
Abstract
Cell metabolism in living organisms is largely regulated at the transcriptional level, and the promoters are regarded as basic regulatory elements responsible for transcription initiation. Promoter engineering is an important technique to regulate gene expression and optimize metabolite biosynthesis in metabolic engineering and synthetic biology. The rational and precise control of gene expression in the multi-gene pathways can significantly affect the metabolic flux distribution and maximize the production of specific metabolites. Thus, many efforts have been made to identify natural promoters, construct inducible or hybrid promoters, and design artificial promoters for fine-tuning specific gene expression at the transcriptional level and improving production levels of the metabolites of interest. In this review, we will briefly introduce the architecture and function of both prokaryotic and eukaryotic promoters, and provide an overview of several major approaches for promoter engineering. The recent achievements and advances by promoter engineering for the optimization of metabolite biosynthetic pathways in multiple widely-used model microorganism, including Escherichia coli, Corynebacterium glutamicum and Saccharomyces cerevisiae, will also be extensively discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metabolic engineering is a powerful tool for the design, construction, modification and optimization of metabolic pathways in biological cells with the goals of improving cellular properties and generating desirable products (Chae et al. 2017; Keasling 2012; Yadav et al. 2012). The optimal flux balance of metabolic pathways is an important prerequisite for the production of various chemicals, bio-fuels or pharmaceuticals in industrial applications (Chen et al. 2018; Raman and Chandra 2009). In recent years, more and more studies have focus on the optimization of metabolic and regulatory processes at different cellular levels (Chen et al. 2018; Engstrom and Pfleger 2017; Lee et al. 2011, 2012; Redden et al. 2015). The engineering toolboxes can be categorized into at least six different aspects: (i) DNA-based engineering strategies, such as promoter engineering and terminator engineering; (ii) RNA-based engineering strategies, such as RNA switch and transcription factor engineering; (iii) protein-based engineering strategies, such as protein engineering and cofactor engineering; (iv) genome-based engineering strategies, such as multiplex genome-scale engineering and genome editing techniques; (v) metabolite-based engineering strategies, such as structural synthetic biotechnology and compartmentalization engineering; (vi) other novel engineering strategies, such as transporter engineering, biosensor engineering, and morphology engineering. Generally speaking, the employment of these powerful engineering tools will be conducive to overcoming metabolic bottlenecks, and thereby improving cell growth and metabolite production (Chen et al. 2018). The promoters with the needed characteristics provide a very powerful tool in genetic engineering, because precise control of key enzymes in specific metabolic pathways will typically maximize microbial growth or product formation. Therefore, promoter engineering is considered as a useful platform for the targeted manipulation of transcriptional activity in biotechnology.
The fine-tuned gene expression of metabolic pathways is a critical step to control the flux of cellular metabolism (Chen et al. 2018; Raman and Chandra 2009; Troein et al. 2007). Since the promoters substantially influence gene expression levels, promoter engineering has been proposed as one of the most efficient strategies of fine-tuning transcriptional control (Alper et al. 2005; Blazeck and Alper 2013; Chen et al. 2018). The promoters can be engineered to achieve precise strengths with a broad range of transcriptional capacities, enabling the tunable gene expression at the transcriptional levels. However, the limited numbers of available endogenous promoters are often bottlenecks for the fine-tuned transcriptional controls. At present, the strategies of promoter engineering are majorly implicated in the construction of promoter library, the replacement of native promoter, the modification of promoter architecture, and the rational design of the hybrid promoter (Blazeck and Alper 2013; Chae et al. 2017; Chen et al. 2018; Deaner and Alper 2018). Promoter engineering has been successfully applied in many industrial model microorganisms, such as Escherichia coli (Hwang et al. 2018), Corynebacterium glutamicum (Yim et al. 2013) and Saccharomyces cerevisiae (Portela et al. 2017).
In this review, we provide an overview of promoter engineering techniques and their biotechnological applications. The review will focus on the diverse nature of various promoter motifs, and the major strategies for promoter engineering. The recent achievements and advances in controlling specific gene expression at the promoter levels are fully discussed in three well-studied model microorganisms.
Promoter architecture and function
Promoters are referred to specific noncoding DNA sequences, which are typically in the range 100–1000 base pairs from the transcriptional start point and can act as indispensable regulatory signals of transcription initiation (Kanhere and Bansal 2005). A specific sequence of the promoter is essential for the efficiency of transcription initiation and then for the expression level of a targeted gene (Solovyev et al. 2010). In prokaryotes, the promoters can be directly recognized by the RNA polymerase with an associated sigma factor. However, several transcription factors are often required for the binding of an RNA polymerase II to promoter in eukaryotes (Butler and Kadonaga 2002; Paget and Helmann 2003). The strength of promoter activity is also affected by the changes in abundance or conformation of regulatory proteins (Hernandez-Garcia and Finer 2014). Considering that the promoters substantially contribute to the levels of transcription initiation, promoter engineering is widely used as a powerful tool for regulating gene expression.
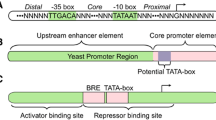
In prokaryotes, the full promoters are typically composed of core promoter and upstream regulatory elements (UP element) (Estrem et al. 1999; Kanhere and Bansal 2005) (Fig. 1). Core promoter is the minimal portion of the promoter required to initiate transcription, which typically consists of two conserved motifs at nucleotide (nt) positions − 35 and − 10 relative to the transcriptional start point (TSP). The statistical consensus sequences within the − 35 and the − 10 region have been widely characterized in detail in the gram-negative bacteria E. coli (Mitchell et al. 2003; Rangel-Chavez et al. 2017). Many promoters of E. coli genes have consensus sequences TATAAT in the − 10 element and TTGACA in the − 35 element, and a typical spacer length between these two motifs is 17 ± 1 nt. However, the nucleotides of the − 10 and − 35 elements are much less conserved in the gram-positive bacteria C. glutamicum (Patek et al. 2013; Patek and Nesvera 2011). The consensus sequences of the C. glutamicum − 35 region and the extended − 10 region are known as ttgnca and gnTAnanTng (capital letter indicates nucleotides occurring at the position in more than 80% of promoters; small letter indicates nucleotides occurring at the position in more than 40% of promoters) (Dostalova et al. 2017; Patek and Nesvera 2011). In addition, one or more UP elements are present in some prokaryotic promoters (Estrem et al. 1999; Ross et al. 1998). The TG dimer positioned 1 nt upstream of the − 10 element and the variable AT-rich region located upstream of the − 35 element are also shown to obviously increase promoter activity in E. coli (Patek and Nesvera 2011). However, the E. coli-type UP-element has not yet been reported in C. glutamcium.

The promoter architectures and putative conserved motifs of three widely-used model organisms. a A schematic diagram of typical E. coli promoters. Five promoters, including rrnB P1, rrnD P1, lacUV5, trp and tac, are selected to show their structural features. These promoters contain the consensus − 10 TATAAT and − 35 TTGACA motifs, and a typical spacer between these two regions is 17 ± 1 nt. b A schematic diagram of typical C. glutamicum promoters. The promoters of four housekeeping genes, including dapA, gdh, ilvA, and gap, are selected to illustrate the consensus domains within the − 35 and extended − 10 motifs. c A schematic diagram of S. cerevisiae promoters. The strong promoter of TDH3 gene (encoding 3-phosphate dehydrogenase) is selected to display the possible motifs of yeast core promoter and upstream activating sequences (UAS). TFBSs indicate the potential binding sites for specific transcription factors, such as RAP1 and GRF2. Two CAAT motif are also found in the UAS region of TDH3 promoter
Eukaryotic promoters usually span a wide range of DNA sequences, which are more complex and diverse than prokaryotic promoters (Blazeck and Alper 2013; Kanhere and Bansal 2005) (Fig. 1). The promoters are majorly recognized by RNA polymerase II and other transcription factors in eukaryotes, typically containing a TATA box (consensus sequence TATAAA). Additionally, the promoter region also has several upstream activating sequences (UASs), such as CAAT-box, GC-box and E-box (Dolfini et al. 2009; Lubliner et al. 2013; Redden and Alper 2015). Interestingly, only 20% of the promoters in the yeast S. cerevisiae contain a consensus TATA box, and about 80% of yeast genes are classified as TATA-less genes (Yang et al. 2014). Although some studies reported that a rigid DNA located 100–200 nt upstream of the start codon or a GAE-containing region can assist in the assembly of the transcriptional machinery at TATA-less promoters (Seizl et al. 2011; Tirosh et al. 2007), more core elements are needed to identify for the better understanding of yeast promoters.
Promoter engineering approaches
It has been reported that the severe disturbances of metabolic homeostasis could not improve product yields, but will increase metabolic burden for the host cells (Chen et al. 2018; Raman and Chandra 2009). In general, the constitutive promoters are considered to give stable gene expression levels and the inducible promoters are only active under the specific circumstances (Patek and Nesvera 2011). However, a precisely temporal and spatial control of gene expression levels requires various promoters with different strengths over a range of several orders of magnitude. Many strategies have been employed for promoter engineering, such as promoter library to screen synthetic promoters with diverse transcriptional strengths, promoter replacement to construct endogenous promoters with desired transcriptional strength, and synthetic ribosome-binding site (RBS) regulation to generate novel promoters with the varying mRNA translation efficiencies (Blazeck and Alper 2013; Chen et al. 2018) (Fig. 2). Moreover, the main approaches for rapid construction of synthetic promoter library include site-directed mutagenesis, error-prone PCR (Ep-PCR), sequence randomization of non-conserved region, hybrid-promoter engineering, and rational design of transcription factor-binding sites (TFBSs). In the past few decades, promoter engineering strategies have been extensively explored in many important industrial strains, including E. coli, C. glutamicum, S. cerevisiae, Streptomyces and lactic acid bacteria. The representative instances are summarized in Table 1.

General scheme of promoter engineering for the optimization of metabolite biosynthesis. Promoter libraries are mainly constructed by site-directed mutagenesis, error-prone PCR (Ep-PCR), sequence randomization of non-conserved region (NCR), hybrid-promoter design, and transcription factor-binding sites (TFBSs) modification. Promoter replacement and synthetic ribosome-binding site (RBS) regulation are also important approaches to achieve desired promoters. Promoters with varying strengths can be assembled with the corresponding genes of the target pathway to generate a random library of combinatorial pathways. The engineered strain can be identified after high-through screening and metabolite analysis. Note P promoter, G gene
Site-directed mutagenesis
The statistical consensus sequence does not guarantee a most efficient promoter, because the promoters ought to be evolved to serve basic physiological requirements rather than achieve the highest transcriptional strengths (Kiryu et al. 2005; Patek and Nesvera 2011). Some less highly conserved nucleotides around the consensus region may significantly affect the activity of the promoter, which could be used as targets for the rapid construction of stronger or weaker promoters. For example, base alterations of the extended − 10 element of C. glutamicum dapA promoter from AGGTAACCT to TGGTATAAT obviously improve promoter activity (Vasicova et al. 1999). Site mutations of the − 35 region of C. glutamicum gdh promoter from TGGTCA to TTGACA or TTGCCA can also significantly increase promoter strengths (Asakura et al. 2007). Moreover, the introduction of a TG dimer 1 nt upstream of the − 10 region contributes to the increase of specific promoter activity both in E. coli and C. glutamicum (Burr et al. 2000; Patek and Nesvera 2011). Although this method may generate specific stronger or weaker promoters, it cannot rapidly build large-scale promoter libraries with desired strength.
Ep-PCR
Ep-PCR is a simple random mutagenesis technique in vitro, which is considered as an effective method to obtain DNA sequence diversity (McCullum et al. 2010). The mutations are randomly inserted into anywhere throughout the promoter region, providing a guarantee for rapid construction of large-scale promoter libraries. For example, the diversified promoter libraries of the constitutive promoters PLtetO-1 in E. coli and TEF1 in S. cerevisiae as well as the inducible oxygen-repressed S. cerevisiae DAN1 promoter are successfully generated by Ep-PCR (Alper et al. 2005; Nevoigt et al. 2006; Redden et al. 2015; Tyo et al. 2011). However, mutational biases exhibited by Ep-PCR often affect sequence diversities of synthetic promoters, and too many missense mutations associated with Ep-PCR method may increase the workload for subsequent screening procedures.
Sequence randomization of non-conserved region
The consensus sequences are involved in the direct binding of core RNA polymerase, and the sequence alterations of these defined DNA regions typically have significant effects on the activity of the promoter (Kanhere and Bansal 2005). Although the non-conserved regions (NCR) are not required for the direct binding of RNA polymerase, the variable sequences may allow the recognition and specific binding of regulatory proteins to promoter, and then change the expression level of targeted gene. Thus, the random mutation of non-conserved region instead of the consensus element might be a more effective approach to yield the promoters with different expression intensities. For example, De Mey et al. construct a synthetic promoter library based on the E. coli − 10 (TATAAT) and − 35 (TTGACAT) consensus regions, 13 semi-conserved and 20 random nucleotides, covering 3 to 4 logs of promoter activity in small steps of activity change (De Mey et al. 2007). Wei et al. design a large-size promoter library based on the C. glutamicum − 10 (NNTANANT) and − 35 (NNGNCN) consensus regions, the conserved RBS (AAAGGA) element and 60 random nucleotides (Wei et al. 2018). The obtained promoters can effectively regulate gene expression and show varying strengths over a wide range. In addition, Jeppsson et al. construct a synthetic promoter library with a combination of conserved structures from several S. cerevisiae promoters, including the regulatory elements CT- box (CTTCC) and RPG-box (ACCCATACA), the consensus TATA box and other random nucleotides (Jeppsson et al. 2003). These promoters are shown to cover approximately three orders of magnitude between the lowest and the highest activity. In addition, Yang et al. design three broad-spectrum promoters based on the minimal yeast promoter elements for E. coli, S. cerevisiae and Bacillus subtilis, expanding the synthetic biology toolbox used for different hosts (Yang et al. 2018). On the whole, this approach is applied to rapidly generate large-scale library of promoters with varying strengths over a wide range, and has been extensively employed for flux optimization in metabolic pathways.
Hybrid-promoter engineering
Hybrid promoter engineering is implicated in the assembly of upstream enhancer element and core promoter region derived from several different promoters (Portela et al. 2017; Pothoulakis and Ellis 2018; Xu et al. 2014). This strategy has been employed to improve the transcription efficiency or enable novel promoter regulation in both prokaryotes and eukaryotes. For example, the commonly used tac promoter of E. coli strain is a hybrid promoter, which combines the − 10 region of the lacUV5 promoter and the − 35 region of the trp promoter (de Boer et al. 1983). The hybrid promoter libraries have also been designed for fine-tuning transcriptional control in S. cerevisiae (Blazeck et al. 2012). The tandem UAS elements of hybrid promoters serve as synthetic transcriptional amplifiers to control expression levels (Blazeck et al. 2011; Guarente et al. 1984). Therefore, the hybrid promoter engineering may be a promising and efficient strategy in achieving stronger promoters compared to native promoters. However, the promoter activities obtained by the hybrid promoter method often alter in a stepwise manner, and more enhancer-core element fusions need to be identified and tested, which often result in negative effects on the precise control of gene expression.
Rational design of transcription factor-binding sites
Both prokaryotic and eukaryotic promoters usually contain several short sequence elements, and these motifs can mediate the binding of specific transcription factors that recruit the transcriptional machinery (Jayaram et al. 2016; Kanhere and Bansal 2005). The binding affinity of transcription factors to specific binding sites plays an important role in promoter strength and regulation (Todeschini et al. 2014). Cox et al. develop a combinatorial library of random promoter architectures in E. coli, in which each promoter contains up to three incorporated operators that correspond to four different transcription factors (Cox et al. 2007). The resulting library shows at least five decades of variation in promoter activity. Murphy et al. also develop a combinatorial promoter design to study the effects of tetO2 operator position and multiplicity within the GAL1 promoter derived from S. cerevisiae (Murphy et al. 2007). The result shows that increasing the number of tetO2 operator sites and/or their proximity to the TATA box result in a stronger transcriptional repression of the GAL1 promoter. Hence the refinement of promoter activity by a direct and rational design of transcription factor-binding sites will be a good alternative to promoter engineering. Relatively little is known, however, about the preferred DNA binding sites and relative binding affinity of specific transcription factors, impeding further application of this method in metabolic engineering.
Promoter replacement
Promoter replacement through selecting specific promoters with various strengths is frequently used to fine-tune gene expression of rate-limiting enzyme (Chen et al. 2018; De Mey et al. 2010). For example, Yuan et al. increase the carotenoid production of E. coli by replacing the native promoter of the isoprenoid pathway with the strong bacteriophage T5 promoter (Yuan et al. 2006). Moreover, the strong promoters of C. glutamicum sod (encoding superoxide dismutase) and tuf (encoding translational elongation factor EF-Tu) genes, are also used for optimizing gene expression (Patek et al. 2013). Several S. cerevisiae strong promoters, such as the promoters of 3-phosphate dehydrogenase TDH3, 3-phosphoglycerate kinase PGK1 and translational elongation factor TEF1, are commonly used for engineering applications (Partow et al. 2010). Furthermore, the heat-induced PRPL promoters of phage λ and the inducible E. coli promoters Plac, Ptac and Ptrc are also efficient tools to regulate gene expression. In brief, the insertion of specific promoters with desired strengths by replacing the native promoter is a very simple, efficient and time-saving method for the optimization of metabolic pathways.
Synthetic RBS regulation
The alternation of RBS strength is also an efficient approach to regulate expression levels, ranging from genetic circuits to production pathways (Chen et al. 2018). The combination of promoter and RBS engineering will contribute to the construction of libraries with a wider range of promoter strengths. Moreover, this strategy can achieve expression regulation at both the transcriptional and translational levels. Kosuri et al. design all combinations of 114 promoters and 111 RBS sites and create a large-scale library of 12,653 synthesized constructs in E. coli (Kosuri et al. 2013). The large dataset shows the expression levels vary over four orders of magnitude, and provides a good resource for researchers seeking to achieve particular regulatory elements.
Applications of promoter engineering in metabolic pathway design
Metabolism is an extensively important and complex cellular process. No matter the modification of specific endogenous pathways or the introduction of heterologous biosynthetic gene clusters, it always disturbs the native metabolism in microbial hosts, and generates flux imbalances of metabolic pathways (Chen et al. 2018; Raman and Chandra 2009). Thus, to balance of the overall metabolic fluxes by orchestrating the expression of multi-genes is one of the major challenges for strain improvement (Biggs et al. 2014). Based on various promoter strengths, the multivariate modular metabolic engineering for pathway optimization has greatly increased our ability to design and generate desired microbial cell factories for industrial applications.
The prokaryotic model microorganisms, such as E. coli and C. glutamicum, are well-known used industrial workhorses for the large-scale production of various added-value metabolites (Du et al. 2011; Lee et al. 2016). The precise gene expression control through promoter engineering is a critical strategy to balance the flux in the metabolic pathways. For example, Wu et al. optimize the resveratrol production of E. coli by regulating the expression strengths of three distinct modules, and the engineered strain exhibits an almost 30-fold increase in the resveratrol production (Wu et al. 2013). Dahl et al. employ the stress-response promoters to regulate farnesyl pyrophosphate (FPP) production by altering the isoprenoid biosynthetic pathway in E. coli, and this strategy improves the production of amorphadiene, the final product, by twofold over that from inducible or constitutive promoters (Dahl et al. 2013). Shen et al. construct a lycopene producer through promoter engineering, and employ promoters with different strengths to balance the expression of the mevalonate pathway (Shen et al. 2015). The engineered E. coli strain produces lycopene of 529.45 mg/L in the fed-batch culture. Hwang et al. show a dissolved oxygen (DO)-dependent nar promoter library with diverse transcriptional strengths, and evaluate the general applications of these synthetic nar promoters in biochemical production (Hwang et al. 2018). By regulating gene expression of specific biosynthesis pathways using the synthetic promoters with different strengths, the production of D-lactate or 2, 3-butanediol is increased by 34% and 72%, respectively. Similarly, promoter engineering strategies are also employed for genetically modifying C. glutamicum strain. For example, Yim et al. isolate synthetic promoters of various strengths, and employ the strongest promoter H36 for the secretory production of endoxylanase, achieving yields of 746 mg/L in the extracellular medium (Yim et al. 2013). By replacing the natural promoter of the genes with a strong promoter and other engineering approaches, Judith et al. report de novo generation of an industrially competitive L-lysine producer (Becker et al. 2011). In addition, we also design an effective promoter library with varying strengths over a wide range, and a promoter library-based module combination (PLMC) technology is established for the optimization of L-threonine biosynthesis pathway (Wei et al. 2018). The threonine titer of engineered strain shows 6.1-fold higher than that of the control strain.
Saccharomyces cerevisiae, the best-characterized eukaryotic organism, is considered the ideal host for microbial production of biofuels, nutraceuticals, and natural products (Du et al. 2011). The fine-tuning of gene expression is also needed to maximize product yields in yeast. In recent years, a strategy named the customized optimization of metabolic pathways by combinatorial transcriptional engineering (COMPACTER) is designed for balancing metabolic fluxes of the heterologous multi-gene pathways (Du et al. 2012). Based on this strategy, the researches successfully obtain a xylose utilizing pathway with near-highest efficiency and a cellobiose utilizing pathway with highest efficiency in S. cerevisiae. Moreover, a strategy combining stepwise metabolic engineering and flux control at the transcriptional level is designed to enhance triterpenoid production, resulting in the β-amyrin yield of 16.30 mg/g dry cell which is best in present reports (Zhang et al. 2015). Perhaps more commonly, promoter engineering merely acts as one of major regulation steps for pathway optimization. For example, by the combinatorial approach gene overexpression under control of the strong constitutive promoters and other disruption strategies, Raphael et al. achieve the highest titer of triacylglycerols in S. cerevisiae, which is about 27.4% of the maximum theoretical yields (Ferreira et al. 2018).
Taken together, as a crucial tool of synthetic biology, promoter engineering has shown great potentials for application in the fine regulation of metabolic pathways. However, the creating of appropriate pathway mutants based on promoter libraries is often combined with high-throughput screening method, which is an inefficient and time-consuming process. With the development of systems biology and bioinformatics technology, much elucidation about the structure and function of promoter core motifs or regulatory elements is still urgently required, and more rational strategies should be developed for the design of promoter sequence. Additionally, using the computer-aided models to predict the expression intensity of promoters in specific metabolic pathways will help to more rationally select proper promoters for pathway optimization.
Conclusion and perspective
Promoters can regulate gene expression of specific metabolic pathways to control the flux of metabolism, which are referred to essential biological elements in synthetic biology. Therefore, the fine-tuning of metabolic flux by promoter engineering provides a powerful strategy for strain and product improvement.
In the past few years, several different promoter engineering strategies have been successfully designed and implemented to allow precise control of gene expression and regulatory circuit for industrial applications in both prokaryotes and eukaryotes. However, the complete rational flux design in metabolic engineering remains difficult to achieve optimal effects since pathway information is frequently not available. Thus, the modularization and multivariate optimization of metabolic pathways by promoters with various strengths are likely to be useful concepts in future (Biggs et al. 2014; Jeschek et al. 2016; Yadav et al. 2012). In addition, with more and more promoter elements have been identified and functionally characterized, individual promoter sequences and regulatory elements can be designed more rationally, thereby reducing the blindness of library construction and the workload of subsequent high-throughput screening process.
References
Alper H, Fischer C, Nevoigt E, Stephanopoulos G (2005) Tuning genetic control through promoter engineering. Proc Natl Acad Sci USA 102:12678–12683. https://doi.org/10.1073/pnas.0504604102
Asakura Y, Kimura E, Usuda Y, Kawahara Y, Matsui K, Osumi T, Nakamatsu T (2007) Altered metabolic flux due to deletion of odhA causes L-glutamate overproduction in Corynebacterium glutamicum. Appl Environ Microbiol 73:1308–1319. https://doi.org/10.1128/AEM.01867-06
Becker J, Zelder O, Hafner S, Schroder H, Wittmann C (2011) From zero to hero–design-based systems metabolic engineering of Corynebacterium glutamicum for L-lysine production. Metab Eng 13:159–168. https://doi.org/10.1016/j.ymben.2011.01.003
Biggs BW, De Paepe B, Santos CNS, De Mey M, Ajikumar PK (2014) Multivariate modular metabolic engineering for pathway and strain optimization. Curr Opin Biotech 29:156–162. https://doi.org/10.1016/j.copbio.2014.05.005
Blazeck J, Alper HS (2013) Promoter engineering: Recent advances in controlling transcription at the most fundamental level. Biotech J 8:46–58. https://doi.org/10.1002/biot.201200120
Blazeck J, Liu L, Redden H, Alper H (2011) Tuning gene expression in Yarrowia lipolytica by a hybrid promoter approach. Appl Environ Microbiol 77:7905–7914. https://doi.org/10.1128/AEM.05763-11
Blazeck J, Garg R, Reed B, Alper HS (2012) Controlling promoter strength and regulation in Saccharomyces cerevisiae using synthetic hybrid promoters. Biotechnol Bioeng 109:2884–2895. https://doi.org/10.1002/bit.24552
Burr T, Mitchell J, Kolb A, Minchin S, Busby S (2000) DNA sequence elements located immediately upstream of the – 10 hexamer in Escherichia coli promoters: a systematic study. Nucleic Acids Res 28:1864–1870
Butler JE, Kadonaga JT (2002) The RNA polymerase II core promoter: a key component in the regulation of gene expression. Gene Dev 16:2583–2592. https://doi.org/10.1101/gad.1026202
Chae TU, Choi SY, Kim JW, Ko YS, Lee SY (2017) Recent advances in systems metabolic engineering tools and strategies. Curr Opin Biotechnol 47:67–82. https://doi.org/10.1016/j.copbio.2017.06.007
Chen X et al (2018) DCEO biotechnology: tools to design, construct, evaluate, and optimize the metabolic pathway for biosynthesis of chemicals. Chem Rev 118:4–72. https://doi.org/10.1021/acs.chemrev.6b00804
Cox RS, Surette III, Elowitz MG MB (2007) Programming gene expression with combinatorial promoters. Mol Syst Biol 3:145. https://doi.org/10.1038/msb4100187
Dahl RH et al (2013) Engineering dynamic pathway regulation using stress-response promoters. Nat biotechnol 31:1039–1046. https://doi.org/10.1038/nbt.2689
de Boer HA, Comstock LJ, Vasser M (1983) The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc Natl Acad Sci USA 80:21–25. https://doi.org/10.1073/pnas.80.1.21
De Mey M, Maertens J, Lequeux GJ, Soetaert WK, Vandamme EJ (2007) Construction and model-based analysis of a promoter library for E. coli: an indispensable tool for metabolic engineering. BMC Biotechnol 7:34. https://doi.org/10.1186/1472-6750-7-34
De Mey M, Maertens J, Boogmans S, Soetaert WK, Vandamme EJ, Cunin R, Foulquie-Moreno MR (2010) Promoter knock-in: a novel rational method for the fine tuning of genes. BMC Biotechnol 10:26. https://doi.org/10.1186/1472-6750-10-26
Deaner M, Alper HS (2018) Promoter and terminator discovery and engineering synthetic biology. Metab Eng 162:21–44. https://doi.org/10.1007/10_2016_8
Dolfini D, Zambelli F, Pavesi G, Mantovani R (2009) A perspective of promoter architecture from the CCAAT box. Cell Cycle 8:4127–4137. https://doi.org/10.4161/cc.8.24.10240
Dostalova H et al (2017) Assignment of sigma factors of RNA polymerase to promoters in Corynebacterium glutamicum. AMB Express 7:133. https://doi.org/10.1186/s13568-017-0436-8
Du J, Shao Z, Zhao H (2011) Engineering microbial factories for synthesis of value-added products. J Ind Microbiol 38:873–890. https://doi.org/10.1007/s10295-011-0970-3
Du J, Yuan Y, Si T, Lian J, Zhao H (2012) Customized optimization of metabolic pathways by combinatorial transcriptional engineering. Nucleic Acids Res 40:e142. https://doi.org/10.1093/nar/gks549
Engstrom MD, Pfleger BF (2017) Transcription control engineering and applications in synthetic biology. Synth Syst Biotechnol 2:176–191. https://doi.org/10.1016/j.synbio.2017.09.003
Estrem ST, Ross W, Gaal T, Chen ZW, Niu W, Ebright RH, Gourse RL (1999) Bacterial promoter architecture: subsite structure of UP elements and interactions with the carboxy-terminal domain of the RNA polymerase alpha subunit. Gene Dev 13:2134–2147
Ferreira R, Teixeira PG, Gossing M, David F, Siewers V, Nielsen J (2018) Metabolic engineering of Saccharomyces cerevisiae for overproduction of triacylglycerols. Metab Eng Commun 6:22–27. https://doi.org/10.1016/j.meteno.2018.01.002
Guarente L, Lalonde B, Gifford P, Alani E (1984) Distinctly regulated tandem upstream activation sites mediate catabolite repression of the CYC1 gene of S. cerevisiae. Cell 36:503–511
Hartner FS, Ruth C, Langenegger D, Johnson SN, Hyka P, Lin-Cereghino GP, Lin-Cereghino J, Kovar K, Cregg JM, Glieder A (2008) Promoter library designed for fine-tuned gene expression in Pichia pastoris. Nucleic Acids Res 36:e76. https://doi.org/10.1093/nar/gkn369
Hernandez-Garcia CM, Finer JJ (2014) Identification and validation of promoters and cis-acting regulatory elements. Plant Sci 217–218:109–119. https://doi.org/10.1016/j.plantsci.2013.12.007
Hwang HJ, Lee SY, Lee PC (2018) Engineering and application of synthetic nar promoter for fine-tuning the expression of metabolic pathway genes in Escherichia coli. Biotechnol Biofuels 11:103. https://doi.org/10.1186/s13068-018-1104-1
Jayaram N, Usvyat D, AC RM (2016) Evaluating tools for transcription factor binding site prediction. BMC Bioinformatics 17:1298. https://doi.org/10.1186/s12859-016-1298-9
Jensen PR, Hammer K (1998) The sequence of spacers between the consensus sequences modulates the strength of prokaryotic promoters. Appl Environ Microbiol 64:82–87
Jeppsson M, Johansson B, Jensen PR, Hahn-Hagerdal B, Gorwa-Grauslund MF (2003) The level of glucose-6-phosphate dehydrogenase activity strongly influences xylose fermentation and inhibitor sensitivity in recombinant Saccharomyces cerevisiae strains. Yeast 20:1263–1272. https://doi.org/10.1002/yea.1043
Jeschek M, Gerngross D, Panke S (2016) Rationally reduced libraries for combinatorial pathway optimization minimizing experimental effort. Nat Commun 7:11163. https://doi.org/10.1038/ncomms11163
Kanhere A, Bansal M (2005) Structural properties of promoters: similarities and differences between prokaryotes and eukaryotes. Nucleic Acids Res 33:3165–3175. https://doi.org/10.1093/nar/gki627
Keasling JD (2012) Synthetic biology and the development of tools for metabolic engineering. Metab Eng 14:189–195. https://doi.org/10.1016/j.ymben.2012.01.004
Kiryu H, Oshima T, Asai K (2005) Extracting relations between promoter sequences and their strengths from microarray data. Bioinformatics 21:1062–1068. https://doi.org/10.1093/bioinformatics/bti094
Kosuri S et al (2013) Composability of regulatory sequences controlling transcription and translation in Escherichia coli. Proc Natl Acad Sci USA 110:14024–14029. https://doi.org/10.1073/pnas.1301301110
Lee JW, Kim TY, Jang YS, Choi S, Lee SY (2011) Systems metabolic engineering for chemicals and materials. Trends Biotechnol 29:370–378. https://doi.org/10.1016/j.tibtech.2011.04.001
Lee JW, Na D, Park JM, Lee J, Choi S, Lee SY (2012) Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Biol 8:536–546. https://doi.org/10.1038/nchembio.970
Lee JY, Na YA, Kim E, Lee HS, Kim P (2016) The Actinobacterium Corynebacterium glutamicum, an Industrial Workhorse. J Microbiol Biotechnol 26:807–822. https://doi.org/10.4014/jmb.1601.01053
Lubliner S, Keren L, Segal E (2013) Sequence features of yeast and human core promoters that are predictive of maximal promoter activity. Nucleic Acids Res 41:5569–5581. https://doi.org/10.1093/nar/gkt256
McCullum EO, Williams BA, Zhang J, Chaput JC (2010) Random mutagenesis by error-prone PCR. Methods Mol Biol 634:103–109. https://doi.org/10.1007/978-1-60761-652-8_7
Mitchell JE, Zheng D, Busby SJ, Minchin SD (2003) Identification and analysis of ‘extended—10’ promoters in Escherichia coli. Nucleic Acids Res 31:4689–4695
Murphy KF, Balazsi G, Collins JJ (2007) Combinatorial promoter design for engineering noisy gene expression. Proc Natl Acad Sci USA 104:12726–12731. https://doi.org/10.1073/pnas.0608451104
Nevoigt E, Kohnke J, Fischer CR, Alper H, Stahl U, Stephanopoulos G (2006) Engineering of promoter replacement cassettes for fine-tuning of gene expression in Saccharomyces cerevisiae. Appl Environ Microbiol 72:5266–5273. https://doi.org/10.1128/AEM.00530-06
Nevoigt E, Fischer C, Mucha O, Matthaus F, Stahl U, Stephanopoulos G (2007) Engineering promoter regulation. Biotechnol Bioeng 96:550–558. https://doi.org/10.1002/bit.21129
Paget MS, Helmann JD (2003) The sigma70 family of sigma factors. Genome Biol 4:203
Partow S, Siewers V, Bjorn S, Nielsen J, Maury J (2010) Characterization of different promoters for designing a new expression vector in Saccharomyces cerevisiae. Yeast 27:955–964. https://doi.org/10.1002/yea.1806
Patek M, Nesvera J (2011) Sigma factors and promoters in Corynebacterium glutamicum. J Biotechnol 154:101–113. https://doi.org/10.1016/j.jbiotec.2011.01.017
Patek M, Holatko J, Busche T, Kalinowski J, Nesvera J (2013) Corynebacterium glutamicum promoters: a practical approach. Microb Biotechnol 6:103–117. https://doi.org/10.1111/1751-7915.12019
Portela RMC, Vogl T, Kniely C, Fischer JE, Oliveira R, Glieder A (2017) Synthetic core promoters as universal parts for fine-tuning expression in different yeast species. ACS Synth Biol 6:471–484. https://doi.org/10.1021/acssynbio.6b00178
Pothoulakis G, Ellis T (2018) Construction of hybrid regulated mother-specific yeast promoters for inducible differential gene expression. PloS ONE 13:e0194588. https://doi.org/10.1371/journal.pone.0194588
Raman K, Chandra N (2009) Flux balance analysis of biological systems: applications and challenges. Brief Bioinform 10:435–449. https://doi.org/10.1093/bib/bbp011
Rangel-Chavez C, Galan-Vasquez E, Martinez-Antonio A (2017) Consensus architecture of promoters and transcription units in Escherichia coli: design principles for synthetic biology. Mol Biosyst 13:665–676. https://doi.org/10.1039/c6mb00789a
Redden H, Alper HS (2015) The development and characterization of synthetic minimal yeast promoters. Nat Commun 6:7810. https://doi.org/10.1038/ncomms8810
Redden H, Morse N, Alper HS (2015) The synthetic biology toolbox for tuning gene expression in yeast. FEMS Yeast Res 15. https://doi.org/10.1111/1567-1364.12188
Ross W, Aiyar SE, Salomon J, Gourse RL (1998) Escherichia coli promoters with UP elements of different strengths: modular structure of bacterial promoters. J Bacteriol 180:5375–5383
Rud I, Jensen PR, Naterstad K, Axelsson L (2006) A synthetic promoter library for constitutive gene expression in Lactobacillus plantarum. Microbiology 152:1011–1019. https://doi.org/10.1099/mic.0.28599-0
Seizl M, Hartmann H, Hoeg F, Kurth F, Martin DE, Soding J, Cramer P (2011) A conserved GA element in TATA-less RNA polymerase II promoters. PloS ONE 6:e27595. https://doi.org/10.1371/journal.pone.0027595
Shen HJ, Hu JJ, Li XR, Liu JZ (2015) Engineering of Escherichia coli for lycopene production through promoter engineering. Curr Pharm Biotechnol 16:1094–1103. https://doi.org/10.2174/1389201016666150731110536
Siegl T, Tokovenko B, Myronovskyi M, Luzhetskyy A (2013) Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab Eng 19:98–106. https://doi.org/10.1016/j.ymben.2013.07.006
Solovyev VV, Shahmuradov IA, Salamov AA (2010) Identification of promoter regions and regulatory sites. Methods Mol Biol 674:57–83. https://doi.org/10.1007/978-1-60761-854-6_5
Tirosh I, Berman J, Barkai N (2007) The pattern and evolution of yeast promoter bendability. Trends Genet 23:318–321. https://doi.org/10.1016/j.tig.2007.03.015
Todeschini AL, Georges A, Veitia RA (2014) Transcription factors: specific DNA binding and specific gene regulation. Trends Genet 30:211–219. https://doi.org/10.1016/j.tig.2014.04.002
Troein C, Ahren D, Krogh M, Peterson C (2007) Is transcriptional regulation of metabolic pathways an optimal strategy for fitness? PloS one 2:e855. https://doi.org/10.1371/journal.pone.0000855
Tyo KE, Nevoigt E, Stephanopoulos G (2011) Directed evolution of promoters and tandem gene arrays for customizing RNA synthesis rates and regulation. Methods Enzymol 497:135–155. https://doi.org/10.1016/B978-0-12-385075-1.00006-8
Vasicova P, Patek M, Nesvera J, Sahm H, Eikmanns B (1999) Analysis of the Corynebacterium glutamicum dapA promoter. J Bacteriol 181:6188–6191
Wang W, Li X, Wang J, Xiang S, Feng X, Yang K (2013) An engineered strong promoter for Streptomycetes. Appl Environ Microbiol 79:4484–4492. https://doi.org/10.1128/AEM.00985-13
Wei L, Xu N, Wang Y, Zhou W, Han G, Ma Y, Liu J (2018) Promoter library-based module combination (PLMC) technology for optimization of threonine biosynthesis in Corynebacterium glutamicum. Appl Microbiol Biotechnol 102:4117–4130. https://doi.org/10.1007/s00253-018-8911-y
Wu J, Liu P, Fan Y, Bao H, Du G, Zhou J, Chen J (2013) Multivariate modular metabolic engineering of Escherichia coli to produce resveratrol from L-tyrosine. J Biotechnol 167:404–411. https://doi.org/10.1016/j.jbiotec.2013.07.030
Xu P, Wang W, Li L, Bhan N, Zhang F, Koffas MA (2014) Design and kinetic analysis of a hybrid promoter-regulator system for malonyl-CoA sensing in Escherichia coli. ACS Chem Biol 9:451–458. https://doi.org/10.1021/cb400623m
Yadav VG, De Mey M, Lim CG, Ajikumar PK, Stephanopoulos G (2012) The future of metabolic engineering and synthetic biology: towards a systematic practice. Metab Eng 14:233–241. https://doi.org/10.1016/j.ymben.2012.02.001
Yang L, Wang J, Lv Y, Hao D, Zuo Y, Li X, Jiang W (2014) Characterization of TATA-containing genes and TATA-less genes in S. cerevisiae by network topologies and biological properties. Genomics 104:562–571. https://doi.org/10.1016/j.ygeno.2014.10.005
Yang S, Liu Q, Zhang Y, Du G, Chen J, Kang Z (2018) Construction and characterization of broad-spectrum promoters for synthetic biology. ACS Synth Biol 7:287–291. https://doi.org/10.1021/acssynbio.7b00258
Yim SS, An SJ, Kang M, Lee J, Jeong KJ (2013) Isolation of fully synthetic promoters for high-level gene expression in Corynebacterium glutamicum. Biotechnol Bioeng 110:2959–2969. https://doi.org/10.1002/bit.24954
Yuan LZ, Rouviere PE, Larossa RA, Suh W (2006) Chromosomal promoter replacement of the isoprenoid pathway for enhancing carotenoid production in E. coli. Metab Eng 8:79–90. https://doi.org/10.1016/j.ymben.2005.08.005
Zhang GL, Cao Q, Liu JZ, Liu BY, Li J, Li C (2015) Refactoring -amyrin synthesis in Saccharomyces cerevisiae. Aiche J 61:3172–3179. https://doi.org/10.1002/aic.14950
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 31500044 and No. 31801526), the Natural Science Foundation of Tianjin (No. 17JCQNJC09600, No. 17JCYBJC24000), the Tianjin Science and Technology Project (15PTCYSY00020), the Key Projects in the Tianjin Science and Technology Pillar Program (14ZCZDSY00058) and “Hundred Talents Program” of Chinese Academy of Sciences for Prof. Jun Liu.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xu, N., Wei, L. & Liu, J. Recent advances in the applications of promoter engineering for the optimization of metabolite biosynthesis. World J Microbiol Biotechnol 35, 33 (2019). https://doi.org/10.1007/s11274-019-2606-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-019-2606-0