
Abstract
In this work, we report a comprehensive study of long terminal repeat retrotransposons of Populus trichocarpa. Our research group studied the retrotransposon component of the poplar genome in 2012, isolating 1479 putative full-length elements. However, in that study, it was not possible to identify the superfamily to which the majority of isolated full-length elements belonged. Moreover, during recent years, the genome sequence of P. trichocarpa has been updated, deciphering thek sequences of a number of previously unresolved loci. In this work, we performed a complete scan of the updated version of the genome sequence to isolate full-length retrotransposons based on sequence and structural features. The new dataset showed a reduced number of elements (958), and 21 full-length elements were discovered for the first time. The majority of retroelements belonged to the Gypsy superfamily (57 %), while Copia elements amounted to 41.1 % of the dataset. Full-length elements were dispersed throughout the chromosomes. However, Gypsy and, to a lesser extent, Copia elements accumulated preferentially at putative centromeres. Gypsy elements were more active in retrotransposition than Copia elements, with the exception of during the past million years, in which Copia elements were the most active. Improved annotation procedures also allowed us to determine the specific lineages to which isolated elements belonged. The three Gypsy lineages, Athila, OGRE, and Chromovirus (in the decreasing order), were by far the most abundant. On the other hand, each identified Copia lineage represented less than 1 % of the genome. Significant differences in the insertion age were found among lineages, suggesting specific activation mechanisms. Moreover, different chromosomal regions were affected by retrotransposition in different ages. In all chromosomes, putative pericentromeric regions were filled with elements older than the mean insertion age. Overall, results demonstrate structural and functional differences among plant retrotransposon lineages and further support the view of retrotransposons as a community of different organisms in the genome.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The LTR-retrotransposons (LTR-REs) (i.e. retrotransposons [REs] characterised by two long terminal repeats [LTRs] at the 5′ and 3′ ends) are a ubiquitous component of plant genomes and are especially abundant in species with large genomes. The sequence of a full-length RE includes a portion encoding two proteins, GAG and POL, flanked by two direct repeats, the LTRs, at both ends. The abundance of LTR-REs in the genome is related to their “copy and paste” mode of replication: LTR-REs are transcribed by the RNA polymerase of the host, then retrotranscribed and inserted into the chromosome by enzymes encoded by the RE. Although this mode of replication is one of the main mechanisms leading to genome obesity, other mechanisms, such as illegitimate recombination between LTRs, prevent unlimited growth of genome size, determining DNA loss (Bennetzen and Kellogg 1997; Devos et al. 2002).
The LTR-REs are primarily distinguished into two superfamilies, Ty1-Copia and Ty3-Gypsy, based on the order of the POL protein domains (protease, retrotranscriptase, integrase, and RNAseH) and on sequence similarity. Sequence similarity is also used to classify REs of a superfamily into specific lineages, which can be recognised in every plant species. Usually, DNA sequence conservation is minimal and limited to some coding regions (Wicker et al. 2007). A number of major evolutionary Copia and six Gypsy lineages have been identified (Wicker and Keller 2007; Llorens et al. 2011). The main Gypsy lineages are OGRE/TAT, large LTR-REs with an open reading frame located upstream of the gag gene (Neumann et al. 2003); Athila, first reported in Arabidopsis thaliana (Wright and Voytas 2002); and Chromovirus, a lineage of REs carrying a chromodomain at the 5′ end of the coding portion, which is especially abundant in centromeres (Gorinsek et al. 2004; Llorens et al. 2011). In certain species, chromoviruses were further subdivided into four sublineages (Galadriel, Tekay, CR, and Reina), based on the relative positions of the chromodomain and a polypurine tract (PPT), and on the LTRs length (Weber et al. 2013). On the other hand, Copia REs can belong to many different lineages, the most frequent being AleI/Retrofit/Hopscotch, AleII, Angela, Bianca, Ivana/Oryco, TAR/Tork, and Maximus/SIRE (Wicker and Keller 2007).
It has been shown that RE sequences can impact the expression of nearby genes (Kashkush et al. 2003) by their presence or absence in the cis-regulatory sequences of genes of the host species. Therefore, the identification and characterisation of LTR-REs are a priority in analysing the genome of crop species.
A survey of the dynamics of different LTR-RE superfamilies and lineages in eukaryotic genomes is facilitated by the availability of the whole-genome sequence or, at least, the sequences of large portions of the genome, such as bacterial artificial chromosome (BAC) clones.
Populus trichocarpa has a relatively small genome (550 Mbp), which has been entirely sequenced (Tuskan et al. 2006). Regarding the repetitive component, in their article on poplar genome sequencing, Tuskan et al. (2006) reported that class-I elements (Ty1-Copia-like, Ty3-Gypsy-like, LINEs, and unidentified retroelements) are the most abundant (more than 5000 copies). Poplar retroelements cover approximately 176 Mbp (32 % of the genome), with a prevalence of Gypsy over Copia RE sequences (Tuskan et al. 2006). A database of repetitive elements (RepPop) was subsequently released (Zhou and Xu 2009).
A comprehensive analysis of full-length LTR-RE dynamics in the poplar genome was first reported by Cossu et al. (2012). A full-length LTR-RE can be defined as one that contains two relatively intact LTRs and identified PPT and PBS sites and is also flanked by target-site duplications (TSDs) (Ma et al. 2004), regardless of whether genes encoding enzymes for retrotransposition are present or not. Cossu et al. (2012) identified 1479 putative full-length LTR-REs using a computational approach based on detection of conserved structural features, on building multiple alignments, and on similarity searches. Ty1-Copia full-length elements were more numerous than Ty3-Gypsy ones. Moreover, the majority of LTR-REs lacked diagnostic features and were non-autonomous; hence, they were not assigned to any superfamily and designated as unknown. The LTR-RE remnants were by far more numerous than full-length elements, indicating that during the evolution of poplar, large amplification of these elements was followed by DNA loss. Ty3-Gypsy full-length REs resulted more redundant than Ty1-Copia REs. Retrotransposition occurred with increasing frequency following the separation of Populus sections, with different waves of retrotransposition activity between Ty3-Gypsy and Ty1-Copia elements (Cossu et al. 2012).
Recently, the genome sequence of P. trichocarpa has been largely revised and extended (Slavov et al. 2012). For this reason, and because in our previous study the majority of full-length elements were of unknown superfamilies, we re-examined the entire genome sequence to confirm previously identified elements or find new ones. In addition to using the LTR-Finder tool (Xu and Wang 2007), during this analysis, each element was carefully checked at the structural level in order to find a targeted site duplication of 4–6 bps. Overall, we could identify 958 full-length elements, of which only 18 were not attributed to a superfamily. All the identified elements were extensively annotated, even at lineage level, and new analyses on poplar RE dynamics were performed.
Materials and methods
Update of the P. trichocarpa RE database
The P. trichocarpa full-length LTR-RE dataset (Cossu et al. 2012) was updated. Putative full-length LTR-REs were identified in the 2013 version of the sequenced genome of P. trichocarpa (Tuskan et al. 2006; Slavov et al. 2012) deposited at the NCBI site (WGS project number AARH02, http://www.ncbi.nlm.nih.gov/assembly/GCF_000002775.3) using LTR-FINDER software (Xu and Wang 2007), under default parameters, using the tRNA library of P. trichocarpa, and looking for typical LTR-RE features of the following: being flanked by the dinucleotides TG and CA at 5′ and 3′ ends, respectively; the presence of a TSD of 4–6 nt in length; a putative 15–18-nt primer binding site (PBS) complementary to a tRNA at the end of the putative 5′-LTR; and a 20–25-nt PPT just upstream of the 5′ end of the 3′ LTR.
All putative LTR-REs were manually validated using DOTTER (Sonnhammer and Durbin 1995), verifying the occurrence of LTRs, dinucleotides TG, and CA at respective 5′ and 3′ ends, and TSDs.
The validated LTR-REs were annotated by BLASTX and BLASTN searches against public sequence databases (non-redundant nucleotide and protein NCBI databases and the RepBase database) and an olive RE dataset (Barghini et al. 2015). To limit false-positive detection, we used a fixed E value threshold of E < 10−5 for BLASTN and E < 10−10 for BLASTX. The full-length LTR-REs that were identified as belonging to Gypsy or Copia superfamilies were then used as a reference dataset for further BLASTN searches in order to classify previously unclassified elements.
Analysis of RE protein domains and lineages
The identified full-length elements were analysed using REPEAT EXPLORER (Novak et al. 2013). We performed searches of protein domains (GAG, protease, retrotranscriptase, RNAseH, integrase, and chromodomain) derived from plant mobile elements against the REPBASE-based database (Jurka et al. 2005) to assign full-length elements to specific Gypsy or Copia lineages. The similarity search was performed using the following parameters: minimum similarity 60 %; minimum identity 40 %; proportion of the hit length from the length of the database sequence 0.8; and allowing for maximus three frameshifts. The occurrence of a protein domain within a RE was reported when domain length was at least 50 amino acids. When a domain length was lower than 50 amino acids, the information was used only for RE annotation.
RE distribution along the poplar genome
Each of the 19 linkage groups (LGs) of the currently available version of the poplar genome, as above, were analysed using RepeatMasker against the updated dataset of poplar full-length REs or against Gypsy or Copia sets of elements, separately, under default parameters but -div 20. All LGs were then subdivided into 200-Kbp-long regions using an in-house perl script. The number of masked bases was then counted for each 200,000-bp fragment using another in-house perl script.
RE redundancy estimation
To estimate the redundancy of the LTR-RE set and of the Gypsy and Copia superfamilies and lineages, a large set of Illumina whole-genome shotgun reads (total coverage 8.1x), cut at 75 nt in length, was mapped onto all isolated elements using CLC-BIO Genomic Workbench 6.5.1, with the following parameters: mismatch cost 1, deletion cost 1, insertion cost 1, similarity 0.7, and length fraction 0.7. Since this tool distributes multireads (i.e. those reads that match multiple distinct sequences) randomly, the number of mapped reads to a single sequence cannot indicate its redundancy. On the other hand, if all sequences of a lineage are taken together, the total number of mapped reads (with respect to total genomic reads) reveals the effective redundancy of that lineage.
In other analyses, mapping onto all isolated elements were performed using CLC-BIO Genomic Workbench 6.5.1, with parameters set at different stringencies. Mismatch cost, deletion cost, and insertion cost were fixed at 1, and similarity and length fraction were both fixed at 0.9, 0.7, or 0.5 to obtain high, medium, or low stringencies, respectively.
The redundancy of each single RE sequence in the genome was analysed by mapping poplar DNA reads (corresponding to two genome equivalents) to full-length REs, one by one, using BWA (alignment via Burrows–Wheeler transformation) version 0.7.5a-r405 (Li and Durbin 2009) with the following parameters: bwaaln -t 4 -l 12 -n 4 -k 2 -o 3 -e 3 -M 2 -O 6 -E 3. The resulting single-end mappings were resolved via the “samse” module of BWA, and the output was converted into a “bam” file using SAMtools version 0.1.19 (Li et al. 2009). SAMtools was used to calculate the number of mapped reads for each alignment using the following parameters: samtools view -c -F 4. The redundancy of each sequence was calculated dividing the number of mapped reads by the sequence length.
Insertion time estimation
Retrotransposon insertion age was estimated by comparing the 5′- and 3′-LTRs of each putative full-length LTR-RE (SanMiguel et al. 1998). An in-house perl script was prepared and used for (i) aligning the two LTRs of each RE using the programme “Stretcher” (EMBOSS package, Rice et al. (2000)); (ii) measuring the nucleotide distance between LTRs using the Kimura two-parameter method (K2P, Kimura (1980)) as implemented in the programme “Distmat” (EMBOSS package, Rice et al. (2000)); and (iii) measuring the insertion time of each RE using a synonymous substitution rate that is twice the one calculated for poplar genes by Cossu et al. (2012), according to SanMiguel et al. (1998) and Ma and Bennetzen (2004).
Correlation analyses and analysis of variance (ANOVA) were performed using Graph-Pad software. After subdividing the sequenced genome into 200-Kbp-long regions as above, the mean insertion age of full-length REs lying in each region was calculated. A smoothed curve was then prepared along poplar LGs using Prism5 (Graph-Pad Software Inc., San Diego), using three neighbours to average.
Results
The updated poplar full-length RE dataset
Putative full-length LTR-REs were identified in the updated 2013 version of the sequenced genome of P. trichocarpa (Slavov et al. 2012). In a different manner from the first version of the RE dataset, each RE was further manually validated according to the occurrence of the TSD. This approach allowed the identification of isolated elements (i.e. apparently adjacent to sequences of the host genome), excluding all LTR-REs interrupted by nested structures, which possibly are present in the poplar genome.
Overall, we collected 958 full-length elements (i.e. LTR-REs with TSD and at least one of the typical LTR-RE features [PPT and PBS]). Their sequences are available at the Department of Agriculture, Food, and Environment of Pisa University repository website (http://www.agr.unipi.it/ricerca/plant-genetics-and-genomics-lab/sequence-repository, see also Supplementary material 1). The mean length of identified full-length LTR-REs was 6736 bp, with a large standard deviation (3700 bp). The recorded putative LTRs had a mean length of 816 bp, with large length variability (up to 4451 bp, standard deviation = 774 bp).
Compared to the previous version of the dataset (Cossu et al. 2012), the availability of an improved version of the poplar genome sequence and the use of more restrictive methods for RE identification (including a careful analysis by dot plotting) determined a reduction in the number of identified LTR-REs (from 1479 to 958), especially of the REs whose superfamily was not identified in the previous work; unidentified LTR-REs amounted to 855/1479 (57.8 %) in the previous version and to 18/958 (1.9 %) in the present version of the dataset. In addition to excluding sequences that were not confirmed as REs, 21 new, putative full-length LTR-REs were identified for the first time during the analyses described in this work (i.e. they were absent in the previous version of our dataset as well as in the other existing database of poplar repeated sequences, RepPop [Zhou and Xu 2009]). A summary of the improvements achieved with this analysis is reported as Supplementary Material 2.
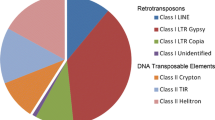
The LTR-REs were first classified as belonging to Ty3-Gypsy or to Ty1-Copia superfamilies by similarity searches against different public RE databases. The full-length REs that were identified as belonging to Gypsy or Copia superfamilies were then used as a reference database for another similarity search. This allowed us to classify almost all full-length elements. Figure 1 shows the number of full-length Ty1-Copia-like, Ty3-Gypsy-like, and unknown LTR-REs identified in the poplar genome. In a different manner from the previous version (Cossu et al. 2012), in this dataset Ty3-Gypsy REs constitute the majority of the REs (546/958), followed by Ty1-Copia-like (394/958).

Number of full-length REs identified in the poplar genome, subdivided into superfamilies (centre). The subdivision of each superfamily into specific lineages is reported for both Copia (left) and Gypsy elements (right)
The collected full-length REs were further analysed for the occurrence of the typical RE protein domains (retrotranscriptase, RNAseH, integrase, protease, and GAG). The similarity to lineage-specific RE protein domains allowed us to subdivide 394 Copia REs into seven lineages and one group whose lineage remained unknown; on the other hand, the Gypsy complement of 546 elements was subdivided into three lineages and one group of unknown elements. Obviously, we cannot exclude that other RE lineages occur in the poplar genome.
Among Copia elements, four lineages were the most frequent (i.e. AleII, Ivana/Oryco, Tork/TAR, and AleI/Retrofit); a few Maximus/SIRE elements also were found, while Angela and Bianca REs were barely represented. Gypsy REs belonged to three main lineages: Athila, Chromovirus, and, at a lesser extent, OGRE/TAT. A number of REs were classified as Gypsy, but they did not show any significant similarity to protein domains of known RE lineages. Hence, they could be classified as LArge Retrotransposon Derivatives (LARDs, Kalendar et al. 2004) and indicated as unknown.
Protein domains (of at least 50 amino acids in length) were recognised in higher numbers in Copia than in Gypsy REs. In Copia REs, POL-related domains were more represented than GAG-related domains. Conversely, GAG domains were slightly more frequent than each POL domain in Gypsy REs (Supplementary material 3).
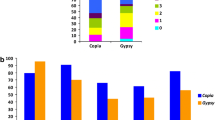
The number of at least 50 amino acids-long domains within each RE (0–5) was counted to deduce the potential autonomy of the RE, and it is reported in Fig. 2, at both the superfamily and lineage levels. It can be observed that the most frequent Copia REs have five protein domains (i.e. they are potentially autonomous); for these elements, a decreasing frequency was observed from those containing five domains to those containing one domain. On the contrary, the most represented group of Gypsy elements showed no protein domains or, at least, protein domains longer than 50 amino acids (i.e. they apparently were non-autonomous elements); in this superfamily, no specific pattern in the number of domains can be inferred.

Number of protein domains per RE, in Copia (left) and Gypsy (right) superfamilies and in the most abundant lineages
Large differences in the frequency of REs with 0–5 detectable domains were observed among lineages (Fig. 3). For example, the vast majority of Ivana/Oryco REs have five protein domains of at least 50 amino acids, while all Maximus/SIRE elements have no long protein domains. The vast majority of Gypsy OGRE/TAT elements are non-autonomous, having 0–2 domains. AleII, Ivana/Oryco, and TAR/Tork lineages (for Copia) and Athila and Chromovirus lineages (for Gypsy) showed a large number of REs with five domains. As reported above, unknown Gypsy elements do not carry any protein domains longer than 50 amino acids.

Distribution of Gypsy, Copia, and putative centromeric sequences along the 19 LGs of the poplar genome. The ratio between Gypsy and Copia relative abundance along LGs is also reported (when Gypsy abundance was higher than that of Copia, it was reported as positive, when it was higher for Copia than for Gypsy as negative)
Chromosomal distribution
Table 1 presents the number of full-length LTR-REs in the 19 LGs of P. trichocarpa. The putative full-length REs identified in our analysis represent 1.70 % of the poplar genome (i.e. a mean of one full-length retroelement every 395,142 bp). The distribution in the 19 LGs ranged from 2.38 % in LGXVI to 0.76 % in LGIX.
The distribution of sequences showing at least 80 % similarity to Gypsy and Copia full-length LTR-REs in the 19 LGs of P. trichocarpa is presented in Fig. 3 together with the distribution of two putative centromeric repeats, C107 and C142 (Rajagopal et al. 1999; Cossu et al. 2012). In the currently available poplar genome sequence, these sequences identify specific regions in each chromosome. In some LGs, they are found at two chromosome positions (LGs IV, V, VI, XII, XVI, and XVIII), suggesting the existence of putative neocentromeres in these chromosomes (see Neumann et al. 2011). It is noteworthy to recall that definition of the centromere position requires biochemical and cytological validation, for example, by BAC in-situ hybridisation (Islam-Faridi et al. 2009).
Poplar REs are generally dispersed throughout the chromosomes (Fig. 3). However, Gypsy REs are usually very abundant at putative centromere (and neocentromere, if any) positions, as frequently observed in plants (Presting et al. 1998; Santini et al. 2002; Neumann et al. 2011). On the contrary, Copia REs are more uniformly dispersed along chromosomes compared to Gypsy REs, although, in some LGs (LGs II, III, VIII, XII, and XIV), the peak redundancy of Copia REs fully matches those of centromeric sequences, suggesting that both superfamilies are prone to accumulation at these chromosome regions.
The ratio between the frequency of Gypsy and Copia REs along chromosomes is also presented in Fig. 3. Generally, Gypsy REs were more represented than Copia REs. This was always true at putative centromere positions, in which the ratios between Gypsy and Copia frequencies can attain values higher than 10 (Table 2). In the other chromosome positions, this ratio is generally higher than 1; Copia elements are more represented than Gypsy ones only in 163 of the 1804 200-Kbp-long fragments, into which the poplar genome was subdivided (Table 2).
Analysis of RE redundancy
The percentage of Illumina reads that match with a class of sequences can be considered an indicator of the proportion of that class in a given genome. After establishing the best parameters for use in the mapping process (i.e. when further relaxation of stringency does not significantly increase the number of mapped reads, Fig. 4a), we used a set of 23,121,470 50-nt-long Illumina sequences of P. trichocarpa (Slavov et al. 2012) to map the set of full-length REs and found that identified full-length REs were mapped by 3,320,456 reads, corresponding to 14.36 % of the genome. Of these, 572,285 mapped to Copia REs (corresponding to 2.48 %), 2,706,615 to Gypsy REs (11.71 %), and 41,556 to unidentified LTR-REs (0.18 %). The ratio between number of mapped reads of Gypsy and Copia REs in the genome was 4.74. Since the ratio between the number of identified full-length elements of Gypsy and Copia superfamilies was 1.39 (546/394), and poplar Gypsy elements were on average 1.68-fold long compared to Copia ones (8180 vs. 4859 bp), this indicates that Gypsy elements are generally more redundant in the genome than Copia ones, especially due to the large number of Gypsy-related RE remnants (i.e. not full-length elements).

a Number of mapped Illumina reads on sets of full-length REs belonging to different lineages at different stringency parameters (see Materials and methods) and b percentages of genomic reads for each RE lineage (calculated at medium stringency [0.7_0.7])
Mapping results of the different RE lineages are summarised in Fig. 4b. The three Gypsy lineages are by far the most redundant, with Athila REs prevailing over OGRE and Chromovirus ones. No single Copia lineage represented more than 1 % of the genome, with three over six lineages that resulted barely represented (Angela, Maximus, and Bianca).
The redundancy was also measured for each full-length RE, mapping REs one by one. The distribution of the number of matching reads per kilobase is reported in Fig. 5 for each RE superfamily and each lineage. The majority of REs showed low numbers of mapping reads; only 16 Copia and 16 Gypsy REs are mapped by more than 120 reads per kilobase (Fig. 5). Often, clear-cut differences in redundancy medians were observed among lineages.

Number of reads per Kbp mapped on individual full-length REs, distinguished into different RE superfamilies or lineages. For each superfamily or lineage, bars represent the median. Tukey’s tests were performed separately among superfamilies, among Copia, and among Gypsy lineages: groups sharing the same letter are not significantly different at p < 0.05
Analysis of RE insertion time
The LTR-RE insertion time can be estimated based on the occurrence of nucleotide substitutions between the LTRs, which should be identical at the retroelement insertion time, using a nucleotide substitution rate suitable for such elements (SanMiguel et al. 1998; Ma and Bennetzen 2004).
Based on the synonymous substitutions between orthologous cDNA sequences of Populus alba and P. trichocarpa and on the estimation of the age at which these two species separated, a synonymous substitution rate of 2.36 × 10−9 substitutions per year was estimated (Cossu et al. 2012). Since it has been suggested that mutation rates for LTR-REs may be approximately twofold higher than silent site mutation rates for protein-coding genes (Xu and Wang 2007), a substitution rate per year of 4.72 × 10−9 was used in our calculations of LTR-RE insertion dates.
When the entire RE set was taken into account, the nucleotide distance (K) between sister LTRs showed large variation among REs, representing a maximum time span of 49 million years (MY). The putative mean age of analysed LTR-REs is 6.1 MY, with much variability (standard deviation = 7.3 MY). The distribution of full-length LTR-REs, according to their putative insertion date, is presented in Fig. 6. Since the most ancient LTR-REs should have accumulated the largest variations in their sequences (being not recognised by LTR-FINDER), the frequency of LTR-REs with older insertion dates decreases progressively, as expected. Analysis of the insertion date profiles provides evidence for overlapping among retrotransposition waves of Gypsy and Copia full-length LTR-REs (Fig. 6). When taking into consideration the past 20 MY (i.e. after the separation of poplar sections), it can be noted that Gypsy elements have been more active in retrotransposition than Copia elements, with the exception of the past 1 MY, during which Copia elements have been more active than Gypsy ones.

Distributions of Copia, Gypsy, and unknown full-length LTR-REs according to their estimated insertion ages (MY). For each superfamily the mean insertion age is reported
The mean insertion dates of the most numerous Gypsy and Copia lineages show that different lineages underwent amplification in different time spans (Fig. 7), as also indicated by one-way ANOVA. For example, TAR/Tork Copia elements are significantly younger than OGRE/TAT, Chromovirus, and Athila REs, suggesting specific activation bursts for the different lineages.

Box and whiskers plot of putative insertion ages (MY) of the most represented poplar RE lineages. The boxes represent the 25–75 %, whiskers represent the whole range of values, and lines in the box represent the medians of the distributions. For each lineage, the mean (±SE) is reported. Lineages sharing the same letter are not significantly different at p < 0.05 according to Tukey’s test
The profiles of LTR-RE insertion ages along the 19 LGs are presented in Fig. 8 and Supplementary material 4. Comparisons among the profiles and the mean poplar RE insertion age suggest that retrotransposition occurred at different times in the different chromosomes and chromosome positions, although the occurrence of changes in mutation rate in different chromosome positions cannot be ruled out.

Mean insertion ages of full-length REs in 200-Kbp-long regions along the 19 poplar LGs. Each point represents one region, and distributions are represented by smoothed curves obtained by averaging three neighbours’ values. The horizontal line represents the poplar RE mean insertion age: grey indicates regions with REs older than the mean; orange indicates regions with REs younger than the mean. The arrow represents the putative position of the centromere as indicated by the occurrence in that position of centromeric repeats
In all chromosomes, putative pericentromeric regions are filled with elements older than the mean insertion age (Fig. 8). This is especially true for Gypsy REs (Supplementary material 4). Regarding Copia REs (Supplementary material 4), in some cases a near-complete chromosome is filled with old elements (e.g. LGVI); others are filled with young elements (e.g. LGs IX, X, and XVII), likely reflecting the most recent mobilisation wave of these REs.
Correlation between RE redundancy and insertion time
In Fig. 9a, the correlation between transposition activity of an RE lineage (as indicated by the mean insertion age of elements belonging to one and the same lineage) and the redundancy of that lineage in the poplar genome is shown. It can be observed that correlation is not significant for both Copia and Gypsy lineages.

a Correlation between mean number of mapped reads per Kbp and mean insertion age and b correlation between median insertion age and whole RE sequence conservation (indicated by 0.7_0.7/0.9_0.9 redundancy ratio [see Materials and methods]: the higher is the value, the less the sequence is conserved) in the most abundant Copia and Gypsy lineages. Each point represents a lineage
To gain insight of RE dynamics of different lineages, we also analysed the curves of Fig. 4a, in which relaxing stringency parameters imply an increase in the number of mapped reads until a plateau is reached. The ratio between the redundancy calculated at medium stringency (with parameters 1_1_1_0.7_0.7, see Materials and methods) and high stringency (1_1_1_0.9_0.9) for a given lineage should indicate the degree of sequence conservation of the elements belonging to that lineage—the lower the ratio, the higher the sequence conservation.
We then studied the correlations between sequence conservation (of full-length REs) and mean insertion age among RE lineages (calculated comparing LTRs, Fig. 9b). The results were highly significant for Copia lineages, of which the most ancient lineages were also the least conserved, as expected. Interestingly, such a relationship was not significant for Gypsy lineages.
Discussion
In this work, we have updated the previously produced poplar LTR-RE dataset (Cossu et al. 2012) based on the last version of the sequenced genome of P. trichocarpa (Slavov et al. 2012). After careful identification and annotation of full-length LTR-REs, the new dataset showed a reduced number of elements and 21 full-length elements were discovered for the first time.
In the updated version of the dataset, Gypsy elements were more redundant in the genome compared to Copia ones. The ratio between redundancy of Gypsy and Copia elements is 4.74. Higher redundancy of Gypsy REs compared to Copia ones already has been reported in P. trichocarpa (Tuskan et al. 2006; Cossu et al. 2012). In angiosperms, different ratios between Gypsy and Copia RE frequencies were reported, ranging from 5:1 in papaya to 1:2 in grapevine (Vitte et al. 2014). Species of the Gossypium genus show a variable proportion of Gypsy versus Copia elements, with Gypsy elements prevailing in species with larger genome sizes (Hawkins et al. 2006).
Full-length elements are distributed differently throughout different LGs, with a percentage per LG ranging from 0.76 (in LG IX) to 2.38 (in LG XVI) (i.e. more than threefold variation). Considering not only full-length LTR-REs but also their related remnants, both Copia and Gypsy REs are widespread along poplar chromosomes; however, a preferential localisation of Gypsy REs is observed in proximity of putative centromeres, as expected (Neumann et al. 2011).
Analysis of sister LTR similarity indicates that, in poplar, both Gypsy and Copia REs have been active during the same period. Obviously, the estimation of insertion time by the number of mutations in sister LTRs is subject to error, because it assumes that the same mutation rates operate in all retroelements and chromosome positions, although this was not proven to be true in, for example, the genus Oryza (Zuccolo et al. 2010). However, this method appears the most suitable for studying LTR-RE dynamics, especially when comparing different superfamilies or lineages within a species. In addition, it is to be considered that all those REs interrupted by other elements (i.e. presumably those older than inserted ones) are not included in our sample.
Using this method, all the identified full-length elements appear to be mobilised in a time span of 49 MY, although it can be presumed that more ancient REs have accumulated too many mutations among sister LTRs to be still recognisable as full-length elements.
The mean poplar RE insertion date is 6.11 MY, i.e. LTR-REs are generally older than those analysed in herbaceous species such as rice, wheat, or sunflower (Ma and Bennetzen 2004; Charles et al. 2008; Buti et al. 2011). On the other hand, in other woody, perennial species as Norway spruce and olive (Nystedt et al. 2013; Barghini et al. 2015), the majority of LTR-REs resulted even older than those analysed in poplar. Probably, this difference between annual and perennial species is related to the different growth habit, being the generation time much larger in woody than herbaceous plants. In the case of Norway spruce, it has also been hypothesised that the occurrence of ancient LTR-REs is related to the absence of efficient recombination mechanisms; in herbaceous species, these mechanisms counteracted genome expansion resulting in younger elements remaining following recent bursts of activity (Nystedt et al. 2013).
The mean insertion date of poplar Copia full-length REs is lower than that of Gypsy REs, as previously observed (Cossu et al. 2012). The insertion date profiles indicate that Copia and Gypsy REs have experienced similar time courses, with Gypsy REs having replicated more than Copia ones, except during the past million years. Copia and Gypsy amplification histories during the evolution of the host have been described in other species, such as, for example, wheat, in which Copia and Gypsy superfamilies are differently represented in the A and B genomes (Charles et al. 2008); rice (Ma et al. 2004); grapevine (Moisy et al. 2008); maize (Brunner et al. 2005; Wang and Dooner 2006); olive (Barghini et al. 2014); sunflower (Ungerer et al. 2009; Vukich et al. 2009; Cavallini et al. 2010; Buti et al. 2011; Natali et al. 2013); and Norway spruce (Nystedt et al. 2013). In general, analysis of plant genomes in a phylogenetic context reveals scarce congruence in RE content and highlights differences in the success of different RE types (Vitte et al. 2014).
The distribution of putative insertion times along chromosomes reveals the existence of chromosome regions that have experienced RE insertions at different times. Putative centromeric regions might have been colonised in more ancient times than non-centromeric ones, or different mean RE insertion age in these regions could reflect the reduced/suppressed recombination activity in centromeric regions (Tian et al. 2009). When separating Gypsy and Copia elements, the occurrence of large regions, in which colonisation has been more recent than the mean insertion time, can be observed for both superfamilies.
In addition to a re-evaluation of poplar RE superfamilies, present analyses allowed for studying the occurrence and dynamics of seven Copia and three Gypsy lineages. Significant differences have been observed among lineages regarding redundancy, sequence conservation, and mean insertion time. Maximus/SIRE and TAR/Tork Copia REs are more redundant than the other Copia lineages, and Athila is the most redundant Gypsy lineage, followed by Gypsy elements whose lineage could not be identified because protein domains were absent or too short.
Regarding insertion ages, calculated analysing the similarity between LTRs, differences between lineages were found. Lineage mean insertion ages are generally paralleled by sequence conservation of the full-length element, mutually supporting each other. For example, Athila, OGRE/Tat, and Chromovirus REs are the most variable and the most ancient lineages; Maximus/SIRE REs are mostly very young and show the largest sequence conservation.
The correlation between lineage mean insertion age and sequence conservation was not significant for the Gypsy superfamily. A possible explanation is that REs of the Gypsy lineages are differently prone to accumulating nucleotide substitutions between LTRs and the RE internal region. In actuality, the ratio between the redundancy calculated at medium stringency and high stringency is by far the highest for three of the four analysed Gypsy lineages, indicating a general tendency of Gypsy elements to accumulate more sequence variation than Copia ones.
Interestingly, sequences of Gypsy unknown elements seem to be the most conserved compared to the Gypsy lineages and also show the lowest mean insertion age (though such difference is not significant). It is presumable that such REs started their transposition activity recently to constitute a new lineage.
When treating lineages separately, absence of correlation between insertion age and redundancy can be observed for both Copia and Gypsy superfamilies. This suggests that LTR-REs lineages have experienced different rates of DNA loss, which were higher in the more ancient lineages compared to the youngest ones. Alternatively, it is possible that RE lineages concurrently started their replication activity, even if such activity showed different insertion time peaks.
In conclusion, our analyses report a re-evaluation and new data on RE dynamics in the evolution of the poplar genome. In general, RE dynamics are similar, including birth through transposition, silencing, and then death by both random mutation and possibly deletion from the genome (Baucom et al. 2009a, b). However, our data support the view that RE dynamics can be different even within superfamilies, i.e. among RE lineages. In this sense, if plant REs can be considered a community of different organisms in a genome (Venner et al. 2009), we can consider RE superfamilies as “species”, and RE lineages, characterised by differences in protein domain sequences and evolutionary history, as “subspecies”, differently adapting to the “ecosystem” in which the REs interact and compete (Le Rouzic et al. 2007).
Abbreviations
- RE:
-
Retrotransposon
- LTR-RE:
-
LTR-retrotransposon
- LTR:
-
Long terminal repeat
- MY:
-
Million of years
References
Barghini E, Natali L, Cossu RM, Giordani T, Pindo M, Cattonaro F, Scalabrin S, Velasco R, Morgante M, Cavallini A (2014) The peculiar landscape of repetitive sequences in the olive (Olea europaea L.) genome. Genome Biol Evol 6:776–791
Barghini E, Natali L, Giordani T, Cossu RM, Scalabrin S, Cattonaro F, Šimková H, Vrána J, Doležel J, Morgante M, Cavallini A (2015) LTR retrotransposon dynamics in the evolution of the olive (Olea europaea) genome. DNA Res 22:91–100
Baucom RS, Estill JC, Chaparro C, Upshaw N, Jogi A, Deragon JM, Westerman RP, SanMiguel PJ, Bennetzen JL (2009a) Exceptional diversity, non-random distribution, and rapid evolution of retroelements in the B73 maize genome. PLoS Genet 5:e1000732. doi:10.1371/journal.pgen.1000732
Baucom RS, Estill JC, Leebens-Mack J, Bennetzen JL (2009b) Natural selection on gene function drives the evolution of LTR retrotransposon families in the rice genome. Genome Res 19:243–254
Bennetzen JL, Kellogg EA (1997) Do plants have a one-way ticket to genomic obesity? Plant Cell 9:1509–1514
Brunner S, Fengler K, Morgante M, Tingey S, Rafalski A (2005) Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 17:343–360
Buti M, Giordani T, Cattonaro F, Cossu RM, Pistelli L, Vukich M, Morgante M, Cavallini A, Natali L (2011) Temporal dynamics in the evolution of the sunflower genome as revealed by sequencing and annotation of three large genomic regions. Theor Appl Genet 123:779–791
Cavallini A, Natali L, Zuccolo A, Giordani T, Jurman I, Ferrillo V, Vitacolonna N, Sarri V, Cattonaro F, Ceccarelli M, Cionini PG, Morgante M (2010) Analysis of transposons and repeat composition of the sunflower (Helianthus annuus L.) genome. Theor Appl Genet 120:491–508
Charles M, Belcram H, Just J, Huneau C, Viollet A, Couloux A, Segurens B, Carter M, Huteau V, Coriton O, Appels R, Samain S, Chalhoub B (2008) Dynamics and differential proliferation of transposable elements during the evolution of the B and A genomes of wheat. Genetics 180:1071–1086
Cossu RM, Buti M, Giordani T, Natali L, Cavallini A (2012) A computational study of the dynamics of LTR retrotransposons in the Populus trichocarpa genome. Tree Genet Genomes 8:61–75
Devos KM, Brown JKM, Bennetzen JL (2002) Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res 12:1075–1079
Gorinsek B, Gubensek F, Kordis D (2004) Evolutionary genomics of chromoviruses in eukaryotes. Mol Biol Evol 21:781–798
Hawkins JS, Kim HR, Nason JD, Wing RA, Wendel JF (2006) Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res 16:1252–1261
Islam-Faridi MN, Nelson CD, DiFazio SP, Gunter LE, Tuskan GA (2009) Cytogenetic analysis of Populus trichocarpa—ribosomal DNA, telomere repeat sequence, and marker-selected BACs. Cytogenet Genome Res 125:74–80
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J (2005) Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110:462–467
Kalendar R, Vicient CM, Peleg O, Anamthawat-Jonsson K, Bolshoyb A, Schulman AH (2004) Large retrotransposon derivatives: abundant, conserved but nonautonomous retroelements of barley and related genomes. Genetics 166:1437–1450
Kashkush K, Feldman M, Levy AA (2003) Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat Genet 33:102–106
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Le Rouzic A, Dupas S, Capy P (2007) Genome ecosystem and transposable elements species. Gene 390:214–220
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25:2078–2079
Llorens C, Futami R, Covelli L, Domínguez-Escribá L, Viu JM, Tamarit D, Aguilar-Rodríguez J, Vicente-Ripolles M, Fuster G, Bernet GP, Maumus F, Munoz-Pomer A, Sempere JM, Latorre A, Moya A (2011) The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res 39:D70–D74
Ma J, Bennetzen JL (2004) Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci U S A 101:12404–12410
Ma J, Devos KM, Bennetzen JL (2004) Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res 14:860–869
Moisy C, Garrison KE, Meredith CP, Pelsy F (2008) Characterization of ten novel Ty1/Copia-like retrotransposon families of the grapevine genome. BMC Genomics 9:469
Natali L, Cossu RM, Barghini E, Giordani T, Buti M, Mascagni F, Morgante M, Gill N, Kane NC, Rieseberg L, Cavallini A (2013) The repetitive component of the sunflower genome as revealed by different procedures for assembling next generation sequencing reads. BMC Genomics 14:686
Neumann P, Požárková D, Macas J (2003) Highly abundant pea LTR-retrotransposon Ogre is constitutively transcribed and partially spliced. Plant Mol Biol 53:399–410
Neumann P, Navrátilová A, Koblížková A, Kejnovský E, Hřibová E, Hobza R, Widmer A, Doležel J, Macas J (2011) Plant centromeric retrotransposons: a structural and cytogenetic perspective. Mob DNA 2:4
Novak P, Neumann P, Pech J, Steinhaisl J, Macas J (2013) RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next generation sequence reads. Bioinformatics 29:792–793
Nystedt B, Nathaniel R, Street NR et al (2013) The Norway spruce genome sequence and conifer genome evolution. Nature 497:579–584
Presting GG, Malysheva L, Fuchs J, Schubert I (1998) A Ty3/gypsy retrotransposon-like sequence localized to the centromeric regions of cereal chromosomes. Plant J 16:721–728
Rajagopal J, Das S, Khurana DK, Srivastava PS, Lakshmikumaran M (1999) Molecular characterization and distribution of a 145-bp tandem repeat family in the genus Populus. Genome 42:909–918
Rice P, Longden I, Bleasby A (2000) EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet 16:276–277
SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL (1998) The paleontology of intergene retrotransposons of maize. Nat Genet 20:43–45
Santini S, Cavallini A, Natali L, Minelli S, Maggini F, Cionini PG (2002) Ty1/Copia- and Ty3/Gypsy-like DNA sequences in Helianthus species. Chromosoma 111:192–200
Slavov GT, DiFazio SP, Martin J, Schackwitz W, Muchero W, Rodgers-Melnick E, Lipphardt MF et al (2012) Genome resequencing reveals multiscale geographic structure and extensive linkage disequilibrium in the forest tree Populus trichocarpa. New Phytol 196:713–725
Sonnhammer EL, Durbin R (1995) A dot-matriprogram with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167:GC1–GC10
Tian Z, Rizzon C, Du JC, Zhu L, Bennetzen JL, Jackson SA, Gaut BS, Ma J (2009) Do genetic recombination and gene density shape the pattern of DNA elimination in rice long terminal repeat retrotransposons? Genome Res 19:2221–2230
Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S et al (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604
Ungerer MC, Strakosh SC, Stimpson KM (2009) Proliferation of Ty3/gypsy-like retrotransposons in hybrid sunflower taxa inferred from phylogenetic data. BMC Biol 7:40
Venner S, Feschotte C, Biemont C (2009) Dynamics of transposable elements: towards a community ecology of the genome. Trends Genet 25:317–323
Vitte C, Fustier MA, Alix K, Tenaillon MI (2014) The bright side of transposons in crop evolution. Brief Funct Genomics. doi:10.1093/bfgp/elu002
Vukich M, Schulman AH, Giordani T, Natali L, Kalendar R, Cavallini A (2009) Genetic variability in sunflower (Helianthus annuus L.) and in the Helianthus genus as assessed by retrotransposon-based molecular markers. Theor Appl Genet 119:1027–1038
Wang Q, Dooner HK (2006) Remarkable variation in maize genome structure inferred from haplotype diversity at the bz locus. Proc Natl Acad Sci U S A 103:17644–17649
Weber B, Heitkam T, Holtgräwe D, Weisshaar B, Minoche AE, Dohm JC, Himmelbauer H, Schmidt T (2013) Highly diverse chromoviruses of Beta vulgaris are classified by chromodomains and chromosomal integration. Mob DNA 4:8
Wicker T, Keller B (2007) Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res 17:1072–1081
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Wright DA, Voytas DF (2002) Athila4 of Arabidopsis and Calypso of soybean define a lineage of endogenous plant retroviruses. Genome Res 12:122–131
Xu Z, Wang H (2007) LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35:W265–W268
Zhou F, Xu Y (2009) RepPop: a database for repetitive elements in Populus trichocarpa. BMC Genomics 10:14
Zuccolo A, Sebastian A, Yu Y, Jackson S, Rounsley S, Billheimer D, Wing RA (2010) Assessing the extent of substitution rate variation of retrotransposon long terminal repeat sequences in Oryza sativa and Oryza glaberrima. Rice 3:242–250
Acknowledgments
Research work was funded by the Department of Agriculture, Food, and Environment, University of Pisa, project PLANTOMICS.
Data archiving statement
All sequences described in this work were isolated from the 2014 version of the sequenced genome of P. trichocarpa (Tuskan et al. 2006; Slavov et al. 2012), deposited at NCBI (project number AARH02, http://www.ncbi.nlm.nih.gov/assembly/GCF_000002775.3). Coordinates and annotation of each sequence are reported in the Supplementary Material 1. Sequences are also available at the Department of Agriculture, Food, and Environment of Pisa University repository website (http://www.agr.unipi.it/Sequence-Repository.358.0.html).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Ingvarsson
This article is part of the Topical Collection on Genome Biology
L. Natali and R. M. Cossu contributed equally to this work.
Rights and permissions
About this article

Cite this article
Natali, L., Cossu, R.M., Mascagni, F. et al. A survey of Gypsy and Copia LTR-retrotransposon superfamilies and lineages and their distinct dynamics in the Populus trichocarpa (L.) genome. Tree Genetics & Genomes 11, 107 (2015). https://doi.org/10.1007/s11295-015-0937-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-015-0937-z