
Abstract
Introduction
Metabolomics is an emerging approach for early detection of cancer. Along with the development of metabolomics, high-throughput technologies and statistical learning, the integration of multiple biomarkers has significantly improved clinical diagnosis and management for patients.
Objectives
In this study, we conducted a systematic review to examine recent advancements in the oncometabolomics-based diagnostic biomarker discovery and validation in pancreatic cancer.
Methods
PubMed, Scopus, and Web of Science were searched for relevant studies published before September 2017. We examined the study designs, the metabolomics approaches, and the reporting methodological quality following PRISMA statement.
Results and Conclusion
The included 25 studies primarily focused on the identification rather than the validation of predictive capacity of potential biomarkers. The sample size ranged from 10 to 8760. External validation of the biomarker panels was observed in nine studies. The diagnostic area under the curve ranged from 0.68 to 1.00 (sensitivity: 0.43–1.00, specificity: 0.73–1.00). The effects of patients’ bio-parameters on metabolome alterations in a context-dependent manner have not been thoroughly elucidated. The most reported candidates were glutamic acid and histidine in seven studies, and glutamine and isoleucine in five studies, leading to the predominant enrichment of amino acid-related pathways. Notably, 46 metabolites were estimated in at least two studies. Specific challenges and potential pitfalls to provide better insights into future research directions were thoroughly discussed. Our investigation suggests that metabolomics is a robust approach that will improve the diagnostic assessment of pancreatic cancer. Further studies are warranted to validate their validity in multi-clinical settings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Pancreatic cancer (PC) is currently one of the leading causes of cancer-related fatality worldwide with the median survival of six months (Oberstein and Olive 2013). Despite the lethal properties of PC, this disease usually has no early warning signs. Hence, a large number of patients are diagnosed at the advanced and incurable stages of the disease. Available screening approaches, however, are only highly capable of detecting PC among high-risk individuals (Shin and Canto et al. 2012). With the advancement of genomics and other post-genomic high-throughput technologies, a tremendous number of genetic molecules and proteins have been reported to be potential biomarkers in the literature. Significantly, an extensive investigation has introduced a compendium that includes 2516 genes and evidence of their expression levels that were associated with PC, which accounted for approximately 12–13% of the known human coding genes (Harsha et al. 2009). Still, there are currently no clinically relevant biomarkers for accurate diagnosis in early stage of PC. Various types of recurrent somatic mutations of PC have been observed in a recent integrated genomics, transcriptomics, and proteomics study, which indicated the remarkable complexity of the disease (Cancer Genome Atlas Research Network 2017). Moreover, the precise mechanisms of PC initiation and progression remain poorly understood despite recent significant progress (Hezel et al. 2006; Makohon-Moore and Iacobuzio-Donahue 2016). This fact has contributed to the unsatisfactory performance of not only the discovery of useful biomarkers for early detection within the general population but also the development of accurate diagnostic tests for the disease in clinical practice (McCormick and Lemoine 1998).
The integration of metabolomics in cancer biomarker research, including PC, is an emerging field (Fan et al. 2012; Halbrook and Lyssiotis 2017; Kumar et al. 2017; Perez-Rambla et al. 2017). This approach is expected to facilitate a comprehensive understanding of the metabolic alterations and to reveal underlying connections between PC and non-PC conditions. General advantages and disadvantages of the employed analytical platforms for PC metabolomics studies as well as a discussion of possible strategies were reviewed intensively (Gangi et al. 2014). Also, several metabolomics-based studies aiming to discover and validate metabolic alterations that are distinctly associated with the status of PC have been conducted (Tumas et al. 2016). The detection of distinct features of PC using biofluids and/or tissue at the metabolome level can be used not only to provide new insights into cancer mechanisms but also to develop new diagnostic approaches (Nguyen et al. 2015).
Increasing interest in applying metabolomics to discover and validate biomarkers for early detection and diagnosis of PC has resulted in a variety of profiles regarding the metabolomics approaches and potential candidates. However, there has been no overall survey of the status of this dominant topic. The complexity of a high-throughput technology like metabolomics combined with the unique challenges of a biomarker study requires considerable effort to translate the findings into meaningful knowledge. Additionally, the clinical applicability of novel diagnostic biomarkers provided by omics-based studies is commonly overstated, possibly due to biases in the study design, small sample size, lack of epidemiological data, inadequate analysis, and/or insufficient validation (Lumbreras et al. 2009). In the current study, we conducted a systematic review to provide a comprehensive discussion on the clinical significance, current perspectives, and potential pitfalls of metabolomics-based biomarkers in PC. Finally, the putative biological relationships of reported biomarker candidates were also explored.
2 Materials and methods
The current systematic review was accomplished according to a predefined reporting protocol recommended by Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA, http://www.prisma-statement.org/). The full checklist is shown in Supplementary file 1.
2.1 Systematic literature search
As shown in Fig. 1, a systematic search of PubMed, Scopus, and Web of Science was carried out to identify studies that evaluated the application of metabolomics in diagnostic biomarker discovery and validation in PC. According to a previously published guideline, the following terms were used: “(pancreas or pancreatic) and (tumor or tumour or malignancy or neoplasm or cancer or carcinoma or adenoma) and (“metabolite profiling” or “metabolite analysis” or “metabolic profiling” or “metabolic fingerprinting” or “metabolic characterization” or metabolome or metabolomics or metabolomic or metabonomics or metabonomic or lipidome or lipidomics or lipidomic)” (Aromataris and Riitano 2014). There was no limit in terms of the search duration. The literature search was conducted on 16 September 2017. In addition, the references to relevant articles were reviewed to identify the suitable studies for further assessment.

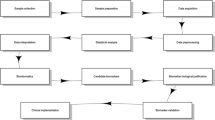
The whole workflow of the systematic review
2.2 Inclusion and exclusion criteria
Data of title, year of publication, authors, and abstracts were extracted to EndNote X6 (Thomson Reuters, NY, USA). Duplicated articles among databases were removed. After that step, an initial screening process was employed by reading the titles and the abstracts of the remaining articles. Potential and suspected articles were further evaluated by reading their full-texts. Eligibility was reviewed by at least two authors to avoid personal biases, and a consensus was made when having inconsistencies.
A study was qualified when it examined the diagnostic biomarker discovery and validation using any metabolomics platforms in PC that described most if not all the following elements: (1) biospecimen (tissue, serum, and/or plasma, etc.); (2) metabolomics platform (GC/MS, LC/MS, and/or NMR, etc.); (3) the statistical biomarker selection and evaluation (univariate analysis, multivariate analysis, or statistical learning) for the discrimination of PC from other cancerous and non-cancerous conditions; (4) suitable prediction metrics of the biomarkers (accuracy, sensitivity, specificity, and area under a receiver operating characteristics curve (AUC-ROC), etc.). We excluded the studies if (1) no suitable control groups were used; (2) the studies were conducted on animal tissues or cell lines; (3) there was duplicated or had no reliable data; and (4) they are letters, reviews, theses, and conference posters or proceedings. There was no restriction to study design, race, geographical area, or certain population.
2.3 Study data extraction
A data extraction form was developed in accordance with previously published systematic reviews and guidelines. Basic information of the study (first author, year of publication), patient characteristics (country, patient participant status, clinical stage, prior treatment), control participant status, age, gender ratio, and follow-up were first extracted. Criteria for diagnosis were examined by three levels: guideline, histological confirmation, or not described. Next, the biological specimen, employed metabolomics or lipidomics platforms, nature of the approach (targeted or untargeted), fasting condition, sample collection and storage condition, sample preparation or pretreatment description, internal standard, analytical validation, and metabolite identification methods were described. Finally, we assessed the validation protocol (external validation or cross-validation), outlier detection, suggestive biomarkers (individual markers or panels of markers), quantitative prediction model, and the measurement of predictive performance (Findeisen and Neumaier 2009). The performances of the biomarker panels were preferably reported. Although R2 (coefficient of determination) and Q2 (cross-validated R2) are not the direct indicators for the clinical utility or the future prediction of the diagnostic models, they are commonly employed in metabolomics biomarker research (Xia et al. 2013). Thus, the R2 and Q2 values of PLS-DA or OPLS-DA models were extracted. For each included article, at least two reviewers read and extracted relevant data for the qualitative synthesis. The reviewers retrieved data independently to avoid unexpected error and/or personal biases. Any inconsistencies were discussed to reach a final consensus.
2.4 Reporting methodological quality assessment
According to the Metabolomics Standards Initiative, a minimum reporting standard of metabolomic experiments includes adequate reports of sample preparation, experimental analysis, quality control, metabolite identification, and data pre-processing (Sumner et al. 2007). For the evaluation of a biomarker study, clear objective, proper clinical context, appropriate data source, robust statistical analysis, and external validation are desired (Azuaje et al. 2009; Bossuyt et al. 2015, 2003; Parker et al. 2010; Turakhia and Sabatine 2017). Collectively, we developed a reporting quality assessment panel of included studies to fully assess the included studies with both diagnostic study and metabolomics study aspects: (1) the study characteristics including study design, characteristics of the studied population, diagnostic criteria, criteria for inclusion and exclusion, prior treatment, follow-up, comparative analysis with reference (e.g., CA19.9); (2) the sampling and experimental analysis including sample collection and storage, sample preparation, experimental condition, the use of internal standard, analytical validation method, outlier detection, metabolite identification; and (3) the computational analysis including data pre-processing, statistical analysis, and quantitative prediction modeling. At least two authors independently assessed the quality of the included studies in accordance with the extracted data. We used the term “low risk of bias” (L) for adequate description, “high risk of bias” (H) for simple description or not described (U) for unknown risk of bias, and we performed the visualization of the quality assessment under QUADAS-2 tool (Whiting et al. 2011). In addition, QUADOMICS, a quality assessment panel specialized for omics-based diagnostic assessment studies was also adopted and applied simultaneously (Lumbreras et al. 2008).
2.5 Pathway enrichment analysis
The compound name standardization was conducted using Human Metabolome Database version 4.0 prior to the analysis (Wishart et al. 2017). The pathway analysis was conducted using MetaboAnalyst version 3.5 (http://www.metaboanalyst.ca) (Xia and Wishart 2016). The Kyoto Encyclopedia of Genes and Genomes (KEGG) and the small molecule pathway (SMP) were used as the knowledgebase to identify relevant pathways. The analysis algorithm was a hypergeometric test for over-representation analysis, and the relative betweenness centrality was selected for pathway topology analysis. The pathway that has an adjusted P-value (false discovery rate, FDR) of less than 0.05 was considered to be significantly enriched.
3 Results
The whole process of the screening step is shown in Fig. 1. From 972 reports of Web of Science, Scopus, and PubMed, 664 studies were included for title and abstract screening after deleting duplications. We then excluded 590 studies that were not related to the study question, conducted on animal tissues or cell lines, or had no suitable control groups or no reliable data. Letters, reviews, theses, conference posters or proceedings were also ruled out. Only 74 studies were eligible for further full-text assessment. We excluded 49 publications because they also met the exclusion criteria. There were no other papers identified by cross-referencing the relevant articles. Eventually, the final list of included studies that contained 25 papers was subjected to systematic review.
3.1 Characteristics of included studies
The main characteristics of included studies are described in Table 1. There were three studies collecting samples from two distinct populations [Xie et al. (2015) from USA and China, Ritchie et al. (2013) from Japan and USA, and Lindahl et al. (2017) from Germany and Sweden]. Biological samples utilized for metabolomic analysis included: serum/plasma (22 studies), urine (two studies), and saliva (one study). Apart from the study of Fukutake et al. (2015) in which 360 cancer and 8400 control cases were involved, the sample size of the remaining studies was relatively small, in which only three studies contained more than 100 PC patients (median of 40, range from 5 to 360). The mean/median age of PC samples seems to be higher than the mean/median age of the control groups. The male/female ratio among studies was greater than one. Nevertheless, 11 studies utilized age- and gender-matched control samples. Healthy controls were used in 22 studies. Twelve studies used one or a mixture of non-cancerous or other cancerous conditions. One study used retrospective, 21 used prospective, and two studies used both retrospective and prospective methods for sample allocation. Noticeably, clinical and laboratory criteria for the diagnosis of PC in metabolomics-based biomarker studies were not adequately reported. Particularly, ten studies (40%) explicitly described that pathological information was used for the diagnosis. Tumor stage was provided in 19 studies, in which the number of samples in the resectable stages (0–II) accounted for approximately nearly a half portion. The training set of Hirata et al. (2017), the test set of Xie et al. (2015), and the whole data set of Suzuki et al. (2017) and Urayama et al. (2010) included PC patients who were only in resectable stages. In the study of Sugimoto et al. (2010), all patients were diagnosed with primary disease. External validation was employed in nine studies (36%). Follow-up of non-cancerous controls was not a common practice as only 24% of the studies applied it to ensure the diagnosis and grouping.
3.2 Metabolomics design properties of included studies
The study design and metabolomics approaches of the included studies can be found in Table 2. Most studies were untargeted metabolomics (14 studies at 56%), lipidomics (two studies at 8%), or both (three studies at 12%). Four studies were targeted metabolomics, which focused on amino acids (Fukutake et al. 2015; Hirata et al. 2017; Leichtle et al. 2013) or fatty acids (Di Gangi et al. 2016). Mass spectrometry (MS) was the most commonly used platform for the assessment of novel biomarkers in PC (19 studies), followed by NMR (four studies), or both (one study). Serum and plasma were the two most important biological specimens in finding potential biomarkers for accurate diagnosis of PC. Thirteen studies employed samples collecting under fasting condition. Samples in the included studies were generally stored at − 80 °C after collection (22 studies). Before the analysis, sample preparation was essentially performed using defined protocols. The use of internal standards was common and observed in 17 studies. However, only 14 studies reported having analytical validation using quality control samples. The compound identification using authentic standards was described in ten studies. Also, in-house library and database matching were the two popular methods. Finally, although outlier detection is useful for the statistical analysis and modeling, only three studies performed outlier detection, mostly using the PCA method.
3.3 Metabolite-based biomarkers are novel for early diagnosis of PC from healthy controls and other non-cancerous conditions
The study design of the included studies focused mainly on the differentiation of PC from healthy controls, PC from non-malignant diseases, and PC from other malignant conditions. There was only one study that applied the “multi”-benign conditions for the control group (Bathe et al. 2011). In general, the reported quantitative values (AUC, accuracy, sensitivity, and specificity) were promising. The AUC of diagnostic models constructed using multivariate analysis ranged from 0.68 to 1.00. The accuracy varied from at least 0.78 to 0.99, while sensitivity and specificity varied from 0.43 to 1.00 and 0.73 to 1.00, respectively. The performance of the biomarker panels in the included studies can be found in Table 3 and the properties of the included studies are extensively depicted in Supplementary File 2.
3.4 Reporting methodological quality assessment
The two quality assessment results of the included studies can be found in Fig. 2 and Supplementary File 3. Our in-house quality assessment contains 16 domains based on both the diagnostic study and the metabolomics study aspects. Twenty of 25 studies (80%) adequately reported at least eight per 16 domains. The remaining five studies reported at least six domains. Characteristics of the diagnostic criteria, criteria for inclusion and exclusion, follow-up, comparative analysis with reference, and outlier detection were among the least adequately reported domains (Fig. 2a). Only five studies had the follow-up program. According to the QUADOMICS tool, 21 of 25 studies (84%) adequately reported at least eight per 16 items (Fig. 2b). All 25 studies were either phase I or phase II. Therefore, items 2 and 14 regarding the use of metabolomics testing in practice were not applicable for all of the studies. According to item 16, the risk of over-fitting presented in 16 of the studies (64%) owing to the lack of an independent validation set.

The two quality assessment results of the included studies. a Our self-developed quality assessment. b QUADOMICS tool. Items 2 and 14 regarding the use of metabolomics testing in practice are not applicable for all of the studies, and then, are not presented in the figure
3.5 Pathway enrichment analysis of potential biomarkers
From the included studies, we extracted 132 potential biomarker candidates. Among them, amino acids were dominant. Glutamic acid and histidine were reported in seven studies, followed by glutamine and isoleucine both of which were revealed in five studies. Only 46 compounds that were estimated more than once were submitted to conduct pathway enrichment analysis. As shown in Fig. 3, alanine, aspartate and glutamate metabolism (impact value of 0.75, FDR of 8.44E−04); glycine, serine and threonine metabolism (impact value of 0.42, FDR of 3.39E−04); and taurine and hypotaurine metabolism (impact value of 0.36, FDR of 3.67E-02) were the most prominent pathways derived from the selected biomarkers. In addition, seven other pathways were also enriched: Arginine and proline metabolism; aminoacyl-tRNA biosynthesis; methane metabolism; valine, leucine and isoleucine biosynthesis; nitrogen metabolism; cyanoamino acid metabolism; and synthesis and degradation of ketone bodies. Detailed information regarding the reported metabolites and enriched pathways can be found in Supplementary File 4.

Pathway enrichment analysis of potential biomarkers. Alanine aspartate and glutamate metabolism; glycine serine and threonine metabolism; and taurine and hypotaurine metabolism were three most prominent pathways
4 Discussion
CA19.9 has been the only approved biomarker for clinical management of PC by Food and Drug Administration (Partyka et al. 2012). However, the level of CA19.9 also increases in other malignancies and even benign conditions while cannot be detected in Lewis antigen-negative patients (Ballehaninna and Chamberlain 2012). Not to mention, CA19.9 had poor clinical utility as a diagnostic biomarker of PC, and its level did not have the power to help clinicians make additional testing or procedures, as well as manage patients (Singh et al. 2011). Although many papers encompassing various disciplines have introduced novel biomarker candidates for early detection and diagnosis of PC, most of the markers have not proceeded beyond the discovery phase (Chan et al. 2013). Moreover, the reporting quality of the development and validation of the proposed markers has been generally poor among medical disciplines, including cancer research (Moons et al. 2015). The important role of cellular metabolism has been recently acknowledged as a hallmark of cancer, which can facilitate the discovery of promising metabolomics-based biomarkers and novel treatments (Halbrook and Lyssiotis 2017; Pavlova and Thompson 2016). A metabolite that significantly alters its expression in PC and can be detected accurately may become a biomarker candidate. Nevertheless, PC cells are highly heterogeneous, and this characteristic hampers the development of a straightforward method for accurate diagnosis of this cancer (Sidaway 2017). In a recent literature review, Tanase et al. (Tanase et al. 2009) concluded that only the combination of soluble biomarkers could provide adequate sensitivity and specificity for clinical applications. Additionally, circulating biomarkers are of great interest for developing promising biomarker panels (Cappelletti et al. 2015; Konforte and Diamandis 2013). In this paper, we examined the status of the oncometabolomics-based biomarker study for the diagnosis of PC. Many individual markers and marker panels were introduced, and some of them were externally validated. Despite the promising results, these studies primarily focused on the identification of the potential biomarkers rather than validation of their predictive capacity. There are also some remaining issues, which may affect the value of the proposed markers for a broader range of clinical use. For example, the sample size was generally small except the study of Fukutake et al. (Fukutake et al. 2015). Concrete epidemiological characteristics, diagnostic criteria, and tumor grading were usually missing. Similar problems were also reported in a systematic review of metabolomic profiling of oesophago-gastric cancer (Abbassi-Ghadi et al. 2013). Moreover, the transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD) statement recommended for multivariate prediction model studies was not yet applied (Moons et al. 2015). Herein, we discuss crucial aspects and provide recommendations for future investigations on metabolomics-based diagnostic biomarker study.
Problems with study design and validation protocol of a biomarker study lead to the suggestion of false positive candidates or rejection of the false negative candidates. In biomarker research, essential information is usually not sufficiently provided in diagnostic studies, which eventually hinders the study assessment, replication, and application (Azuaje et al. 2009; Bossuyt et al. 2015). The lack of clinically relevant biomedical context in the investigations on diagnostic accuracy is also a major shortcoming (Irwig et al. 2002; Pepe et al. 2008). According to our assessment, the quality of scientific reporting regarding the metabolomic aspects was not strictly followed. Hence, there is an urgent need to improve important reported elements in diagnostic accuracy research. The Standards for Reporting of Diagnostic Accuracy (STARD), updated in 2015, is an important source for reference to improve the accurate reporting of diagnostic studies (Bossuyt et al. 2015). It is also worth mentioning that a biomarker study for diagnostic purpose may require different criteria in terms of clinical indicators than a screening-based biomarker study (Pepe et al. 2008). This domain-specific property requires intensive considerations regarding the particular purpose of the biomarker research. For instance, the prospective-specimen-collection, retrospective-blinded-evaluation (ProBE) study design guideline suggests that a diagnostic study should demand a very high sensitivity while the acceptable value of a false-positive rate (1—specificity) can be up to 75% (Pepe et al. 2008). In addition, a diagnostic biomarker does not have to be the gold standard for the diagnosis of PC to be valuable. Instead, it can be a companion tool to assist other clinically relevant biomarker panels or diagnostic tests, such as endoscopy ultrasound, enhanced MRI, and ultrasound-guided fine-needle aspiration (Ryan et al. 2014; Takhar et al. 2004).
Surgical resection of the primary tumor followed by adjuvant chemotherapy is the only curable option for PC. The resectable PC is mainly regarded as stage I and stage II (Ryan et al. 2014). Therefore, biomarker panels that can accurately diagnose PC at those stages are essential. There is an agreement that an effective approach for early diagnosis should be able to detect the PC at pancreatic intraepithelial neoplasia or T1N0M0 stage (Canto et al. 2013). However, our findings show that none of the included metabolomics-based biomarker studies focused on the premalignant lesions, such as pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasm, and only five studies paid attention to the early stages of PC when introducing new biomarkers (Fukutake et al. 2015; Hirata et al. 2017; Suzuki et al. 2017; Urayama et al. 2010; Zhang et al. 2014). This may come from the fact that PC is usually asymptomatic and not visible on conventional imaging studies at early stages (Kamisawa et al. 2016). Furthermore, the inclusion of more PC patients of stage III or IV may cause an overoptimistic estimation in terms of early diagnosis (Kobayashi et al. 2013). A similar problem was also observed in diagnostic biomarkers of bladder cancer (Rodrigues et al. 2016).
Errors and biases can occur at all phases of biomarker discovery and validation studies that eventually increase the risk of misleading results (Diamandis 2010). As an example, the significant difference of the age of patients and controls may become the confounding factor that affects the reliability of the biomarker panels (Kuan 2014). Hence, the effects of suspect confounding factors should be measured using proper analyses. In addition, there are several studies that applied a gender- and/or age-matched control approach. Although this method is statistically efficient and helps control potential confounders, it is usually problematic and is not preferred (Pepe et al. 2008).
Among statistical approaches, multivariate regression analysis, partial least squares discriminant analysis, and orthogonal projections to latent structures discriminant analysis have been the commonly applied methods in PC biomarker research. Nonetheless, there is an increasing trend of using the so-called “black-box” machine learning models. On the one hand, machine learning methods substantially increase the effectiveness of discriminant analysis, but on the other hand, they raise considerable concerns in the biomedical community, especially because of its lack of interpretability (Foster et al. 2014). However, lack of interpretability can partially be solved by applying local interpretable model-agnostic explanations (LIME) method to readily interpret how a particular model works (Ribeiro et al. 2016). It is important to note that the consecutive analyses using different statistical and machine learning methods on the same data set, in which the later analyses use the outcome of the previous models as the supervised methods for feature selection, may eventually provide overfitting results (Smialowski et al. 2010). These data over-interpretation practice should be avoided. The clinical value of a set of biomarkers is reflected by positive predictive value, negative predictive value, positive likelihood ratio, and negative likelihood ratio; these parameters, when applicable, are encouraged to be reported in future studies (Usher-Smith et al. 2016). Additionally, there is no universal rule to develop criteria for determining potential biomarkers. Our results show that most of the studies applied univariate and/or multivariate analysis for this purpose. However, the relative difference of a particular compound of different groups (fold change) is rarely employed as a criterion. A small change, such as 0.9 or 1.1, may be purely due to the disturbance of the data-gathering process and may not be reproducible. However, there is no consensus on what cut-off should be applied. For instance, Crews et al. (2009) suggested a fold change of greater than two in their variability analysis of human plasma while other authors applied a fold change of 1.5 for their analyses (Patti et al. 2012; Vinaixa et al. 2012). Therefore, researchers may flexibly consider this indicator as an additional criterion when interpreting the metabolomics results.
The class-imbalanced problem, even though is usually neglected in biomarker research, often results in poor performance of a diagnostic model for the minority class in clinical utility (Kondo 2014; Saito and Rehmsmeier 2015). This issue becomes even more problematic when it comes up with the high-dimensional nature of a high-throughput study (Yin et al. 2013). Besides, some additional factors that affect the performance of the prediction include, but are not limited to the inadequate numbers of the exploratory data (e.g., small sample size), data complexity, and the considerable similarity of the metabolomes of different conditions. Excellent discussions of this particular issue can be found elsewhere (Chen et al. 2014; Lin and Chen 2013). It is of importance to mention that untargeted metabolomics data contain many redundant and noisy features that negatively affect the performance of the classification models (Grissa et al. 2016). Feature selection aims to reduce the data complexity derived from a high-throughput experiment by selecting the discriminatory features between cases and control samples. The smaller subset of valuable features can be used to build different supervised learning models for the classification purpose. However, a separate portion of data for feature selection may be required before typical training and test sets are used to search for the optimal predictive models to avoid overfitting results. Besides, the integration of demographic information, clinical signs and typical laboratory tests, and the metabolomics-based biomarker panels may significantly enhance the diagnostic performance (Liesenfeld et al. 2013). For instance, hyperglycemia, increased CA19-9, and elevated branched-chain amino acids (BCAAs) are strongly associated with the status of the PC (Pannala et al. 2009). However, this approach is currently not widely applied to metabolomics-based biomarker discovery and validation studies.
Lately, common pre-analytical, analytical, and data processing pitfalls and best practices for different platforms of metabolite measurement are intensively addressed (Baran 2017; Lu et al. 2017). Nevertheless, the application of reporting standards has not been strictly followed in metabolomics studies. According to The Metabolomics Society Data Quality Task Group, there is still a lack of a consensus on quality assurance (QA) and quality control (QC) practices among metabolomics laboratories, suggesting the need of courses, specialist meetings and reports, and expert panels to create practical QA and QC recommendations and guidelines as well as to enhance the practice in the metabolomics community (Dunn et al. 2017). Besides the lack of QC methods, errors in sample processing and storage can also cause misleading results in biomarker discovery. Moreover, the metabolic perturbation during quenching and extraction can be avoided by rapid manipulation, or the metabolite degradation and interconversion after extraction can be addressed by shortening the time interval between sample preparation and analysis or by using preservatives (Lu et al. 2017). One prominent problem is the short-term and long-term stability of metabolites in biospecimens since the process of sample acquisition in a biomarker study usually takes months or years (Dunn et al. 2011; Yang et al. 2013). Yang et al. indicated that the concentration of some common metabolites in plasma was altered as rapidly as one hour when stored at 4 °C. Moreover, the metabolome of the specimens could be changed significantly even when stored at − 80 °C in a 5-year period (Yang et al. 2013). The vulnerable metabolites, therefore, should be considered for exclusion from the biomarker panel. The harmonization in quantitative untargeted lipidomics has been an issue that prevents inter-study and inter-laboratory comparisons (Bowden et al. 2017). However, a recent comparative investigation on 126 identical plasma and 29 quality control samples of nine MS instruments has demonstrated that the untargeted lipidomics analysis using different instruments can achieve almost comparable results. The study partly answered a long-standing question of whether different conclusions in biological studies are mainly affected by the MS instruments (Cajka et al. 2017).
Peak misidentification of significantly different compounds in untargeted metabolomics settings, especially in LC–MS based untargeted metabolomics, is currently a bottleneck that prevents the application of metabolomics in clinical settings (Kind et al. 2017). Peak misidentification may be brought about by co-eluting isomers, similar molecular-weight interferences, or in-source fragmentation products (Lu et al. 2017). The estimation of false discovery rate is recommended to improve the robustness of the metabolite annotation in untargeted MS experiments (Scheubert et al. 2017). In silico prediction algorithms and computational annotation solutions for putatively identifying compounds have been recently become an active research area (Aksenov et al. 2017; Domingo-Almenara et al. 2017). Besides, the aid of ion mobility spectrometry to the typical LC–MS-based metabolomics or lipidomics study will also improve the reliability of the metabolite identification (Paglia and Astarita 2017). Another consideration that needs to be mentioned is that the authors are also encouraged to include the corresponding Human Metabolome Database (HMDB), PubChem, KEGG, and Chemical Entities of Biological Interest (ChEBI) Compound Identifier number of the identified metabolites for further analysis.
The repository of raw data will greatly contribute to the development of biomarker study. Thus, there is an urgent need for publishing the paper together with the incorporation of data on MetaboLights, Metabolomics Workbench, Metabolomics Repository Bordeaux, XCMS online, or MetabolomeXpress to facilitate the validation, comparison, and meta-analysis of proposed biomarkers (Spicer and Steinbeck 2017). Along with data sharing, national and international efforts to establish and maintain repositories or biobanks of specimens will hopefully accelerate the discovery and validation of new biomarkers.
Among differentially expressed metabolites, amino acids were mostly reported. This suggested that amino acid-related pathways might play an important role in the differentiation between PC and non-malignant conditions. Alanine, aspartate and glutamate metabolism and taurine and hypotaurine metabolism pathways were also enriched in lung cancer (Kumar et al. 2017). Taurine and hypotaurine metabolism pathway was also associated with renal clear cell carcinoma (Yang et al. 2014). In addition, the role of serine and glycine metabolism in cancer cell growth was thoroughly reviewed and discussed (Amelio et al. 2014). Noticeably, our study indicated that the valine, leucine and isoleucine biosynthesis pathway was significantly enriched in PC, agreeing with a previous study stating that the concentrations of BCAAs (valine, leucine, and isoleucine) were elevated in the early stage of PDAC development (Mayers et al. 2014). Insulin resistance-related conditions, such as diabetes and obesity, are well-known risk factors of PC, and the circulating biomarkers of peripheral insulin resistance were found to be significantly associated with a higher risk of developing PC (Bao et al. 2013; Li 2012; Wolpin et al. 2013). The increased level of BCAAs was associated with type 2 diabetes mellitus and metabolic syndrome that may eventually contribute to the PC carcinogenesis (Lynch and Adams 2014). Indeed, a recent study suggested that the elevated level of BCAAs in plasma was an independent risk factor of PC (Leake 2014). Furthermore, the altered metabolism of BCAAs was observed in many other cancer types such as glioblastoma, lung cancer, and breast cancer (Ananieva and Wilkinson 2018). It is worthy to note that we only performed pathway enrichment analysis for biomarkers reported in all groups of patients. At this time, it was not feasible to conduct subgroup pathway analysis of each patient characteristic such as age, tumor stage, or prior treatment.
5 Conclusions
The specific properties of PC have made it a legitimate target for numerous efforts to improve the current diagnostic approaches. Most of the current studies suggest that the accurate diagnosis of PC using metabolic alterations of biospecimens is a practical certainty, although there is a need for further improvements and standardizations. Our systematic review and discussion on the current issues of the oncometabolomics-based diagnostic biomarker discovery and validation may assist the advancement in this area. Given the strong potential of the metabolomics approach, further investigations on the multi-center scale with rigorous study designs should be conducted to fully explore the potential of the metabolomics-based diagnostic approach.
Abbreviations
- AUC:
-
Area under the curve
- BCAAs:
-
Branched-chain amino acids
- CP:
-
Chronic pancreatitis
- GC–MS:
-
Gas chromatography–mass spectrometry
- LC–MS:
-
Liquid chromatography–mass spectrometry
- MRI:
-
Magnetic resonance imaging
- MS/MS:
-
Tandem mass spectrometry
- NMR:
-
Nuclear magnetic resonance
- OPLS-DA:
-
Orthogonal projections to latent structures discriminant analysis
- PC:
-
Pancreatic cancer
- PCA:
-
Principal component analysis
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PLS-DA:
-
Partial least squares discriminant analysis
- PLS-DF:
-
Partial least squares discriminant function
- Q2 :
-
Cross-validated coefficient of determination
- R2 :
-
Coefficient of determination
- ROC:
-
Receiver operating characteristic curve
- SFE-SFC/MS/MS:
-
Supercritical fluid extraction-supercritical fluid chromatography coupled with tandem mass spectrometry
References
Abbassi-Ghadi, N., Kumar, S., Huang, J., Goldin, R., Takats, Z., & Hanna, G. B. (2013). Metabolomic profiling of oesophago-gastric cancer: A systematic review. European Journal of Cancer, 49, 3625–3637.
Akita, H., Ritchie, S. A., Takemasa, I., Eguchi, H., Pastural, E., Jin, W., et al. (2016). Serum metabolite profiling for the detection of pancreatic cancer: Results of a large independent validation study. Pancreas, 45, 1418–1423.
Aksenov, A. A., da Silva, R., Knight, R., Lopes, N. P., & Dorrestein, P. C. (2017). Global chemical analysis of biology by mass spectrometry. Nature Reviews Chemistry, 1, 0054.
Amelio, I., Cutruzzola, F., Antonov, A., Agostini, M., & Melino, G. (2014). Serine and glycine metabolism in cancer. Trends in Biochemical Sciences, 39, 191–198.
Ananieva, E. A., & Wilkinson, A. C. (2018). Branched-chain amino acid metabolism in cancer. Current Opinion in Clinical Nutrition and Metabolic Care, 21, 64–70.
Aromataris, E., & Riitano, D. (2014). Constructing a search strategy and searching for evidence. A guide to the literature search for a systematic review. AJN The American Journal of Nursing, 114, 49–56.
Azuaje, F., Devaux, Y., & Wagner, D. (2009). Challenges and standards in reporting diagnostic and prognostic biomarker studies. Clinical and Translational Science, 2, 156–161.
Ballehaninna, U. K., & Chamberlain, R. S. (2012). The clinical utility of serum CA 19–9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: An evidence based appraisal. Journal of Gastrointestinal Oncology, 3, 105–119.
Bao, Y., Giovannucci, E. L., Kraft, P., Stampfer, M. J., Ogino, S., Ma, J., et al. (2013). A prospective study of plasma adiponectin and pancreatic cancer risk in five US cohorts. Journal of the National Cancer Institute, 105, 95–103.
Baran, R. (2017). Untargeted metabolomics suffers from incomplete raw data processing. Metabolomics, 13(9), 107
Bathe, O. F., Shaykhutdinov, R., Kopciuk, K., Weljie, A. M., McKay, A., Sutherland, F. R., et al. (2011). Feasibility of identifying pancreatic cancer based on serum metabolomics. Cancer Epidemiology and Prevention Biomarkers, 20, 140–147.
Beger, R. D., Schnackenberg, L. K., Holland, R. D., Li, D., & Dragan, Y. (2006). Metabonomic models of human pancreatic cancer using 1D proton NMR spectra of lipids in plasma. Metabolomics, 2, 125–134.
Bossuyt, P. M., Reitsma, J. B., Bruns, D. E., Gatsonis, C. A., Glasziou, P. P., Irwig, L., et al. (2015). STARD 2015: An updated list of essential items for reporting diagnostic accuracy studies. Radiology, 277, 826–832.
Bossuyt, P. M., Reitsma, J. B., Bruns, D. E., Gatsonis, C. A., Glasziou, P. P., Irwig, L. M., et al. (2003). Towards complete and accurate reporting of studies of diagnostic accuracy: The STARD initiative. BMJ, 326, 41.
Bowden, J. A., Heckert, A., Ulmer, C. Z., Jones, C. M., Koelmel, J. P., Abdullah, L., et al. (2017). Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-metabolites in frozen human plasma. Journal of Lipid Research, 58, 2275–2288.
Cajka, T., Smilowitz, J. T., & Fiehn, O. (2017). Validating quantitative untargeted lipidomics across nine liquid chromatography–high-resolution mass spectrometry platforms. Analytical Chemistry, 89, 12360–12368.
Cancer Genome Atlas Research Network. (2017). Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell, 32, 185–203 e13.
Canto, M. I., Harinck, F., Hruban, R. H., Offerhaus, G. J., Poley, J. W., Kamel, I., et al. (2013). International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut, 62, 339–347.
Cappelletti, V., Appierto, V., Tiberio, P., Fina, E., Callari, M., & Daidone, M. G. (2015). Circulating biomarkers for prediction of treatment response. Journal of the National Cancer Institute Monographs, 2015, 60–63.
Chan, A., Diamandis, E. P., & Blasutig, I. M. (2013). Strategies for discovering novel pancreatic cancer biomarkers. Journal of Proteomics, 81, 126–134.
Chen, J. J., Lu, T.-P., Chen, D.-T., & Wang, S.-J. (2014). Biomarker adaptive designs in clinical trials. Translational Cancer Research, 3(3), 279–292
Crews, B., Wikoff, W. R., Patti, G. J., Woo, H. K., Kalisiak, E., Heideker, J., et al. (2009). Variability analysis of human plasma and cerebral spinal fluid reveals statistical significance of changes in mass spectrometry-based metabolomics data. Analytical Chemistry, 81, 8538–8544.
Davis, V. W., Schiller, D. E., Eurich, D., Bathe, O. F., & Sawyer, M. B. (2013). Pancreatic ductal adenocarcinoma is associated with a distinct urinary metabolomic signature. Annals of Surgical Oncology, 20(Suppl 3), S415–S423.
Di Gangi, I. M., Mazza, T., Fontana, A., Copetti, M., Fusilli, C., Ippolito, A., et al. (2016). Metabolomic profile in pancreatic cancer patients: A consensus-based approach to identify highly discriminating metabolites. Oncotarget, 7, 5815–5829.
Diamandis, E. P. (2010). Cancer biomarkers: Can we turn recent failures into success? Journal of the National Cancer Institute, 102, 1462–1467.
Domingo-Almenara, X., Montenegro-Burke, J. R., Benton, H. P., & Siuzdak, G. (2017). Annotation: A computational solution for streamlining metabolomics analysis. Analytical Chemistry, 90(1), 480–489.
Dunn, W. B., Broadhurst, D., Begley, P., Zelena, E., Francis-McIntyre, S., Anderson, N., et al. (2011). Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nature Protocols, 6, 1060–1083.
Dunn, W. B., Broadhurst, D. I., Edison, A., Guillou, C., Viant, M. R., Bearden, D. W., et al. (2017). Quality assurance and quality control processes: Summary of a metabolomics community questionnaire. Metabolomics, 13, 50.
Fan, L., Zhang, W., Yin, M., Zhang, T., Wu, X., Zhang, H., et al. (2012). Identification of metabolic biomarkers to diagnose epithelial ovarian cancer using a UPLC/QTOF/MS platform. Acta Oncology, 51, 473–479.
Findeisen, P., & Neumaier, M. (2009). Mass spectrometry based proteomics profiling as diagnostic tool in oncology: Current status and future perspective. Clinical Chemistry and Laboratory Medicine, 47, 666–684.
Foster, K. R., Koprowski, R., & Skufca, J. D. (2014). Machine learning, medical diagnosis, and biomedical engineering research—Commentary. Biomedical Engineering Online, 13, 94.
Fukutake, N., Ueno, M., Hiraoka, N., Shimada, K., Shiraishi, K., Saruki, N., et al. (2015). A novel multivariate index for pancreatic cancer detection based on the plasma free amino acid profile. PLoS ONE 10, e0132223.
Gangi, I. M. D., Vrhovsek, U., Pazienza, V., & Mattivi, F. (2014). Analytical metabolomics-based approaches to pancreatic cancer. TrAC Trends in Analytical Chemistry, 55, 94–116.
Grissa, D., Petera, M., Brandolini, M., Napoli, A., Comte, B., & Pujos-Guillot, E. (2016). Feature selection methods for early predictive biomarker discovery using untargeted metabolomic data. Frontiers in Molecular Biosciences, 3, 30.
Halbrook, C. J., & Lyssiotis, C. A. (2017). Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell, 31, 5–19.
Harsha, H. C., Kandasamy, K., Ranganathan, P., Rani, S., Ramabadran, S., Gollapudi, S., et al. (2009). A compendium of potential biomarkers of pancreatic cancer. PLoS Med 6, e1000046.
He, X., Zhong, J., Wang, S., Zhou, Y., Wang, L., Zhang, Y., et al. (2017). Serum metabolomics differentiating pancreatic cancer from new-onset diabetes. Oncotarget, 8, 29116–29124.
Hezel, A. F., Kimmelman, A. C., Stanger, B. Z., Bardeesy, N., & Depinho, R. A. (2006). Genetics and biology of pancreatic ductal adenocarcinoma. Genes and Development, 20, 1218–1249.
Hirata, Y., Kobayashi, T., Nishiumi, S., Yamanaka, K., Nakagawa, T., Fujigaki, S., et al. (2017). Identification of highly sensitive biomarkers that can aid the early detection of pancreatic cancer using GC/MS/MS-based targeted metabolomics. Clinica Chimica Acta, 468, 98–104.
Irwig, L., Bossuyt, P., Glasziou, P., Gatsonis, C., & Lijmer, J. (2002). Designing studies to ensure that estimates of test accuracy are transferable. BMJ, 324, 669–671.
Itoi, T., Sugimoto, M., Umeda, J., Sofuni, A., Tsuchiya, T., Tsuji, S., et al. (2017). Serum Metabolomic profiles for human pancreatic cancer discrimination. International Journal of Molecular Sciences, 18(4), 767.
Kamisawa, T., Wood, L. D., Itoi, T., & Takaori, K. (2016). Pancreatic cancer. Lancet, 388, 73–85.
Kind, T., Tsugawa, H., Cajka, T., Ma, Y., Lai, Z., Mehta, S. S., et al. (2017). Identification of small molecules using accurate mass MS/MS search. Mass Spectrometry Reviews, 9999, 1–20.
Kobayashi, T., Nishiumi, S., Ikeda, A., Yoshie, T., Sakai, A., Matsubara, A., et al. (2013). A novel serum metabolomics-based diagnostic approach to pancreatic cancer. Cancer Epidemiology Prevention Biomarkers, 22, 571–579.
Kondo, T. (2014). Inconvenient truth: cancer biomarker development by using proteomics. Biochimica et Biophysica Acta, 1844, 861–865.
Konforte, D., & Diamandis, E. P. (2013). Is early detection of cancer with circulating biomarkers feasible? Clinical Chemistry, 59, 35–37.
Kuan, P. F. (2014). Propensity score method for partially matched omics studies. Cancer Informatics, 13, 1–10.
Kumar, N., Shahjaman, M., Mollah, M. N. H., Islam, S. M. S., & Hoque, M. A. (2017). Serum and plasma metabolomic biomarkers for lung cancer. Bioinformation, 13, 202–208.
Leake, I. (2014). Early events in pancreatic cancer. Nature Reviews Gastroenterology & Hepatology, 11, 703.
Leichtle, A. B., Ceglarek, U., Weinert, P., Nakas, C. T., Nuoffer, J.-M., Kase, J., et al. (2013). Pancreatic carcinoma, pancreatitis, and healthy controls: Metabolite models in a three-class diagnostic dilemma. Metabolomics, 9, 677–687.
Li, D. (2012). Diabetes and pancreatic cancer. Molecular Carcinogenesis, 51, 64–74.
Liesenfeld, D. B., Habermann, N., Owen, R. W., Scalbert, A., & Ulrich, C. M. (2013). Review of mass spectrometry-based metabolomics in cancer research. Cancer Epidemiology and Prevention Biomarkers, 22, 2182–2201.
Lin, W. J., & Chen, J. J. (2013). Class-imbalanced classifiers for high-dimensional data. Brief Bioinformatics, 14, 13–26.
Lindahl, A., Heuchel, R., Forshed, J., Lehtio, J., Lohr, M., & Nordstrom, A. (2017). Discrimination of pancreatic cancer and pancreatitis by LC-MS metabolomics. Metabolomics, 13, 61.
Lu, W., Su, X., Klein, M. S., Lewis, I. A., Fiehn, O., & Rabinowitz, J. D. (2017). Metabolite measurement: Pitfalls to avoid and practices to follow. Annual Review of Biochemistry, 86, 277–304.
Lumbreras, B., Parker, L. A., Porta, M., Pollan, M., Ioannidis, J. P., & Hernandez-Aguado, I. (2009). Overinterpretation of clinical applicability in molecular diagnostic research. Clinical Chemistry, 55, 786–794.
Lumbreras, B., Porta, M., Marquez, S., Pollan, M., Parker, L. A., & Hernandez-Aguado, I. (2008). QUADOMICS: An adaptation of the Quality Assessment of Diagnostic Accuracy Assessment (QUADAS) for the evaluation of the methodological quality of studies on the diagnostic accuracy of ‘-omics’-based technologies. Clinical Biochemistry, 41, 1316–1325.
Lynch, C. J., & Adams, S. H. (2014). Branched-chain amino acids in metabolic signalling and insulin resistance. Nature Reviews Endocrinology, 10, 723.
Makohon-Moore, A., & Iacobuzio-Donahue, C. A. (2016). Pancreatic cancer biology and genetics from an evolutionary perspective. Nature Reviews Cancer, 16, 553–565.
Mayerle, J., Kalthoff, H., Reszka, R., Kamlage, B., Peter, E., Schniewind, B., et al. (2018). Metabolic biomarker signature to differentiate pancreatic ductal adenocarcinoma from chronic pancreatitis. Gut, 67, 128–137.
Mayers, J. R., Wu, C., Clish, C. B., Kraft, P., Torrence, M. E., Fiske, B. P., et al. (2014). Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nature Medicine, 20, 1193–1198.
McCormick, F. C., & Lemoine, N. R. (1998). Molecular basis of pancreatic cancer: Strategies for genetic diagnosis and therapy. In: C. D. Johnson & C. W. Imrie (Eds.), Pancreatic disease: Towards the year 2000 (2nd ed.). New York: Springer.
Moons, K. G., Altman, D. G., Reitsma, J. B., Ioannidis, J. P., Macaskill, P., Steyerberg, E. W., et al. (2015). Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD): Explanation and elaboration. Annals of Internal Medicine, 162, W1–W73.
Napoli, C., Sperandio, N., Lawlor, R. T., Scarpa, A., Molinari, H., & Assfalg, M. (2012). Urine metabolic signature of pancreatic ductal adenocarcinoma by 1H nuclear magnetic resonance: Identification, mapping, and evolution. Journal of Proteome Research, 11, 1274–1283.
Nguyen, V., Hurton, S., Ayloo, S., & Molinari, M. (2015). Advances in pancreatic cancer: The role of metabolomics. JOP: Journal of the Pancreas, 16, 244–248.
Nishiumi, S., Shinohara, M., Ikeda, A., Yoshie, T., Hatano, N., Kakuyama, S., et al. (2010). Serum metabolomics as a novel diagnostic approach for pancreatic cancer. Metabolomics, 6, 518–528.
Oberstein, P. E., & Olive, K. P. (2013). Pancreatic cancer: Why is it so hard to treat? Therapeutic Advances in Gastroenterology, 6, 321–337.
Paglia, G., & Astarita, G. (2017). Metabolomics and lipidomics using traveling-wave ion mobility mass spectrometry. Nature Protocols, 12, 797.
Pannala, R., Basu, A., Petersen, G. M., & Chari, S. T. (2009). New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncology, 10, 88–95.
Parker, L. A., GómezSaez, N., Lumbreras, B., Porta, M., & Hernández-Aguado, I. (2010). Methodological deficits in diagnostic research using ‘-Omics’ technologies: Evaluation of the QUADOMICS tool and quality of recently published studies. PLoS ONE 5, e11419.
Partyka, K., Maupin, K. A., Brand, R. E., & Haab, B. B. (2012). Diverse monoclonal antibodies against the CA 19–9 antigen show variation in binding specificity with consequences for clinical interpretation. Proteomics, 12, 2212–2220.
Patti, G. J., Tautenhahn, R., & Siuzdak, G. (2012). Meta-analysis of untargeted metabolomic data: combining results from multiple profiling experiments. Nature Protocols, 7, 508–516.
Pavlova, N. N., & Thompson, C. B. (2016). The emerging hallmarks of cancer metabolism. Cell Metabolism, 23, 27–47.
Pepe, M. S., Feng, Z., Janes, H., Bossuyt, P. M., & Potter, J. D. (2008). Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: standards for study design. Journal of the National Cancer Institute, 100, 1432–1438.
Perez-Rambla, C., Puchades-Carrasco, L., Garcia-Flores, M., Rubio-Briones, J., Lopez-Guerrero, J. A., & Pineda-Lucena, A. (2017). Non-invasive urinary metabolomic profiling discriminates prostate cancer from benign prostatic hyperplasia. Metabolomics, 13, 52.
Ribeiro, M. T., Singh, S., & Guestrin, C. (2016). “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. arXiv:1602.04938v3.
Rios Peces, S., Diaz Navarro, C., Marquez Lopez, C., Caba, O., Jimenez-Luna, C., Melguizo, C., et al. (2017). Untargeted LC-HRMS-based metabolomics for searching new biomarkers of pancreatic ductal adenocarcinoma: A pilot study. SLAS Discovery, 22, 348–359.
Ritchie, S. A., Akita, H., Takemasa, I., Eguchi, H., Pastural, E., Nagano, H., et al. (2013). Metabolic system alterations in pancreatic cancer patient serum: Potential for early detection. BMC Cancer, 13, 416.
Rodrigues, D., Jeronimo, C., Henrique, R., Belo, L., de Lourdes Bastos, M., de Pinho, P. G., et al. (2016). Biomarkers in bladder cancer: A metabolomic approach using in vitro and ex vivo model systems. International Journal of Cancer, 139, 256–268.
Ryan, D. P., Hong, T. S., & Bardeesy, N. (2014). Pancreatic adenocarcinoma. New England Journal of Medicine, 371, 1039–1049.
Saito, T., & Rehmsmeier, M. (2015). The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS ONE, 10, e0118432.
Sakai, A., Suzuki, M., Kobayashi, T., Nishiumi, S., Yamanaka, K., Hirata, Y., et al. (2016). Pancreatic cancer screening using a multiplatform human serum metabolomics system. Biomarkers in Medicine, 10, 577–586.
Scheubert, K., Hufsky, F., Petras, D., Wang, M., Nothias, L.-F., Dührkop, K., et al. (2017). Significance estimation for large scale metabolomics annotations by spectral matching. Nature Communications, 8, 1494.
Shin, E. J., & Canto, M. I. (2012). Pancreatic cancer screening. Gastroenterology Clinics of North America, 41, 143–157.
Sidaway, P. (2017). Pancreatic cancer: TCGA da ta reveal a highly heterogeneous disease. Nature Reviews Clinical Oncology, 14, 648.
Singh, S., Tang, S. J., Sreenarasimhaiah, J., Lara, L. F., & Siddiqui, A. (2011). The clinical utility and limitations of serum carbohydrate antigen (CA19-9) as a diagnostic tool for pancreatic cancer and cholangiocarcinoma. Digestive Diseases and Science, 56, 2491–2496.
Smialowski, P., Frishman, D., & Kramer, S. (2010). Pitfalls of supervised feature selection. Bioinformatics, 26, 440–443.
Spicer, R. A., & Steinbeck, C. (2017). A lost opportunity for science: Journals promote data sharing in metabolomics but do not enforce it. Metabolomics, 14, 16.
Sugimoto, M., Wong, D. T., Hirayama, A., Soga, T., & Tomita, M. (2010). Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics, 6, 78–95.
Sumner, L. W., Amberg, A., Barrett, D., Beale, M. H., Beger, R., Daykin, C. A., et al. (2007). Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics, 3, 211–221.
Suzuki, M., Nishiumi, S., Kobayashi, T., Sakai, A., Iwata, Y., Uchikata, T., et al. (2017). Use of on-line supercritical fluid extraction-supercritical fluid chromatography/tandem mass spectrometry to analyze disease biomarkers in dried serum spots compared with serum analysis using liquid chromatography/tandem mass spectrometry. Rapid Communications in Mass Spectrometry, 31, 886–894.
Takhar, A. S., Palaniappan, P., Dhingsa, R., & Lobo, D. N. (2004). Recent developments in diagnosis of pancreatic cancer. BMJ, 329, 668–673.
Tanase, C. P., Neagu, M., Albulescu, R., Codorean, E., & Dima, S. O. (2009). Biomarkers in the diagnosis and early detection of pancreatic cancer. Expert Opinion on Medical Diagnostics, 3, 533–546.
Tumas, J., Kvederaviciute, K., Petrulionis, M., Kurlinkus, B., Rimkus, A., Sakalauskaite, G., et al. (2016). Metabolomics in pancreatic cancer biomarkers research. Medical Oncology, 33, 133.
Turakhia, M. P., & Sabatine, M. S. (2017). How we evaluate biomarker studies. JAMA Cardiology, 2, 524–524.
Urayama, S., Zou, W., Brooks, K., & Tolstikov, V. (2010). Comprehensive mass spectrometry based metabolic profiling of blood plasma reveals potent discriminatory classifiers of pancreatic cancer. Rapid Communications in Mass Spectrometry, 24, 613–620.
Usher-Smith, J. A., Sharp, S. J., & Griffin, S. J. (2016). The spectrum effect in tests for risk prediction, screening, and diagnosis. BMJ, 353, i3139.
Vinaixa, M., Samino, S., Saez, I., Duran, J., Guinovart, J. J., & Yanes, O. (2012). A guideline to univariate statistical analysis for LC/MS-based untargeted metabolomics-derived data. Metabolites, 2, 775–795.
Whiting, P. F., Rutjes, A. W., Westwood, M. E., Mallett, S., Deeks, J. J., Reitsma, J. B., et al. (2011). QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Annals of Internal Medicine, 155, 529–536.
Wishart, D. S., Feunang, Y. D., Marcu, A., Guo, A. C., Liang, K., Vázquez-Fresno, R., et al. (2017). HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Research, 46(D1), D608–D617.
Wolpin, B. M., Bao, Y., Qian, Z. R., Wu, C., Kraft, P., Ogino, S., et al. (2013). Hyperglycemia, insulin resistance, impaired pancreatic β-cell function, and risk of pancreatic cancer. JNCI: Journal of the National Cancer Institute, 105, 1027–1035.
Xia, J., Broadhurst, D. I., Wilson, M., & Wishart, D. S. (2013). Translational biomarker discovery in clinical metabolomics: an introductory tutorial. Metabolomics, 9, 280–299.
Xia, J., & Wishart, D. S. (2016). Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr Protoc Bioinformatics, 55(1), 14–10.
Xie, G., Lu, L., Qiu, Y., Ni, Q., Zhang, W., Gao, Y. T., et al. (2015). Plasma metabolite biomarkers for the detection of pancreatic cancer. Journal of Proteome Research, 14, 1195–1202.
Yang, W., Chen, Y., Xi, C., Zhang, R., Song, Y., Zhan, Q., et al. (2013). Liquid chromatography-tandem mass spectrometry-based plasma metabonomics delineate the effect of metabolites’ stability on reliability of potential biomarkers. Analytical Chemistry, 85, 2606–2610.
Yang, W., Yoshigoe, K., Qin, X., Liu, J. S., Yang, J. Y., Niemierko, A., et al. (2014). Identification of genes and pathways involved in kidney renal clear cell carcinoma. BMC Bioinformatics, 15(Suppl 17), S2.
Yin, L., Ge, Y., Xiao, K., Wang, X., & Quan, X. (2013). Feature selection for high-dimensional imbalanced data. Neurocomputing, 105, 3–11.
Zhang, H., Wang, Y., Gu, X., Zhou, J., & Yan, C. (2011). Metabolomic profiling of human plasma in pancreatic cancer using pressurized capillary electrochromatography. Electrophoresis, 32, 340–347.
Zhang, L., Jin, H., Guo, X., Yang, Z., Zhao, L., Tang, S., et al. (2012). Distinguishing pancreatic cancer from chronic pancreatitis and healthy individuals by (1)H nuclear magnetic resonance-based metabonomic profiles. Clinical Biochemistry, 45, 1064–1069.
Zhang, Y., Qiu, L., Wang, Y., Qin, X., & Li, Z. (2014). High-throughput and high-sensitivity quantitative analysis of serum unsaturated fatty acids by chip-based nanoelectrospray ionization-Fourier transform ion cyclotron resonance mass spectrometry: early stage diagnostic biomarkers of pancreatic cancer. Analyst, 139, 1697–1706.
Funding
The Bio-Synergy Research Project of the Ministry of Science, ICT and Future Planning through the National Research Foundation of Korea (NRF-2012M3A9C4048796), the National Research Foundation of Korea (NRF-2017R1E1A2A02022658, NRF-2018R1A5A2024425), and the BK21 Plus Program in 2017.
Author information
Authors and Affiliations
Contributions
SWK and SSH supervised the project. SWK, SSH, and NPL contributed to the study design. NPL, SJY, NHA, TDN, DKL, and YJH searched and collected the data. NPL, SJY, NHA, TDN, DKL, and YJH performed data processing and interpretation. NPL, SJY, NHA, and TDN prepared the first draft of the manuscript. All authors have read, revised critically, and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Long, N.P., Yoon, S.J., Anh, N.H. et al. A systematic review on metabolomics-based diagnostic biomarker discovery and validation in pancreatic cancer. Metabolomics 14, 109 (2018). https://doi.org/10.1007/s11306-018-1404-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-018-1404-2