
Abstract
With increased attention to excellent biocatalysts, evolving methods based on nature or unnatural amino acid (UAAs) mutagenesis have become an important part of enzyme engineering. The emergence of powerful method through expanding the genetic code allows to incorporate UAAs with unique chemical functionalities into proteins, endowing proteins with more structural and functional features. To date, over 200 diverse UAAs have been incorporated site-specifically into proteins via this methodology and many of them have been widely exploited in the field of enzyme engineering, making this genetic code expansion approach possible to be a promising tool for modulating the properties of enzymes. In this context, we focus on how this robust method to specifically incorporate UAAs into proteins and summarize their applications in enzyme engineering for tuning and expanding the functional properties of enzymes. Meanwhile, we aim to discuss how the benefits can be achieved by using the genetically encoded UAAs. We hope that this method will become an integral part of the field of enzyme engineering in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Enzymes are important biocatalysts that catalyze a variety of chemical reactions found in nature, which have been extensively applied in the field of fine chemical, agrochemical and pharmaceutical (Naowarojna et al. 2021; Zheng and Kwon 2012). Owing to the growing market demand for biocatalysts with ideal properties such as high activity, stability and selectivity, enzyme engineering techniques are going on to be applied to modulate their properties for industrial applications (Zeymer and Hilvert 2018). Although numerous achievements have been made through traditional techniques such as directed evolution, they are limited to only 20 proteinogenic amino acids building blocks for changing biochemical properties and physiological functions of enzymes (Gargiulo and Soumillion 2021). To overcome such limitations, the development of more innovative methodologies is essential to give rapid access to made-to-order biocatalysts with desired functions.
With unlocking of two rare proteinogenic amino acids (selenocysteine, and pyrrolysine) in some organisms, the concept that proteins composed of only 20 natural amino acids (NAAs) was rethought (Nikic-Spiegel 2020). Replacing the NAAs with unnatural amino acids (UAAs) containing various chemical groups (such as bipyridine, alkynyl, azide) has emerged as a powerful mean to evolve enzymes with ideal physicochemical properties and biological functions (Drienovská et al. 2020; Gao et al. 2019; Jin et al. 2019; Mayer 2019). Generally, incorporation of UAAs can be achieved by two different manners including residue-specific incorporation and site-specific incorporation (Won et al. 2019), which are based on “misacylation” of tRNA with UAAs (Agostini et al. 2017).
The residue-specific manner exploits endogenous translation machinery of auxotrophic host cells to mischarge UAAs into proteins in the absence of the cognate amino acid (Link et al. 2006). Notably, the most important prerequisite for residue-specific incorporation is that UAAs must be recognized by endogenous aminoacyl tRNA synthetase (aaRS), enabling its use by the native translational machinery; meanwhile, intracellular cognate amino acids of auxotrophic host cells is removed extremely to avoid the competition with the UAAs (isostructural analogs) (Agostini et al. 2017). The residue-specific manner is straightforward via selective pressure incorporation and allows the global replacement of one cognate amino acid with one UAA. The most prominent characteristic of this manner is the multi-site incorporation of UAA into the enzyme, resulting in the synergistic effects (favorable or detrimental). To date, many successful cases have been reported through residue-specific manner to evolve enzymes. For example, the fluorinated ω-transaminase (FY-ω-TA) displayed improved thermostability and organic solvent tolerance by the global replacement of tyrosine with 3-fluorotyrosine (Deepankumar et al. 2014). Although the residue-specific incorporation of UAAs seems as an amazing strategy for manipulating biological functions and properties, its utility for engineering enzymes is still limited since it often cause large perturbations in the folded structure (Won et al. 2019). In addition, residue-specific incorporation is restricted to the chemical structure of UAAs that must be highly similar to NAAs and required available auxotrophic host cells that can use these UAAs in the absence of cognate NAAs (Agostini et al. 2017).
A powerful alternative is the site-specific manner, which allows diverse chemistries to be precisely introduced into a protein of interest (Wang and Schultz 2002). This method relies on introducing an orthogonal translation system (OTS) into the host cells for the incorporation of UAAs (Ravikumar et al. 2015). The OTS requires an orthogonal aaRS/tRNA pair which is specific for UAAs in response to the reassigned codon. Compared with residue-specific manner, the site-specific manner enables the incorporation of a UAA into the target protein with high fidelity. For instance, the incorporation of O-methyl-L-tyrosine into dihydrofolate reductase exhibited the fidelity of translation up to over 99% (Wang et al. 2001). The site-specific incorporation of UAAs was an innovative way for engineering of enzymes. In this review, we offered a brief introduction of site-specific incorporation of UAA and highlighted its application in enzyme engineering.
Site-specific incorporation of UAAs
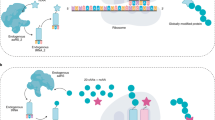
In the native translation machinery, a transfer RNA (tRNA) is aminoacylated specifically with its cognate amino acid by the corresponding aaRS and then delivered to the ribosome for peptide chain extension (Liu and Schultz 2010). The site-specific incorporation of UAAs relies on an engineered translation system which is orthogonal to the native one to warrant a reassigned codon to specify the target UAAs (Fig. 1) (Wals and Ovaa 2014). In the orthogonal system, the exploitation of the desirable orthogonal aaRS/tRNA pair with reassigned codon has been a challenge. An ideal orthogonal aaRS/tRNA pair is that imported aaRS can effectively charge cognate tRNA with a given UAA in response to a unique codon while not cross-react with native pairs (Nikic-Spiegel 2020). To avoid the competition with endogenous tRNA, the reassignment of the stop codons including amber (TAG), ochre (TAA) and opal (TGA) is regarded as a good strategy which provide the stop signal to terminate the translation process (Kisselev et al. 2003) but not used for encoding any of the NAAs in the endogenous translation systems (Dumas et al. 2015). Typically, the amber codon has attracted a great deal of attention for UAAs incorporation due to low abundance in both prokaryotes and eukaryotes. In addition, the use of opal and ochre stop codons, as well as quadruplet codons has also been reported, especially in the application for incorporation of multiple distinct UAAs into one protein (Neumann et al. 2010; Wan et al. 2010; Yu et al. 2015).

Schematic representation of UAA incorporation using an orthogonal translation system by amber codon suppression. aaRS, aminoacyl tRNA synthetase; o-aaRS, orthogonal aminoacyl tRNA synthetase; o-tRNA, orthogonal tRNA; Amino acids, natural (circle) and unnatural (star)
The orthogonal aaRS/tRNA pair is a prerequisite to achieve high efficiency and specificity for UAAs incorporation. To date, numerous orthogonal aaRS/tRNA pairs such as the TyrosylRS/tRNA pair from Methanocaldococcus jannaschi (MjTyrRS/tRNACUA pair), the TyrosylRS/tRNA pair from E. coli (EcTyrRS/tRNACUA pair), the LeucylRS/tRNA pairs from E. coli (EcLeuRS/tRNACUA pair) and the pyrrolysylRS/tRNA pair from Methanosarcina mazei (MmPylRS/tRNACUA pair) and Methoansoarcina barkeri (MbPylRS/tRNACUA pair) have been commonly applied (Chin 2017; Krauskopf and Lang 2020). In general, the orthogonal aaRS/tRNA pairs are only applicable to the corresponding hosts. The MjTyrRS/tRNACUA pair is suitable for E. coli system whereas the EcTyrRS/tRNACUA pair and EcLeuRS/tRNACUA pair are fit for eukaryotes. Comparatively, the PylRS/tRNACUA pair manifests more flexibility due to its orthogonality in both prokaryotes and eukaryotes (Hancock et al. 2010; Wan et al. 2014). In addition, the orthogonal pairs can be engineered for UAAs incorporation by successive rounds of positive selection (based on the chloramphenicol-resistance gene) and negative selection (based on lethal barnase gene) (Young and Schultz 2010). With these wild-type or evolved orthogonal aaRS/tRNA pairs, more than 200 UAAs could successfully be introduced into proteins, which now is becoming a valuable tool kit in the application of protein engineering and enzyme engineering (Vargas-Rodriguez et al. 2018).
Enzyme engineering via site-specific incorporation method
In addition to traditional enzyme engineering techniques, genetically encoded UAAs incorporation has emerged as a powerful technology to open new avenues for improving enzyme catalytic properties, exploring enzyme mechanisms and even creating enzymes with new catalytic activity, explicitly delivering their values for a variety of desired applications in biocatalysis (Gargiulo and Soumillion 2021; Mayer et al. 2019). In the following text, an overview of many successful attempts of UAAs mutagenesis-based engineering enzymes is presented and these examples are grouped based on the effect on enzyme properties such as enhanced activity, enhanced stability and altered selectivity, which are also displayed in Table 1. The chemical structures of UAAs used in these examples are mentioned by number (bold font); structures 1–24 is depicted in Fig. 2. In addition, we described a schematic representation of the distribution of introduced UAAs into different positions of enzymes (I–VI) which are also displayed in Fig. 3.

The structures of UAAs discussed in this Minireview. p-nitrophenylalanine (pNF),1; L-(7-hydroxycoumarin-4-yl) ethylglycine (Hco), 2; Nδ-methyl histidine (NMH), 3; 3-chloro-L-tyrosine (ClTyr), 4; 3-methoxytyrosine (OMeY), 5; p-acrylamido-phenylalanine (AcrF), 6; 3-bromo-L-tyrosine, 7; 4-benzoyl-L-phenylalanine (pBzF), 8; O-(3bromoethyl)-L-tyrosine (BprY), 9; O-(4-mercaptobutyl)-L-tyrosine (SbuY), 10; p-amino-phenylalanine (pAmF), 11; p-acetyl-phenylalanine (pAcF), 12; 3-(2-napthyl) alanine (NapA), 13; O-benzyl-tyrosine (OBnY), 14; O-tert-butyl-L-tyrosine (BuOF), 15; 4-cyanophenylalanine (CNF), 16; 4-methoxy-L-phenylalanine (OMeF), 17; 4-phenyl-L-phenyalanine, 18; o-chlorophenylalanine, 19; o-bromophenylalanine (oBrF), 20; p-bromophenylalanine (pBrF), 21; (2,2’-bipyridin-5yl)alanine (BpyA), 22; p-azido-L-phenylalanine, 23; L-3,4-dihydroxyphenylalanine (DOPA), 24

Schematic representation of the distribution of genetically encoded UAAs in different positions of enzymes
Engineering of enzyme with improved activity
The catalytic capacity of enzymes has attracted great interest for chemists and biologists during the past decades. The main source of catalytic power of an enzyme is its active site, the pocket (often buried) in which the catalytic reaction occurs, involving in a small subset of residues that participate in substrates (and/or cofactor) binding and the catalytic reaction (Holliday et al. 2009). The conformation of active site is susceptible to changes in the global protein structure, and sometimes even imperceptible changes caused by residues remotion from the active site. Introducing genetically encoded UAAs will endow the enzymes with unprecedented catalytic behavior by altering the active site environment such as steric and electronic interactions either directly or indirectly, which may not be achieved by substitution of any of NAAs.
The site-specific incorporation of UAAs into substrate binding regions of enzymes may be beneficial for enzyme activity. For example, replacing phenylalanine (Phe) 124, a critical residue for substrate binding, with a diverse set of UAAs in the prodrug activator nitroreductase (NTR) prominently increased its catalytic properties. Among those variants, the NTR containing p-nitrophenylalanine, 1, (pNF-NTR) exhibiting more than 30-fold and 2.3-fold improvement in the catalytic efficiency (kcat/KM), over that of the native NTR and the best natural mutants at the same site, respectively. The improved activity of pNF-NTR might be attribute to the effect of pi-stacking interaction between polarized aromatic ring of pNF and aromatic substrate (Jackson et al. 2006). In addition, Tyr309, a residue lies in substrate binding region of Agrobacterium radiobacter phosphotriesterase (arPTE), was replaced by the L-(7-hydroxycoumarin-4-yl) ethylglycine (Hco), 2, generating an over 8-fold improvement in turnover rate towards the paraoxon hydrolysis compared to the native one. The electrostatic repulsion between negative charge associated with Hco and the product (4-nitrophenolate) results in an accelerated release of product, revealing the importance of a trade-off between efficient binding and turnover (Ugwumba et al. 2011).
Moreover, the adjustment of metal coordination environments in tunable protein scaffolds through incorporating UAAs can contribute to improving the catalytic properties of the enzyme. Histidine is an important residue in the catalysis mechanisms of many enzymes such as serine peptidases (Polgar 2005), arylamine N-acetyltransferase (Sim et al. 2008) and heme enzymes (Roach et al. 2000). It participates in forming the important interactions with neighboring functional residues and substrates, and also act as a metal ion chelating ligand (Green et al. 2016; Hongsthong et al. 2004). The site-specific incorporation of UAAs into heme peroxidases, which catalyze various oxidative transformations with a histidine as the axial ligand, can improve the catalytic properties of the enzyme. For example, the introduction of Nδ-methyl histidine (NMH), 3, into the engineered ascorbate peroxidase (APX2) at position 163 and sperm whale myoglobin (Mb) at position 93 lead to the substantial increase in total turnover number (5-fold) (Green et al. 2016) and catalytic efficiency (kcat/KM) (3.7-fold) (Pott et al. 2018), respectively, toward oxidation of guaiacol compared to the parent enzymes. Notably, manipulating coordination ligand environments in metalloenzymes through genetically encoded UAAs could be realized to evolve biocatalysts with augmented catalytic properties.
In addition, tyrosine (Tyr) is found to be highly conserved in numerous enzymes and is a crucial residue at active center of enzyme due to its ability to donate electron and proton in catalytic mechanism (Jackson et al. 2007; Kong et al. 1992). It’s reported that fine-tuning Tyr residues redox potential by genetically incorporating Tyr analogs may enhance the oxidase activity. For example, the introduction of 3-chlorotyrosine (ClTyr), 4, in the active site of a myoglobin-based functional oxidase models (Phe33Tyr-CuBMb) at positon 33 caused a significant increase of the turnover number (>2.0 fold) (Yu et al. 2015). Similarly, the substitution of 3-methoxytyrosine (OMeY), 5, also led to an improvement of turnovers (>2.0 fold) (Yang et al. 2015). The results may be attribute to a change in proton-donating or electron-donating ability of the phenol ring by the substitution of ClTyr or OMeY.
Except for the introduction of UAAs at predetermined positions, the genetic code expansion is considered as a potential tool for use in directed enzyme evolution. In term of catalytic activity, a single substituted mutation library of TEM-1 β-lactamase variants with 10 structurally distinct UAAs randomly throughout the protein was reported (Xiao et al. 2015). The β-lactamase variant with p-acrylamido-phenylalanine (AcrF), 6, at position of 216 (near the active site) was obtained through a growth-based screening, leading to a substantial improvement in catalytic efficiency (∼8.0-fold). In comparison, it was not available with any of NAAs at this position to achieve similar improvement.
Engineering of enzyme with enhanced stability
The stability of protein relies on plenty of van der Waals, electrostatic interactions, hydrogen bonding and a handful of disulfide bonds (Gromiha 2010). The ability to maintain enzyme stability under harsh conditions is one of major challenges in the enzyme engineering. Despite a lot of efforts on enhancing enzyme stability through the traditional approaches such as directed evolution have been achieved, the application of genetically encoded UAAs by introducing unusual noncovalent or covalent interactions has provided a new avenue to evolve enzyme with improved stability.
The incorporation of halogenated amino acids has proved to be a very useful strategy for modifying the properties of proteins, especially in terms of stability (Ohtake et al. 2015). The UAAs with the bulky halogens such as chlorine and bromine can be site-specific incorporated into proteins, and several successful cases have been reported. For instance, Ohtake et al. (2015) have reported that incorporation of ClTyr, 4, and 3-bromo-L-tyrosine, 7, into glutathione S-transferase at seven selected positions exhibited significantly enhanced structural stability with an increase of 5.2 and 5.6 kcal/mol, respectively. A tightly packed protein interior was observed from crystal structure of the variants, most likely deriving from the additional halogen bond interactions between inter-residue and the bulky halogen atoms. Similarly, engineering the T4 lysozyme by site-specific introducing ClTyr, 4, offered an increased thermal stability (∼1 °C for melting temperature and 3 kcal/mol for the melting enthalpy) (Carlsson et al. 2018). Hence, the site-specific incorporation of halogenated UAAs seem to be an effective strategy for enhancing enzyme stability as larger halogens may fill the void to exert stabilizing effects and form a distinctive halogen bond interaction with adjacent residues.
The disulfide bond that generated between two cysteine (Cys) residues is vital for the function and stability of numerous enzymes (Rietsch and Beckwith 1998). However, the reversibility and inherent redox sensitivity, as well as the constraints of distance and dihedral angle restrict its applications in enzyme engineering (Berkmen 2012). As such, selective introduction of unusual covalent bonds provides new avenues to address such issues. Owning the unique reactivity of cysteine thiol, the design of a new covalent bond between a genetically encoded UAA and an adjacent Cys residues can be readily achieved. More recently, various achievements have been witnessed by using this strategy. For instance, an evolved homoserine O-succinyltransferase (metA) equipped with (p-benzoylphenyl) alanine (pBzF), 8, at position 21, resulting in a 21 °C improvement of melting temperature. Phe 21 lies in an extremely flexible N-terminal domain and its substitute with pBzF may lead to a stabilization of the native dimeric configuration of metA due to the cross-link between the aryl keto group of pBzF 21 and thiol group of Cys 90 (Li et al. 2018). The schematic diagram of the engineered cysteine cross-links was shown in Fig. 4a. In addition, the site-specific introduction of haloalkane UAAs into ZSPA affibody (Afb) also exhibited significantly improved thermal stability, because of the formation of intramolecular covalent linkage between alkyl halides and an adjacent Cys (Fig. 4b). Among those variants, the replacement of Phe 30 with O-(3bromoethyl)-L-tyrosine (BprY), 9, offered an obvious increased melting temperature, from 46 oC up to 60 oC (Xiang et al. 2014). Similarly, the β-lactamase mutants equipping with UAAs with long side-chain thiols manifested markedly improved thermostability due to the formation of an unusual disulfide bond between Cys and UAAs (Fig. 4c). In particular, the variant R65C/A184SbuY containing O-(4-mercaptobutyl)-L-tyrosine (SbuY), 10, showed a 9 °C enhancement in thermal stability, which was higher than most of the engineered Cys disulfide bonds with a common Tm increase around 5 °C (Liu et al. 2016). These results suggested that introducing an unusual covalent bond between a genetically encoded UAA and a NAA exhibit a tremendous potential for the enhancement of enzyme stability.

The schematic diagram of the engineered cysteine cross-links. a Genetically encoded UAAs containing aryl keto group. b Genetically encoded haloalkane UAAs. c Genetically encoded UAAs containing long-side-chain thiols
Engineering of enzyme with altered regioand stereo-selectivity
Apart from enhancement of activity and stability, altering regioand stereo-selectivity is also important for the application of enzymes. To date, controlling regioand stereo-selectivity of enzymes remains a great challenge in the field of biocatalysts. In terms of technologies, the site-specific incorporation of UAAs is considered as an effective complement in enzyme engineering for altering such properties. For example, incorporating four Tyr analogs including p-amino-phenylalanine (pAmF), 11, p-acetyl-phenylalanine (pAcF), 12, 3-(2-napthyl) alanine (NapA), 13, and O-benzyl-tyrosine (OBnY), 14, into a cytochromes P450 enzymes (P450s) at 11 active-site positions resulted in large shifts in regioselectivity of these engineered variants (Kolev et al. 2014). The native enzyme converts (S)-ibuprofen methylester to benzylic alcohol (62%) and allylic alcohol (38%) derivatives. Comparatively, the variant (Ala78pAcF) containing pAcF at position 78 obtained 88% of benzylic alcohol and 12% of allylic alcohol derivatives, while another variant (Ala328NapA) incorporated with NapA at position 328 produced 5% of alcohol and 95% of allylic alcohol derivatives. As such, introducing UAAs specifically into the active sites of enzymes seem to be a useful strategy in altering regio-selectivity of the enzyme.
Also, the method allows to fine-tune enantioselectivity of enzymes to an ideal level. Diketoreductase (DKR), a homodimeric protein, is a useful biocatalyst that can reduce various ketones to chiral alcohols. Trp222 of DKR lie in the C-terminal hydrophobic dimeric interface and is crucial to structural integrity and dimerization of the enzyme (Huang et al. 2012). Replacing Trp222 with Tyr analogs, O-tert-butyl-L-tyrosine (BuOF), 15, 4-cyanophenylalanine (CNF), 16, 4-methoxy-L-phenylalanine, 17, 4-phenyl-L-phenyalanine, 18, alter the enantiopreference of DKR toward 2-chloro-L-phenylethanone (Ma et al. 2014a). The parent enzyme presented a Re-preference that converts 2-chloro-L-phenylethanone into R-alcohols with a 9.1% enantiomeric excess (e.e.) value. The replacement with BuOF, 15, causes a notable change in the conformation, leading to an enhanced enantioselectivity of DKR with a 33.7% e.e. value. Interestingly, the substitution of Trp222 with CNF, 16, yields an enantiopreference inversion of DKR from Re- to Si-preference.
Except for enantioselectivity of enzymes, the diastereoselectivity is also an important property when multiple chiral centers exist in the substrates. Genetically encoded UAAs apply equally to manipulation of enzyme diastereoselectivity. Yu et al. (2021) proposed that the diastereoselectivity of Pseudomonas alcaligenes lipase (PaL) may be determined by certain key factors such as the flexibility of the active center and steric hindrance after analyzing the interactions between PaL active sites and the substrate L-menthol propionate with three chiral centers. Thereafter, four UAAs with diverse size and polarity including pAmF, 11, CNF, 16, o-chlorophenylalanine, 19, and o-bromophenylalanine (oBrF), 20, were introduced in 9 selected sites at the active center of PaL, generating a series of variants that obviously enhanced diastereopreference. Among those variants, the substitution of Ala253 with all four UAAs exhibited higher conversion rate and concurrently the diastereomeric excess (90%–95%) was higher than parent enzyme over 40%.
Engineering of enzyme with altered substrate specificity
Manipulating the substrate specificity of enzymes has always been the research topic for enzyme engineering (Zheng et al. 2015). In general, the synergism of substrate access and enzyme-substrate recognition interactions within the active center can affect the substrate specificity of the enzymes (Stevenson et al. 2000). Nowadays, various techniques utilizing NAAs or UAAs, particularly genetically encoded UAAs, have been applied to re-design the active site of enzymes to fit new substrates or poor substrates. The murine dihydrofolate reductase (mDHFR) readily catalyzes dihydrofolate (DHF) into tetrahydrofolate, whereas folate (FOL) is regard as a relatively poor substrate for the enzyme. Zheng et al. introduced NapA, 13, into the active site of mDHFR at position 31 to explored whether the enzyme substrate specificity can be changed (Zheng et al. 2015). The variant exhibits a 2-fold improvement in binding affinity (KM) toward the substrate FOL. Meanwhile, it also showed a 7.6-fold increase in catalytic efficiency ratio ((kcat/KM for FOL)/ (kcat/KM for DHF)) compared with the native mDHFR, demonstrating a dramatically change in the substrate specificity of the variant toward FOL.
The inhibitory effects of substrates, products, and other compounds on enzyme activities are a rather common phenomenon in the field of biocatalysis. In particular, the competitive inhibitors usually compete with the substrates for the active sites of an enzyme due to highly structural similarities between substrate and inhibitor (Alam et al. 2017). To selectively weaken the binding of inhibitors without compromising enzyme substrate binding is a huge challenging. Rationally introducing UAAs into the active sites of enzyme can be a promising strategy to control the binding affinity of inhibitors. For instance, Zheng and his co-workers set a model system including the enzyme mDHFR, its substrate DHF and inhibitor methotrexate to selectively control the inhibitory effect (Zheng and Kwon 2013). Replacing the key phe31 with NapA, 13, and p-bromophenylalanine (pBrF), 21, resulted in a significantly reduced binding affinity toward the inhibitor with a negligible impact on the catalytic efficiency toward the substrate DHF.
Expanding the reaction scope of enzyme
The posttranslational modification (PTM) such as glycosylation, acetylation, phosphorylation and methylation of the amino acid side chains is a conventional approach to expand the catalytic repertoire and regulate the function of enzymes (Okeley and van der Donk 2000; Walsh et al. 2005; Zhang et al. 2017; Zheng et al. 2018). However, it is difficult to introduce the specific modifications into the desired sites of the enzymes via PTM. Catalytic promiscuity, an inherent property of natural enzymes, is a catalytic ability of enzyme for markedly different chemical transformations (Bornscheuer and Kazlauskas 2004). Making use of the catalytic promiscuity provides access to create reactivities unrelated to the native function of an enzyme and even generate new biocatalysts with the ability to catalyze abiological transformation. The catalytic promiscuity of enzymes can sometimes be augmented with the re-design of the hydrophobic pore through enzyme engineering technology. Since site-specific incorporation allow various UAAs with diverse functional groups into enzymes at any desired site, it is considered as a promising approach for mimicking nature PTM and affording novel functions that would not be readily achievable with NAAs.
Embedding genetically encoded UAAs as a catalytic residue into the enzyme may endow it with novel catalytic ability that are naturally unavailable (Drienovská and Roelfes 2020). For instance, the incorporation of pAmF, 11, into the hydrophobic pore of Lactococcus lactis multidrug transcriptional regulator (LmrR), enables the enzyme to catalyze the abiological hydrazone and oxime formation reaction. The catalytic proficiency is attribute to a promiscuous binding pocket generating from the combination effects of the reactive pAmF residue (a potentially novel catalytic residue) and the hydrophobic interactions in proteinaceous scaffold (Drienovská et al. 2018). Repurposing solar energy into chemical energy has attracted a tremendous interest. However, it is hard to improve and expand the functions of photosensitizers through enzyme engineering. Liu et al. (2018) utilize the genetic codon expansion technology to incorporate a UAA pBzF, 8, into a superfolder yellow fluorescent protein (sfYFP) at position 66. Tyr66 is an important chromophore residue in the native sfYFP that generate a highly fluorescent p-hydroxybenzylidene-5-imidizolinone species through the spontaneous transformation of its tripeptide, Gly65-Tyr66-Gly67. Then, the variant was followed by multisite mutations and combined with organic nickel-terpyridine complex and the final variant PSP2T2 was obtained. This designed CO2-reducing enzyme exhibited a CO2/CO conversion quantum efficiency of 2.6%, which is over most reported CO2 photoreduction catalysts.
Cyclopropane compounds can be chemically synthesized through the metal-catalyzed cyclopropanation of olefins with diazo compounds (Nakagawa et al. 2015). More recently, several engineered enzymes such as haem proteins have emerged as promising alternatives for catalyzing the abiological reactions such as carbene-transfer reaction (Carminati and Fasan 2019). In terms of enzyme engineering technology, the genetically encoded UAAs has been proved to be effective to realize the non-natural reactions. For example, the substitution of a His residue of the myoglobin variant Mb (H64V, V68A) at position 93 with the NMH, 3, augments the enzyme’s promiscuous carbene-transfer chemistry, endowing the ability of the enzyme to convert the electron-rich styrene to the cyclopropane product without reductant and under aerobic conditions (Hayashi et al. 2018). The use of NMH as the axial haem ligand enables to capture an unusual bridging Fe (III)–C(carbene)–N(pyrrole) configuration, which participates in the cyclopropanation of substrate. The electron-rich olefins can be readily converted to cyclopropane compounds by using these engineered biocatalysts. However, the cyclopropanation of electron-deficient olefins remains an unmet goal in nature. To overcome such limitation, introducing unnatural components (cofactors and UAAs) into the enzyme may enable it to be a best-of-both-worlds catalyst (Agostini et al. 2017). A variant Mb (H64V, V68A) was designed by site-specific incorporating a UAA NMH and an unnatural iron-porphyrin cofactor to form a variant (Mb (H64V, V68A, H93NMH) [Fe (DADP)]). The variant exhibited high efficiency for the asymmetric cyclopropanation of electron-rich olefins, even for the electron-deficient alkenes (Carminati and Fasan 2019). These results suggest that introducing a metal coordinating residue into the active sites of the enzyme by modulating metal coordination environments may be a practical strategy to extend the capabilities of enzymes for abiological chemical transformations.
Apart from a metal coordinating residue, incorporating a metal-binding residues may also be another strategy to achieve this goal (Drienovská and Roelfes 2020; Drienovská et al. 2015) designed an artificial metalloenzymes by introducing of a metal-binding amino acid (2, 2’-bipyridin-5yl) alanine (BpyA), 22, at various positions, which was able to bind a transition metal ion into the LmrR. The resulting artificial metalloenzymes offered a novel catalytic activity, enabling the asymmetric vinylogous Friedel–Crafts alkylation reactions of indoles derivatives with α, β-unsaturated 2-acylimidazoles. The best variant (LmrR_LM_M89X_F93W) showed an 83% e.e. value for the product. It contained a BpyA at position 89, which lie in the hydrophobic pore of the enzyme.
Overall, incorporating UAAs into enzymes through the genetically encoded technology has a considerable effect on the field of enzyme engineering to acquire make-to-order biocatalysts with novel catalytic properties that have no equivalent in nature nowadays.
Application of site-specific incorporation in enzyme immobilization
Amino acids featuring reactive functional groups offer a chemical handle to immobilize it to a solid support. Immobilizing enzymes with various advantages such as improved longevity, enhanced stability, excellent reusability and easy separation from the reaction mixtures, have received great attention on industrial applications. In general, the enzyme immobilization methods are divided into four types including adsorption, covalent bonding, entrapment and cross-linking (Nguyen and Kim 2017). Among them, covalent immobilization is the preferred method to form covalent bonds leading to the stability of enzyme. Although the conventional covalent immobilization has been frequently applied, its randomization may cause unwanted covalent linkage between enzyme and matrix. To overcome this issue, site-specific immobilization by incorporating UAAs into the enzyme is easy to install the reactive handle at a predefined position (Wang et al. 2016). For example, p-azido-L-phenylalanine (pAzF), 23, was firstly inserted at different sites of lipase to make it easy to be coupled with support using strain-promoted azide-alkyne cycloaddition. Among these rational immobilized lipases, several variants presented better thermo-stability and higher relative activity such as AzPhe-Lip243 (6.0-fold improvement) and AzPhe-Lip274 (6.8-fold improvement) than those lipases with traditional immobilization using glutaraldehyde (Wang et al. 2016). In addition, L-3,4‐dihydroxyphenylalanine (DOPA), 24, is regard as a selective chemical cross-linker and the site-specifc incorporation of DOPA into enzymes enables the enzymes to specifically crosslink with polysaccharides or amino-group containing materials. The ω-transaminases (ω-TA) equipped with DOPA were successfully anchored in chitosan or polystyrene beads, resulting in a great reusability (10 cycles) in the kinetic resolution of chiral amines (Deepankumar et al. 2015). These reports manifested that UAA-mediated bioconjugation can be an efficient and unique tool addressing unmet needs in enzyme engineering, especially rational immobilization.
Challenges and outlook
The incorporation of UAAs into proteins has proved to be a prospective strategy to improve and alter the properties of enzymes such as activity, stability, selectivity. Despite UAAs incorporation for enzyme engineering would increase versatility and possibilities of enzyme catalysis, there are many issues waiting to be coped. How to rapidly and precisely predict the “hot spots” to evolve the enzyme with ideal properties remains a formidable challenge in enzyme engineering. Therefore, it is of great significance to sum up some practical strategies based on the existing successful examples. With these exciting examples, some superficial rules may be helpful for engineering enzymes. For instance, introducing a metal-binding UAA and a UAA possessing a unique reactive side chains as a catalytic residue were demonstrated to be good tactics to extend the reaction scope of enzyme. In addition, introducing halogenated UAAs or UAAs that can spontaneously cross-link with Cys may be beneficial to enhance stability. The positions outside the active sites may fit better for enzyme immobilization.
Nowadays, continued efforts have been invested in accelerating the evolution of enzyme through incorporation of UAAs (Gargiulo and Soumillion 2021; Mayer 2019; Ribeiro et al. 2019). Nevertheless, the applications of UAA mutagenesis-based enzyme engineering are still restricted. There are significant challenges currently being addressed including the expensive and commercial unavailable UAAs and poor protein yields as well as unattainable incorporation of multiple UAAs (Gao et al. 2019).
The high cost of UAAs and their commercial unavailability can preclude researchers from considering the use of this method to engineer enzymes, let alone for further utilization of UAAs in industrial applications. It is reported that the total synthesis cost of green fluorescent protein containing p-Propargyloxyphenylalanine is up to USD 0.658 per 100 µg protein (Shrestha et al. 2014). One valuable way to overcome such limitations is exploiting synthetic methodologies for chiral amino acids. Notably, enzymatic asymmetric synthesis of UAAs has attracted extensive attention (Xue et al. 2018). For example, tyrosine phenol lyase exhibits its potential for synthesis of tyrosine analogs, such as L-DOPA (Kim et al. 2018), halogenated tyrosine, methoxytyrosine (Nagasawa et al. 1981) and 2-amino-3-(8-hydroxyquinolin-5-yl) propanoic acid (Liu et al. 2013). With ongoing efforts on asymmetric synthesis of UAAs via chemical and enzymatic methods, it will be a cheap and accessible commodity, which will greatly promote the application of the incorporation of UAAs into proteins. Another way to address such limitation is designing the host cells to enable biosynthesis of UAAs in vivo. Evolving metabolic pathways of host cells to produce UAAs from cheap sources and simultaneously fetching them into desired enzymes will facilitate the applications of engineered enzymes (Chen et al. 2018; Ma et al. 2014b).
Although great value of genetically encoded UAAs has been exhibited in the field of biocatalysis, the limitation of poor protein expression levels was still not be tackled. In view of this problem, the coordinate optimization of aaRS, tRNA, codons, the ribosome and elongation factor may improve the UAAs incorporation efficiency (Jin et al. 2019; Park et al. 2011; Young and Schultz 2010). Of particular importance to incorporate UAAs is the optimization of o-aaRS/tRNA pairs and codon utilization (Gao et al. 2019). The common OTS have depended on the reassignment of ‘non-sense’ codons, however the competitions are unavoidable between this commonly used system and termination machinery of the host cell, leading to truncated proteins and lower protein yields. In prokaryote, the incorporation efficiency of UAAs at the amber codon is robustly repressed by termination machinery, due to recognition of amber stop codon by release factor 1 (RF1) in protein translation (Yanagisawa et al. 2014). To relieve the dependence on RF1 for termination, Lajoie et al. (2013) engineered a E. coli MG1655 strain by converting all 321 amber codons to synonymous ochre codons and concurrently deleting prfA gene (encodes RF1). Finally, this genomically recoded strain exhibits a higher UAAs incorporation efficiency than the native one. Another valuable method to improve incorporation efficiency of UAAs is using cell-free protein synthesis system which is a protein synthesis methodology without living cells (Lu 2017). The system requires the purified or unpurified transcriptional and translational components from cells, and also contains other essential components, including energy sources, cofactors, salts, buffers, nucleotides, and amino acids for protein production (Gao et al. 2019). To enable UAAs incorporation into proteins, UAA and its corresponding orthogonal aaRS/tRNA pair are also needed (Cui et al. 2020). The cell-free protein synthesis system has paved the way to efficiently incorporate of UAAs into proteins as it abolishes the transmembrane transport and cytotoxicity of UAAs.
The incorporation of UAAs through genetic codon expansion approach provides a useful toolbox for tailoring the protein properties and biochemical studies of protein function. However, introduction of multiple unique UAAs simultaneously into a single protein is still a formidable obstacle (Dumas et al. 2015). In general, the site-specific manner enables the incorporation of a UAA into one protein relying on the amber codon suppression. To date, tremendous efforts have been focused on introduction of multiple UAAs, and the uses of distinct stop codons and evolved quadruplet codons as well as mining new aaRS/tRNA pairs seem to be potential strategies to increase diversity of protein modifications. For example, Wan et al. (2010) developed a handy system for incorporation of two distinct UAAs (Nε-acetyl-L-lysines and p-azido-L-phenylalanine) at two defined sites of one protein in E. coli by combining the mutated PylRS–tRNAPyl pair to suppress the ochre (UAA) codon and an evolved MjTyrRS–tRNACUA pair.
In conclusion, the site-specific incorporation of UAAs is proved to open a promising new window in enzymes engineering. With future development of such technology, it will act as a standardized toolkits to evolve enzyme with desired properties and has a big impact on industrial applications including biopharmaceuticals, drug discovery and clinical trials.
References
Agostini F, Voller JS, Koksch B, Acevedo-Rocha CG, Kubyshkin V, Budisa N (2017) Biocatalysis with unnatural amino acids: enzymology meets xenobiology. Angew Chem Int Ed Engl 56:9680–9703. https://doi.org/10.1002/anie.201610129
Alam MT, Olin-Sandoval V, Stincone A, Keller MA, Zelezniak A, Luisi BF, Ralser M (2017) The self-inhibitory nature of metabolic networks and its alleviation through compartmentalization. Nat Commun 8:16018. https://doi.org/10.1038/ncomms16018
Berkmen M (2012) Production of disulfide-bonded proteins in Escherichia coli. Protein Expr Purif 82:240–251. https://doi.org/10.1016/j.pep.2011.10.009
Bornscheuer UT, Kazlauskas RJ (2004) Catalytic promiscuity in biocatalysis: using old enzymes to form new bonds and follow new pathways. Angew Chem Int Ed Engl 43:6032–6040. https://doi.org/10.1002/anie.200460416
Carlsson AC, Scholfield MR, Rowe RK, Ford MC, Alexander AT, Mehl RA, Ho PS (2018) Increasing enzyme stability and activity through hydrogen bond-enhanced halogen bonds. Biochemistry 57:4135–4147. https://doi.org/10.1021/acs.biochem.8b00603
Carminati DM, Fasan R (2019) Stereoselective cyclopropanation of electron-deficient olefins with a cofactor redesigned carbene transferase featuring radical reactivity. ACS Catal 9:9683–9697. https://doi.org/10.1021/acscatal.9b02272
Chen Y, Loredo A, Gordon A, Tang J, Yu C, Ordonez J, Xiao H (2018) A noncanonical amino acid-based relay system for site-specific protein labeling. Chem Commun (Camb) 54:7187–7190. https://doi.org/10.1039/c8cc03819h
Chin JW (2017) Expanding and reprogramming the genetic code. Nature 550:53–60. https://doi.org/10.1038/nature24031
Cui Z, Johnston WA, Alexandrov K (2020) Cell-free approach for non-canonical amino acids incorporation into polypeptides. Front Bioeng Biotechnol 8:1031. https://doi.org/10.3389/fbioe.2020.01031
Deepankumar K, Shon M, Nadarajan SP, Shin G, Mathew S, Ayyadurai N, Kim BG, Choi SH, Lee SH, Yun H (2014) Enhancing thermostability and organic solvent tolerance of ω-transaminase through global incorporation of fluorotyrosine. Adv Synth Catal 356:993–998. https://doi.org/10.1002/adsc.201300706
Deepankumar K, Nadarajan SP, Mathew S, Lee SG, Yoo TH, Hong EY, Kim BG, Yun H (2015) Engineering transaminase for stability enhancement and site-specific immobilization through multiple noncanonical amino acids incorporation. ChemCatChem 7:417–421. https://doi.org/10.1002/cctc.201402882
Drienovská I, Roelfes G (2020) Expanding the enzyme universe with genetically encoded unnatural amino acids. Nat Catal 3:193–202. https://doi.org/10.1038/s41929-019-0410-8
Drienovská I, Rioz-Martinez A, Draksharapu A, Roelfes G (2015) Novel artificial metalloenzymes by in vivo incorporation of metal-binding unnatural amino acids. Chem Sci 6:770–776. https://doi.org/10.1039/c4sc01525h
Drienovská I, Mayer C, Dulson C, Roelfes G (2018) A designer enzyme for hydrazone and oxime formation featuring an unnatural catalytic aniline residue. Nat Chem 10:946–952. https://doi.org/10.1038/s41557-018-0082-z
Drienovská I, Scheele RA, Gutierrez de Souza C, Roelfes G (2020) A hydroxyquinoline-based unnatural amino acid for the design of novel artificial metalloenzymes. Chembiochem 21:3077–3081. https://doi.org/10.1002/cbic.202000306
Dumas A, Lercher L, Spicer CD, Davis BG (2015) Designing logical codon reassignment expanding the chemistry in biology. Chem Sci 6:50–69. https://doi.org/10.1039/c4sc01534g
Gao W, Cho E, Liu Y, Lu Y (2019) Advances and challenges in cell-free incorporation of unnatural amino acids into proteins. Front Pharmacol 10:611. https://doi.org/10.3389/fphar.2019.00611
Gargiulo S, Soumillion P (2021) Directed evolution for enzyme development in biocatalysis. Curr Opin Chem Biol 61:107–113. https://doi.org/10.1016/j.cbpa.2020.11.006
Green AP, Hayashi T, Mittl PR, Hilvert D (2016) A chemically programmed proximal ligand enhances the catalytic properties of a heme enzyme. J Am Chem Soc 138:11344–11352. https://doi.org/10.1021/jacs.6b07029
Gromiha MM (2010) Protein stability. Protein BioinformaticsDoi. https://doi.org/10.1016/b978-8-1312-2297-3.50006-0
Hancock SM, Uprety R, Deiters A, Chin JW (2010) Expanding the genetic code of yeast for incorporation of diverse unnatural amino acids via a pyrrolysyl-tRNA synthetase/tRNA pair. J Am Chem Soc 132:14819–14824. https://doi.org/10.1021/ja104609m
Hayashi T, Tinzl M, Mori T, Krengel U, Proppe J, Soetbeer J, Klose D, Jeschke G, Reiher M, Hilvert D (2018) Capture and characterization of a reactive haem–carbenoid complex in an artificial metalloenzyme. Nat Catal 1:578–584. https://doi.org/10.1038/s41929-018-0105-6
Holliday GL, Mitchell JB, Thornton JM (2009) Understanding the functional roles of amino acid residues in enzyme catalysis. J Mol Biol 390:560–577. https://doi.org/10.1016/j.jmb.2009.05.015
Hongsthong A, Subudhi S, Sirijuntarat M, Cheevadhanarak S (2004) Mutation study of conserved amino acid residues of Spirulina delta 6-acyl-lipid desaturase showing involvement of histidine 313 in the regioselectivity of the enzyme. Appl Microbiol Biotechnol 66:74–84. https://doi.org/10.1007/s00253-004-1655-x
Huang Y, Lu Z, Ma M, Liu N, Chen Y (2012) Functional roles of Tryptophan residues in diketoreductase from Acinetobacter baylyi. BMB Rep 45:452–457. https://doi.org/10.5483/BMBRep.2012.45.8.064
Jackson JC, Duffy SP, Hess KR, Mehl RA (2006) Improving nature’s enzyme active site with genetically encoded unnatural amino acids. J Am Chem Soc 128:11124–11127. https://doi.org/10.1021/ja061099y
Jackson JC, Hammill JT, Mehl RA (2007) Site-specific incorporation of a (19)F-amino acid into proteins as an NMR probe for characterizing protein structure and reactivity. J Am Chem Soc 129:1160–1166. https://doi.org/10.1021/ja064661t
Jin X, Park OJ, Hong SH (2019) Incorporation of non-standard amino acids into proteins: challenges, recent achievements, and emerging applications. Appl Microbiol Biotechnol 103:2947–2958. https://doi.org/10.1007/s00253-019-09690-6
Kim S, Sung BH, Kim SC, Lee HS (2018) Genetic incorporation of L-dihydroxyphenylalanine (DOPA) biosynthesized by a tyrosine phenol-lyase. Chem Commun (Camb) 54:3002–3005. https://doi.org/10.1039/c8cc00281a
Kisselev L, Ehrenberg M, Frolova L (2003) Termination of translation: interplay of mRNA, rRNAs and release factors? EMBO J 22:175–182. https://doi.org/10.1093/emboj/cdg017
Kolev JN, Zaengle JM, Ravikumar R, Fasan R (2014) Enhancing the efficiency and regioselectivity of P450 oxidation catalysts by unnatural amino acid mutagenesis. Chembiochem 15:1001–1010. https://doi.org/10.1002/cbic.201400060
Kong K-H, Nishida M, Inoue H, Takahashi K (1992) Tyrosine-7 is an essential residue for the catalytic activity of human class Pi glutathione S-transferase: Chemical modification and site-directed mutagenesis studies. Biochem Bioph Res Co 182:1122–1129
Krauskopf K, Lang K (2020) Increasing the chemical space of proteins in living cells via genetic code expansion. Curr Opin Chem Biol 58:112–120. https://doi.org/10.1016/j.cbpa.2020.07.012
Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, Rohland N, Schultz PG, Jacobson JM, Rinehart J, Church GM, Isaacs FJ (2013) Genomically recoded organisms expand biological functions. Science 342:357–360. https://doi.org/10.1126/science.1241459
Li JC, Liu T, Wang Y, Mehta AP, Schultz PG (2018) Enhancing protein stability with genetically encoded noncanonical amino acids. J Am Chem Soc 140:15997–16000. https://doi.org/10.1021/jacs.8b07157
Link AJ, Vink MKS, Agard NJ, Prescher JA, Bertozzi CR, Tirrell DA (2006) Discovery of aminoacyl-tRNA synthetase activity through cell-surface display of noncanonical amino acids. Proc Natl Acad Sci 103:10180–10185. https://doi.org/10.1073/pnas.0601167103
Liu CC, Schultz PG (2010) Adding new chemistries to the genetic code. Annu Rev Biochem 79:413–444. https://doi.org/10.1146/annurev.biochem.052308.105824
Liu X, Li J, Hu C, Zhou Q, Zhang W, Hu M, Zhou J, Wang J (2013) Significant expansion of the fluorescent protein chromophore through the genetic incorporation of a metal-chelating unnatural amino acid. Angew Chem Int Ed Engl 52:4805–4809. https://doi.org/10.1002/anie.201301307
Liu T, Wang Y, Luo X, Li J, Reed SA, Xiao H, Young TS, Schultz PG (2016) Enhancing protein stability with extended disulfide bonds. Proc Natl Acad Sci 113:5910–5915. https://doi.org/10.1073/pnas.1605363113
Liu X, Kang F, Hu C, Wang L, Xu Z, Zheng D, Gong W, Lu Y, Ma Y, Wang J (2018) A genetically encoded photosensitizer protein facilitates the rational design of a miniature photocatalytic CO2-reducing enzyme. Nat Chem 10:1201–1206. https://doi.org/10.1038/s41557-018-0150-4
Lu Y (2017) Cell-free synthetic biology: engineering in an open world. Synth Syst Biotechnol 2:23–27. https://doi.org/10.1016/j.synbio.2017.02.003
Ma H, Yang X, Lu Z, Liu N, Chen Y (2014a) The “gate keeper” role of Trp222 determines the enantiopreference of diketoreductase toward 2-chloro-L-phenylethanone. PLoS One 9:e103792. https://doi.org/10.1371/journal.pone.0103792
Ma Y, Biava H, Contestabile R, Budisa N, di Salvo ML (2014b) Coupling bioorthogonal chemistries with artificial metabolism: intracellular biosynthesis of azidohomoalanine and its incorporation into recombinant proteins. Molecules 19:1004–1022. https://doi.org/10.3390/molecules19011004
Mayer C (2019) Selection, addiction and catalysis: emerging trends for the incorporation of noncanonical amino acids into peptides and proteins in vivo. Chembiochem 20:1357–1364. https://doi.org/10.1002/cbic.201800733
Mayer C, Dulson C, Reddem E, Thunnissen AWH, Roelfes G (2019) Directed evolution of a designer enzyme featuring an unnatural catalytic amino acid. Angew Chem Int Ed Engl 58:2083–2087. https://doi.org/10.1002/anie.201813499
Nagasawa T, Utagawa T, Goto J, Kim CJ, Tani Y, Kumagai H, Yamada H (1981) Syntheses of L-tyrosine-related amino acids by tyrosine phenol-lyase of Citrobacter intermedius. Eur J Biochem 117:33–40. https://doi.org/10.1111/j.1432-1033.1981.tb06299.x
Nakagawa Y, Chanthamath S, Shibatomi K, Iwasa S (2015) Ru(II)-pheox-catalyzed asymmetric intramolecular cyclopropanation of electron-deficient olefins. Org Lett 17:2792–2795. https://doi.org/10.1021/acs.orglett.5b01201
Naowarojna N, Cheng R, Lopez J, Wong C, Qiao L, Liu P (2021) Chemical modifications of proteins and their applications in metalloenzyme studies. Synth Syst Biotechnol 6:32–49. https://doi.org/10.1016/j.synbio.2021.01.001
Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW (2010) Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 464:441–444. https://doi.org/10.1038/nature08817
Nguyen HH, Kim M (2017) An overview of techniques in enzyme immobilization. Appl Sci Converg Tec 26:157–163. https://doi.org/10.5757/asct.2017.26.6.157
Nikic-Spiegel I (2020) Expanding the genetic code for neuronal studies. Chembiochem 21:3169–3179. https://doi.org/10.1002/cbic.202000300
Ohtake K, Yamaguchi A, Mukai T, Kashimura H, Hirano N, Haruki M, Kohashi S, Yamagishi K, Murayama K, Tomabechi Y, Itagaki T, Akasaka R, Kawazoe M, Takemoto C, Shirouzu M, Yokoyama S, Sakamoto K (2015) Protein stabilization utilizing a redefined codon. Sci Rep 5:9762. https://doi.org/10.1038/srep09762
Okeley NM, van der Donk WA (2000) Novel cofactors via post-translational modifications of enzyme active sites. Chem BiolDoi. https://doi.org/10.1016/s1074-5521(00)00140-x
Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, Soll D (2011) Expanding the genetic code of Escherichia coli with phosphoserine. Science 333:1151–1154. https://doi.org/10.1126/science.1207203
Polgar L (2005) The catalytic triad of serine peptidases. Cell Mol Life Sci 62:2161–2172. https://doi.org/10.1007/s00018-005-5160-x
Pott M, Hayashi T, Mori T, Mittl PRE, Green AP, Hilvert D (2018) A noncanonical proximal heme ligand affords an efficient peroxidase in a globin fold. J Am Chem Soc 140:1535–1543. https://doi.org/10.1021/jacs.7b12621
Ravikumar Y, Nadarajan SP, Yoo TH, Lee CS, Yun H (2015) Unnatural amino acid mutagenesis-based enzyme engineering. Trends Biotechnol 33:462–470. https://doi.org/10.1016/j.tibtech.2015.05.002
Ribeiro LF, Amarelle V, Alves LF, Viana de Siqueira GM, Lovate GL, Borelli TC, Guazzaroni ME (2019) Genetically engineered proteins to improve biomass conversion: new advances and challenges for tailoring biocatalysts. Molecules 24:2879. https://doi.org/10.3390/molecules24162879
Rietsch A, Beckwith J (1998) The genetics of disulfide bond metabolism. Annu Rev Genet 32:163–184. https://doi.org/10.1146/annurev.genet.32.1.163
Roach MP, Ozaki S, Watanabe Y (2000) Investigations of the myoglobin cavity mutant H93G with unnatural imidazole proximal ligands as a modular peroxide O-O bond cleavage model system. Biochemistry 39:1446–1454. https://doi.org/10.1021/bi992054v
Shrestha P, Smith MT, Bundy BC (2014) Cell-free unnatural amino acid incorporation with alternative energy systems and linear expression templates. Nat Biotechnol 31:28–34. https://doi.org/10.1016/j.nbt.2013.09.002
Sim E, Walters K, Boukouvala S (2008) Arylamine N-acetyltransferases: from structure to function. Drug Metab Rev 40:479–510. https://doi.org/10.1080/03602530802186603
Stevenson J-A, Jones JP, Wong LL (2000) Mutations of phenylalanine-193 in the putative substrate access channel alter the substrate specificity of cytochrome P450cam. Isr J Chem 40:55–62. https://doi.org/10.1560/npyw-gu7v-nrqu-r7t6
Ugwumba IN, Ozawa K, Xu ZQ, Ely F, Foo JL, Herlt AJ, Coppin C, Brown S, Taylor MC, Ollis DL, Mander LN, Schenk G, Dixon NE, Otting G, Oakeshott JG, Jackson CJ (2011) Improving a natural enzyme activity through incorporation of unnatural amino acids. J Am Chem Soc 133:326–333. https://doi.org/10.1021/ja106416g
Vargas-Rodriguez O, Sevostyanova A, Soll D, Crnkovic A (2018) Upgrading aminoacyl-tRNA synthetases for genetic code expansion. Curr Opin Chem Biol 46:115–122. https://doi.org/10.1016/j.cbpa.2018.07.014
Wals K, Ovaa H (2014) Unnatural amino acid incorporation in E. coli: current and future applications in the design of therapeutic proteins. Front Chem 2:15. https://doi.org/10.3389/fchem.2014.00015
Walsh CT, Garneau-Tsodikova S, Gatto GJ (2005) Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl 44:7342–7372. https://doi.org/10.1002/anie.200501023
Wan W, Huang Y, Wang Z, Russell WK, Pai PJ, Russell DH, Liu WR (2010) A facile system for genetic incorporation of two different noncanonical amino acids into one protein in Escherichia coli. Angew Chem Int Ed Engl 49:3211–3214. https://doi.org/10.1002/anie.201000465
Wan W, Tharp JM, Liu WR (2014) Pyrrolysyl-tRNA synthetase: an ordinary enzyme but an outstanding genetic code expansion tool. Biochim Biophys Acta 1844:1059–1070. https://doi.org/10.1016/j.bbapap.2014.03.002
Wang L, Schultz PG (2002) Expanding the genetic code. Chem Commun 1-11. https://doi.org/10.1039/b108185n
Wang L, Brock A, Herberich B, Schultz PG (2001) Expanding the genetic code of Escherichia coli. Science 292:498–500. https://doi.org/10.1126/science.1060077
Wang A, Du F, Pei X, Chen C, Wu SG, Zheng Y (2016) Rational immobilization of lipase by combining the structure analysis and unnatural amino acid insertion. J Mol Catal B-Enzym 132:54–60. https://doi.org/10.1016/j.molcatb.2016.06.015
Won Y, Pagar AD, Patil MD, Dawson PE, Yun H (2019) Recent advances in enzyme engineering through incorporation of unnatural amino acids. Biotechnol Bioproc E 24:592–604. https://doi.org/10.1007/s12257-019-0163-x
Xiang Z, Lacey VK, Ren H, Xu J, Burban DJ, Jennings PA, Wang L (2014) Proximity-enabled protein crosslinking through genetically encoding haloalkane unnatural amino acids. Angew Chem Int Ed Engl 53:2190–2193. https://doi.org/10.1002/anie.201308794
Xiao H, Nasertorabi F, Choi SH, Han GW, Reed SA, Stevens RC, Schultz PG (2015) Exploring the potential impact of an expanded genetic code on protein function. Proc Natl Acad Sci 112:6961–6966. https://doi.org/10.1073/pnas.1507741112
Xue YP, Cao CH, Zheng YG (2018) Enzymatic asymmetric synthesis of chiral amino acids. Chem Soc Rev 47:1516–1561. https://doi.org/10.1039/c7cs00253j
Yanagisawa T, Umehara T, Sakamoto K, Yokoyama S (2014) Expanded genetic code technologies for incorporating modified lysine at multiple sites. Chembiochem 15:2181–2187. https://doi.org/10.1002/cbic.201402266
Yang Y, Zhou Q, Wang L, Liu X, Zhang W, Hu M, Dong J, Li J, Xiaoxuan L, Ouyang H, Li H, Gao F, Gong W, Lu Y, Wang J (2015) Significant improvement of oxidase activity through the genetic incorporation of a redox-active unnatural amino acid. Chem Sci 6:3881–3885. https://doi.org/10.1039/C5SC01126D
Young TS, Schultz PG (2010) Beyond the canonical 20 amino acids: expanding the genetic lexicon. J Biol Chem 285:11039–11044. https://doi.org/10.1074/jbc.R109.091306
Yu Y, Lv X, Li J, Zhou Q, Cui C, Hosseinzadeh P, Mukherjee A, Nilges MJ, Wang J, Lu Y (2015) Defining the role of tyrosine and rational tuning of oxidase activity by genetic incorporation of unnatural tyrosine analogs. J Am Chem Soc 137:4594–4597. https://doi.org/10.1021/ja5109936
Yu Z, Yu H, Tang H, Wang Z, Wu J, Yang L, Xu G (2021) Site-specifically incorporate non‐canonical amino acids into pseudomonas alcaligenes lipase to hydrolyze L‐menthol propionate among the eight isomers. ChemCatChem 13:2691–2701. https://doi.org/10.1002/cctc.202100358
Zeymer C, Hilvert D (2018) Directed evolution of protein catalysts. Annu Rev Biochem 87:131–157. https://doi.org/10.1146/annurev-biochem-062917-012034
Zhang MS, Brunner SF, Huguenin-Dezot N, Liang AD, Schmied WH, Rogerson DT, Chin JW (2017) Biosynthesis and genetic encoding of phosphothreonine through parallel selection and deep sequencing. Nat Methods 14:729–736. https://doi.org/10.1038/nmeth.4302
Zheng S, Kwon I (2012) Manipulation of enzyme properties by noncanonical amino acid incorporation. Biotechnol J 7:47–60. https://doi.org/10.1002/biot.201100267
Zheng S, Kwon I (2013) Controlling enzyme inhibition using an expanded set of genetically encoded amino acids. Biotechnol Bioeng 110:2361–2370. https://doi.org/10.1002/bit.24911
Zheng S, Lim SI, Kwon I (2015) Manipulating the substrate specificity of murine dihydrofolate reductase enzyme using an expanded set of amino acids. Biochem Eng J 99:85–92. https://doi.org/10.1016/j.bej.2015.03.011
Zheng Y, Gilgenast MJ, Hauc S, Chatterjee A (2018) Capturing post-translational modification-triggered protein-protein interactions using dual noncanonical amino acid mutagenesis. ACS Chem Biol 13:1137–1141. https://doi.org/10.1021/acschembio.8b00021
Author information
Authors and Affiliations
Contributions
All authors contributed to the manuscript and have approved the submitted version. HQZ: Literature Search, Data Collection and Analysis, Writing—original draft. RCZ, YGZ and XLT: Conceptualization, revising, editing, supervision and validation.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no financial or proprietary interests in any material discussed in this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhu, HQ., Tang, XL., Zheng, RC. et al. Recent advancements in enzyme engineering via site-specific incorporation of unnatural amino acids. World J Microbiol Biotechnol 37, 213 (2021). https://doi.org/10.1007/s11274-021-03177-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-021-03177-1