metabolomics approach for predicting response to neoadjuvant chemotherapy for breast cancer. Molecular Oncology, 7(3), 297–307.
Xia, J., Broadhurst, D. I., Wilson, M., & Wishart, D. S. (2013). Translational biomarker discovery in clinical metabolomics: An introductory tutorial. Metabolomics, 9(2), 280–299.
Xia, J., & Wishart, D. S. (2010). MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Research, 38(SUPPL. 2), W71.
Xia, J., Wishart, D. S., Xia, J., & Wishart, D. S. (2016). Using metaboanalyst 3.0 for comprehensive metabolomics data analysis. In Current Protocols in Bioinformatics (pp. 14.10.1–14.10.91). Wiley.
Zhang, A., Sun, H., Qiu, S., & Wang, X. (2013). Metabolomics in noninvasive breast cancer. Clinica Chimica Acta, 424, 3–7.
Acknowledgements
This work was supported by FCT-Fundação para a Ciência e a Tecnologia (Project PEstOE/QUI/UI0674/2019, CQM, UID/MAT/00006/2019, Portuguese Government funds, and INNOINDIGO/0001/2015), Madeira 14-20 Program (Project PROEQUIPRAM—Reforço do Investimento em Equipamentos e Infraestruturas Científicas na RAM—M1420-01-0145-FEDER-000008) and by ARDITI-Agência Regional para o Desenvolvimento da Investigação Tecnologia e Inovação through the Project M1420-01-0145-FEDER-000005—Centro de Química da Madeira—CQM+ (Madeira 14-20). Catarina Silva acknowledge the FCT for the Ph.D Grant SFRH/BD/97039/2013.
Author information
Authors and Affiliations
Contributions
CS: conception of studies and experimental design, urines, BC and CF tissue extractions, sample analysis by HS-SPME/GC–qMS, NMR analysis, statistical data analysis/interpretation and manuscript preparation. RP: design of experiments and data analysis/interpretation. FC: histological classification of BC tissues. HT: co-supervisor of the work, conception of studies, experimental design, and manuscript preparation. JSC: main supervisor of the work, conception of studies, experimental design, and manuscript preparation.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare there is no conflict of interest.
Ethical approval
The research was approved by the Ethics Committee of Funchal Central Hospital Dr. Nélio Mendonça (Approval no. S.1708625/2017) and has been performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. All the participants were fully informed of the objectives of the study and signed the informed consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Silva, C.L., Perestrelo, R., Capelinha, F. et al. An integrative approach based on GC–qMS and NMR metabolomics data as a comprehensive strategy to search potential breast cancer biomarkers. Metabolomics 17, 72 (2021). https://doi.org/10.1007/s11306-021-01823-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-021-01823-1