
Abstract
Due to the lack of efficient control elements and tools, the fine-tuning of gene expression in the multi-gene metabolic pathways is still a great challenge for engineering microbial cell factories, especially for the important industrial microorganism Corynebacterium glutamicum. In this study, the promoter library-based module combination (PLMC) technology was developed to efficiently optimize the expression of genes in C. glutamicum. A random promoter library was designed to contain the putative − 10 (NNTANANT) and − 35 (NNGNCN) consensus motifs, and refined through a three-step screening procedure to achieve numerous genetic control elements with different strength levels, including fluorescence-activated cell sorting (FACS) screening, agar plate screening, and 96-well plate screening. Multiple conventional strategies were employed for further precise characterizations of the promoter library, such as real-time quantitative PCR, sodium dodecyl sulfate polyacrylamide gel electrophoresis, FACS analysis, and the lacZ reporter system. These results suggested that the established promoter elements effectively regulated gene expression and showed varying strengths over a wide range. Subsequently, a multi-module combination technology was created based on the efficient promoter elements for combination and optimization of modules in the multi-gene pathways. Using this technology, the threonine biosynthesis pathway was reconstructed and optimized by predictable tuning expression of five modules in C. glutamicum. The threonine titer of the optimized strain was significantly improved to 12.8 g/L, an approximate 6.1-fold higher than that of the control strain. Overall, the PLMC technology presented in this study provides a rapid and effective method for combination and optimization of multi-gene pathways in C. glutamicum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
With development of synthetic biology and metabolic engineering, many microbial cell factories have been genetically engineered for the conversion of renewable feedstocks into valuable compounds (Keasling 2012; Mao et al. 2017). Due to the complexity of metabolic networks, whether the engineering of endogenous target pathways or the introduction of heterologous biosynthesis pathways always disturbs the native metabolism in the hosts, and generates flux imbalances of metabolic pathways (Lo et al. 2013). One of the major challenges for the engineering of microbial cell factories is to balance the overall metabolic fluxes by orchestrating the expression of genes in the multi-gene pathways (Xu et al. 2012). Several previous studies have attempted to develop a series of precisely controlled elements to fine-tune gene expression, including multifarious plasmid libraries with different copy numbers (Juminaga et al. 2012), promoter strengths (Du et al. 2012; Islam et al. 2017), ribosome binding sites (RBSs) (Nowroozi et al. 2014), and terminators (Jeschek et al. 2017). Overall, based on these endeavors, the multivariate modular metabolic engineering for pathway and strain optimization has greatly increased our ability to design and generate microbial cell factories for industrial applications.
Corynebacterium glutamicum, a nonpathogenic Gram-positive bacterium, has been widely used for the industrial production of various amino acids, vitamins, and nucleotides (Zhang et al. 2017). Owing to the lack of well-characterized genetic control elements and efficient approaches for the modular assembly, each module of multi-gene pathways needs to be individually combined and optimized to satisfy practical requirements for high yields in most metabolic engineering projects (Feng et al. 2016; Zhang et al. 2015a; Zhang et al. 2015b). The cumbersome and laborious optimization procedures severely limit the applications of this important industrial organism. Recently, several regulatable gene expression systems have been reported to achieve the desired expression level in C. glutamicum (Rytter et al. 2014; Yim et al. 2013). However, due to PCR amplification bias of fully random templates (Kanagawa 2003) or the lack of proper screening methods, the diversity and precision of the promoter library is limited. Ideally, the promoter elements should be standardized by engineering and mechanical-streamlining principles, and be designed as plug-and-play biological parts for the predictable tuning expression of target genes (Engler et al. 2014; Xu et al. 2012).
Generally, promoters in bacteria require the assembly and action of the RNA polymerase (RNAP) for initiation of transcription (Patek et al. 2013). The σ factor mediates the recognition of specific promoter sequences and the activation of RNAP holoenzyme (Patek and Nesvera 2011). In C. glutamicum, seven σ factors are identified and divided into three groups, the primary factor σA, the primary-like factor σB, and the alternative factors σC, σD, σE, σH, and σM (Patek et al. 2013). The σA and σB factors are present in all sequenced corynebacterial species and form the largest described group (Patek et al. 2013; Patek and Nesvera 2011). Therefore, functional synthetic promoter elements should contain the same core elements of σA- and σB-dependent promoters. However, the nucleotide sequences in the − 35 and − 10 regions of these native promoters derived from C. glutamicum genome are much less conserved than those of Escherichia coli and other bacteria (Patek et al. 2013). In C. glutamicum, the consensus core sequences of σA-dependent promoters were presumed to contain TTGNCA for the − 35 motif and GNTANANTNG for the − 10 motif, and the consensus core sequences of σB-dependent promoters were speculated to contain GNGNCN for the − 35 motif and TAMAATTGA for the − 10 motif, according to statistical sequence analysis of the promoters of housekeeping genes (Patek et al. 2013).
L-threonine, an essential branched-chain amino acid, is widely used in the food, pharmaceutical, and cosmetic industries (Liu et al. 2015). The large market and valuable applications have provided a strong impetus to further improve the large-scale production of L-threonine. C. glutamicum is regarded as a food-safe organism, and exhibits great potential for the industrial production of threonine (Dong et al. 2011; Zhang et al. 2015a). In C. glutamicum, five important enzymes are responsible for the biosynthesis of threonine from aspartate, including aspartate kinase (LysC), aspartyl-semialdehyde dehydrogenase (Asd), homoserine dehydrogenase (Hom), homoserine kinase (ThrB), and threonine synthase (ThrC) (Dong et al. 2011) (Fig. S1). The inappropriate modulation of gene expression in this metabolic pathway usually leads to the accumulation of an intermediate metabolite L-homoserine, which is detrimental for host cell growth (Dong et al. 2011; Lee et al. 2013). Thus, fine-tuned expression of genes in the pathway to balance metabolic fluxes has become an important aspect for further improvement of threonine production.
In this study, a synthetic promoter library was rationally designed by statistical analysis of nucleotide distributions in the native promoters of housekeeping genes in C. glutamicum, and was sorted by a three-step screening procedure, including the fluorescence-activated cell sorting (FACS) screening, agar plate screening, and 96-well plate screening. Subsequently, a rapid and efficient multi-module combination and optimization technology was developed based on the convenient and well-characterized promoter elements. Using this technology, the threonine biosynthesis pathway was reconstructed and optimized. Finally, a more efficient threonine biosynthesis pathway was obtained, which reduced the accumulation of undesired by-products and greatly increased threonine production.
Materials and methods
Strains, plasmids, media, and culture conditions
The strains used in this study were listed in Table 1. E. coli DH5α was used as the host for gene cloning and plasmid construction. The restriction-deficient C. glutamicum RES 167 was applied to improve the efficiency of transformation for promoter library construction and screening (Tauch et al. 2002). C. glutamicum ATCC 13032 was used as the parent strain for threonine production.
E. coli was grown at 37 °C in LB medium (10 g/L tryptone, 10 g/L NaCl, and 5 g/L yeast extract), and C. glutamicum was cultivated at 30 °C in LBHIS medium (5 g/L tryptone, 5 g/L NaCl, 2.5 g/L yeast extract, 18.5 g/L brain-heart infusion powder and 91 g/L sorbitol) (Becker et al. 2011). When needed, chloramphenicol was added to a final concentration of 12 or 34 mg/L for C. glutamicum or E. coli, respectively. For fermentative production of threonine, the seed medium contained the components as follows: 30 g/L glucose, 30 g/L corn steep liquor, 5 g/L (NH4)2SO4, 2 g/L urea, 1 g/L KH2PO4, and 0.5 g/L MgSO4. The fermentation medium contained the components as follows: 100 g/L glucose, 20 g/L corn steep liquor, 20 g/L (NH4)2SO4, 1 g/L KH2PO4, 1 g/L K2HPO4, 0.5 g/L MgSO4, 0.01 g/L MnSO4, 0.01 g/L FeSO4, 0.2 g/L L-isoleucine, 0.2 g/L methionine, 2 mg/L vitamin B1, 4 mg/L vitamin B6, and 0.1 mg/L biotin.
Construction of the random promoter library in C. glutamicum
The plasmids used in this study were listed in Table 2. All DNA manipulations of E. coli and C. glutamicum were carried out according to standard methods (Sambrook et al. 2001). All primers used in this study were listed in Table S1. The reporter plasmid pXMJ21 was created to determine the strength of the promoters by inserting the enhanced green fluorescent protein (EGFP) coding sequence into pXMJ20 backbone, which was generated by removing the BsaI recognition site from pXMJ19 using the primers J19mut-F/J19mut-R (Fig. S2). The Trc promoter Ptrc was amplified from pTrc99a using the primers pTrc-F/pTrc-R, and cloned into pXMJ21 to yield plasmid p-Trc, which was used as the positive control. Moreover, the native promoters of the genes cg2195, cg1791, cg3219, cg0587, and cg3237 were selected to demarcate the expression strength of the combinatorial promoters. These promoters were amplified from the genomic DNA of C. glutamicum ATCC 13032 and individually inserted into pXMJ21 to generate the plasmids p-2195, p-1791, p-3219, p-0587, and p-3237.
For the construction of promoter library, the 120-bp random primer (RDP) containing an 80-bp designed promoter sequence, the BsaI restriction enzyme recognition site, and the EGFP forward primer was chemically synthesized by Genewiz Biotech Co., Ltd. (Suzhou, China) (Fig. 1a). Subsequently, an EGFP fragment including the varied promoters was obtained by PCR amplification using the primers RDB/EGFP-R, and cloned into pXMJ20 via Golden Gate assembly. After transformation and plasmid extraction from E. coli DH5α, the purified plasmid library was used to electro-transform C. glutamicum RES 167 to construct the promoter library as described previously (Kirchner and Tauch 2003).

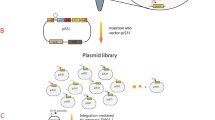
Design and construction of the promoter library. a Schematic diagram of the designed random promoters. The promoters with an overall 80-bp length were designed with random bases (indicated with N) at strategic positions. The yellow, red, and green regions indicate the putative − 35, − 10, and ribosome binding site (RBS) regions, respectively. b Integration of the promoter library within the reporter plasmid pXMJ20 for promoter screening
High-throughput screening of the promoter library
The transformed cells were streaked onto LBHIS agar plates containing 12 mg/L chloramphenicol, and grown at 30 °C for 48 h to avoid negative interference. The resulting cells were washed from the agar plates with phosphate-buffered saline (PBS), and harvested by centrifugation at 8000×g for 10 min at 4 °C. For sample preparation, the cells were washed twice and resuspended in PBS buffer, and then vortexed for 5 min to obtain a uniform suspension. Before the screening procedure, the cell suspension was diluted to an optical density at 600-nm wavelength (OD600) of 0.1 to avoid internal interference. The cells were screened using the MoFlo XDP Flow Cytometry Sorter (Beckman Coulter Inc., Fullerton, CA, USA). The fluorescence signal of the EGFP reporter was excited at 488 nm and emission was detected using a 530/40 band-pass filter. The “Purify model” of the FACS was used as the sorting model to improve the ratio of positive cells in the library. All isolated cells were cultivated on LBHIS agar plates at 30 °C for 48 h for further analysis. The resulting cells were selected according to the fluorescence intensity of EGFP excited by blue light (476–495 nm). Part of the cells were selected and transferred onto fresh LBHIS agar plates. Finally, the 96-well plate screening was implemented to further accurately characterize the isolated promoters of various strengths. The cells were cultivated in LBHIS liquid medium for 18 h in the 96-well plate, and the fluorescence intensity was measured using a multi-detection microplate reader (SpectraMax M2, Molecular Devices, CA, USA) at excitation and emission wavelengths of 488 and 530 nm, respectively.
Real-time quantitative PCR analysis
The transcription strengths of the promoters were determined by quantitative PCR (qPCR). The samples were cultivated in LBHIS liquid medium for 18 h, and harvested by centrifugation at 10,000×g for 10 min at 4 °C. The total RNA was isolated using the RNA-prep Pure Plant Kit (Tiangen Biotech Co. Ltd., Beijing, China) according to the manufacturer’s protocol. The quantity of harvested RNA was measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). Quantitative reverse transcription PCR was performed according to a previously described method (Du et al. 2012). The transcription level of the 16S rRNA gene was used as internal control to normalize the samples. For comparison of the transcription strengths of different promoters, the transcription level of the positive control, Trc promoter, was defined as 1. The primers used for qPCR amplification were listed in Table S1.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was carried out to analyze the protein expression of the screened promoters according to the method described previously (Yim et al. 2013). For protein preparation, the cells were cultivated in LBHIS liquid medium at 30 °C for 48 h. Then, the samples were centrifuged at 8000×g for 10 min at 4 °C and resuspended in Tris-NaCl lysis buffer (200 mM NaCl, 20 mM Tris HCl, pH 7.5). Following, the cells were sonicated for 15 min on ice, and centrifuged at 15,000×g for 20 min at 4 °C. The supernatants were analyzed by electrophoresis on an SDS-PAGE gel.
Construction of lacZ reporter system
The lacZ reporter system was also constructed to benchmark the promoter library. The H36 and p7 promoters, previously shown to be strong promoters (Rytter et al. 2014; Yim et al. 2013), were embedded into the lacZ gene forward primer and chemical synthesized by Genewiz Biotech Co., Ltd. (Suzhou, China). After fusion of the selected promoter elements with lacZ reporter gene, the fragments were cloned into pXMJ20 by Golden Gate assembly, respectively. Then, these plasmids were transformed into the C. glutamicum RES 167 by electroporation, respectively. The β-galactosidase activity was measured using the method described previously (Israelsen et al. 1995), and calculated as Miller units (MU) (Rytter et al. 2014).
Gene deletion and mutation in C. glutamicum
The genetic manipulation of C. glutamicum was performed according to the standard methods (Sambrook et al. 2001). For construction of the ilvA deletion strain, the upstream and downstream flanking regions were, respectively, amplified from the genomic DNA of C. glutamicum using the primers ilvAUP-F/ilvAUP-R and ilvADn-F/ilvADn-R, and fused by overlap extension PCR using the primers ilvAUP-F/ilvADn-R. The final PCR product was digested with BamHI and XbaI, ligated into the pCRD206 treated with the same enzyme to yield the plasmid pCRDilvA. C. glutamicum mutant strain was achieved by a two-step homologous recombination using the temperature-sensitive plasmid pCRD206 as described previously (Okibe et al. 2011). The ilvA deletion strain was confirmed by PCR with the primer pair ilvACF/CR. The metX and tdcB deletion strains were created using the same strategy. The point mutation of LysC (T311I) was carried out to remove the feedback inhibition by threonine. The upstream and downstream flanking regions containing the point mutation at T311I site were amplified from the genomic DNA of C. glutamicum using the primers lysCUp-F/lysCUp-R and lysCDn-F/LysCDn-R, respectively. Then, the fused fragment was digested with BamHI and XbaI, and inserted into the same sites of pCRD206 via to obtain the mutant plasmid pCRMlysC. The point mutation of Hom (G378E) was implemented according to the same method.
Promoter library-based module combination for the optimization of threonine biosynthesis pathway
For the stable expression of genes in C. glutamicum, the low-copy plasmid pXMJ22 was developed by replacing the pBL1 replicon of pXMJ20 with the antisense RNA-mediated pCGR2 replicon, the copy number of which has been demonstrated to be less than three copies per chromosome (Okibe et al. 2010) (Fig. S2). Taking into account the combination of modules via Golden Gate assembly, the restriction enzyme BsaI recognition sites of the relevant genes, asd, homR (homoserine dehydrogenase threonine-feedback-resistant mutant), and thrC, were silently mutated using the primers listed in Table S1 and cloned into pXMJ20. For application of the promoter library-based module combination (PLMC) technology, the long primer pairs contained varied promoter sequences and T7 terminator were chemically synthesized and used to amplify the threonine biosynthesis modules. The vector backbone pXMJ22 and various obtained modules were mixed at equimolar concentrations in a 20 μL Golden Gate reaction solution. The reaction was implemented with the standard procedure as reported previously (Peccoud et al. 2011), and the ligation product was transformed into E. coli DH5α to obtain the recombinant plasmids. Subsequently, the recombinant plasmids were electro-transformed into the threonine-producing C. glutamicum rLHdAXB strain to determine the optimal combination of multiple modules for better productive performance.
Homoserine dehydrogenase activity assay
The activity of homoserine dehydrogenase was measured by monitoring the absorbance of NADPH at 340 nm according to the method published previously (Dong et al. 2016). The sample was harvested at 48 h, separated from the fermentation medium by centrifugation, and resuspended in Tris-NaCl lysis buffer. Subsequently, the harvested cells were sonicated for 15 min on ice and centrifuged at 15,000×g for 20 min at 4 °C. The resulting supernatants were used for enzyme activity assays. The protein concentration was determined by bicinchoninic acid (BCA) protein assay (Pierce, USA) according to the manufacturer’s protocol.
Analysis of fermentation profiles
For fermentation analysis, threonine-producing strains were activated in LBHIS liquid medium and cultivated in the seed medium for 18 h at 30 °C. Then, the cells were inoculated into 50 mL of fermentation medium in a 500-mL Erlenmeyer flask, and cultivated at 30 °C for 72 h. The initial OD600 was adjusted to 1. The concentrations of amino acids were assayed using a high-performance liquid chromatography (HPLC) system (Agilent 1260, USA) with a ZORBAX Eclipse AAA column (4.6 × 150 mm, 5 μm; Agilent, USA), according to the standard methods described previously (Becker et al. 2011).
Results
Design and construction of the promoter library
The consensus core elements of C. glutamicum promoter were designed to contain NNGNCN for the − 35 motif and NNTANANT for the − 10 motif according to the key elements of σA- and σB-dependent promoters, and the ribosome binding site (RBS), speculated as a common AAAGGA motif (Zhang et al. 2015a), was also included. For the convenient application of the synthetic promoter library, the BsaI recognition sites and the forward primer of the EGFP reporter gene were also fused into the random promoter (Fig. 1a). The random synthetic promoter library with the EGFP reporter gene was achieved by PCR amplification using the designed primers, and inserted into pXMJ20 to construct the promoter library (Fig. 1b). In order to improve the plasmid transformation efficiency, the restriction-deficient C. glutamicum RES167 was used as the host for analysis of the promoter library (Tauch et al. 2002). Finally, a large-size promoter library (about 5 × 106 clones) was constructed.
High-throughput screening of the promoter library
A three-step high-throughput screening method was established for screening the promoter library. The samples of library were firstly examined using FACS analysis to verify the practicability of the library. As shown in Fig. 2a, the fluorescence intensity of the positive control, Cg-Trc, was significantly higher than that of the negative control, Cg-pXMJ20. Moreover, the fluorescence intensity of samples in the library had a wide range, and was obviously higher than that of the negative control, indicating that the FACS-based screening strategy was reliable. In order to ensure the capacity and coverage of the promoter library, about 5 × 107 cells were screened by FACS. According to the fluorescence intensity, the top 0.04% of the screened cells was sorted and cultivated on LBHIS agar plates (Fig. 2b). Subsequently, the agar plate screening was carried out for further evaluation of the screened library based on the visible fluorescence signal of EGFP reporter under excitation by blue light (Yang et al. 1996). As shown in Fig. 2c, the cells containing promoters with various strengths were emitted distinctly different visible fluorescent signal. Finally, the fluorescent intensity of the selected cells was measured by the 96-well plate assay to determine the precise expression strength. The fluorescence intensities of the sorted cells displayed a considerable range from 300 to 14,000 a.u. (Fig. 2d), suggesting that the three-step screening process yielded a promoter library with a wide range.

Sorting of the synthetic promoter library using a three-step screening procedure. a FACS analysis of the promoter library. Cg-pXMJ20 (black curve) was used as the negative control, and Cg-Trc (red curve) was used as the positive control. The blue curve represents samples from the synthetic promoter library. Of the samples, 60,000 clones were used for FACS analysis. b FACS screening of the promoter library. The blue square refers to the top 0.04% of all screened cells. c Visual agar-plate screening. The cells were cultivated on LBHIS agar plates for 48 h before detection. d Ninety-six-well-plate screening. The fluorescence intensities of 760 selected strains were measured by a multi-detection microplate reader
Detailed analysis of the promoter library
According to the fluorescence intensity measured, the sorted promoter library was classified into three groups: high-strength group (more than 8000 a.u.), medium-strength group (3000–6000 a.u.), and low-strength group (200–2000 a.u.) (Figs. 2d and 3a). A total of 30 promoters were randomly selected from these three groups for further individual analysis. Sequence analysis was carried out using BPROM software (http://linux1.softberry.com/berry.phtml) (Fig. S3). The result indicated that the − 10 motifs of the promoter library were mostly conformed to the designed sequence (NNTANANT) despite the different locations, whereas the − 35 motifs of the promoter library were less conserved. The distance between − 10 and − 35 motif was predicted to be 10–16 bp, which was not quite consistent with the designed 17 bp. Furthermore, the nucleotide usage frequency analysis of the promoters from high-strength group exhibited that the whole artificial 80-bp sequence was AT-rich, especially from 9 to 67 bp (Fig. S4), which was consistent with the promoters from other prokaryotic bacteria (Patek et al. 2013). Finally, an archetypical strong promoter sequence was proposed based on these statistics, which can be regarded as a universal promoter element for the high regulatable expression of target genes in C. glutamicum (Fig. S4).
In order to calibrate the expression strength of these artificial promoters, several native promoters of C. glutamicum were selected and compared, including the promoters from cg2195 encoding a putative membrane protein, cg1791 encoding glyceraldehyde 3-phosphate dehydrogenase, cg3219 encoding lactic dehydrogenase, cg0587 encoding elongation factor Tu, and cg3237 encoding superoxide dismutase (Kim et al. 2016). As shown in Fig. 3a, most of the sorted promoters displayed higher fluorescence intensity than that of the positive control (PTrc). Moreover, most of the promoters from high-strength group exhibited higher fluorescence intensities than those of the commonly strong native promoters, suggesting that the artificial promoters from high group were more suitable for high-regulated gene expression. Furthermore, the expression strengths of these artificial promoters were also determined by qPCR, FACS analysis, and SDS-PAGE (Figs. 3b, c and S5). The relative expression of the promoters at the transcriptional level exhibited the same trend as the fluorescence intensity data (Fig. 3b). The transcriptional levels of promoters from high-strength group were obviously higher than those of the promoters from medium-strength group, which were also greatly stronger than those of the promoters from low-strength group. The protein levels of EGFP reporter were positively correlated with the strength of promoters, and the EGFP reporter from the high-strength group showed the highest protein concentration at the translational level (Fig. S5). These observations were also demonstrated by the mean fluorescence of promoters from different groups (Fig. 3c). Finally, the lacZ reporter system was used to benchmark the promoter library. The β-galactosidase activity of H10 strain was 8978.6 ± 531.3 MU, while the positive control pTrc only had an activity of 512.8 ± 50.2 MU (Fig. 3d). As a comparison, the expression levels of previously described strong promoters H36 and P7 were also measured (Rytter et al. 2014; Yim et al. 2013). The activities of H36 and P7 promoters were 6887.6 ± 401 and 5798.3 ± 312.5 MU, respectively, which were obviously lower than that of H10 promoter.

Analysis of the synthetic promoter library in C. glutamicum. a Analysis of fluorescence intensities of the promoter library. Cg-pXMJ20 was used as the negative control, and Cg-Trc was used as the positive control. The native promoters of cg3219 (lactic dehydrogenase), cg0587 (Tu elongation factor), cg3237 (superoxide dismutase), cg2195 (membrane protein), and cg1791 (glyceraldehyde 3-phosphate dehydrogenase) were used to calibrate the synthetic promoter strengths. b Real-time quantitative PCR analysis of the promoter library. The expression level of the Trc promoter was normalized as 1 as the positive control. c FACS analysis of three promoters from the different groups. The fluorescence signals of L6, M6, and H6 were shown in green, blue, and red, respectively. The Trc promoter shown in black was used as the control. d The activities of β-galactosidase under the control of different promoters were measured. All data and standard errors were derived from three independent biological replicates
PLMC technology for the optimization of multi-gene biosynthesis pathways in C. glutamicum
Generally, unbalanced pathway fluxes are severe obstacles for the high-level production of target chemicals (Ajikumar et al. 2010). Thus, the PLMC technology was developed for rapid and efficient combination and optimization of modules in multi-gene pathway in this study (Fig. 4). As shown in Fig. 4a, a pair of long primers, containing the varied promoters at the 5′ end of the forward primer and the terminator at the 5′-end of the reverse primer, was designed to facilitate the assembly. With these primers, various modules regulated by the designed promoters were achieved by one-step PCR amplification (Fig. 4b). Subsequently, the multiple genetic modules in the pathway were seamlessly assembled via Golden Gate assembly to directly form an entire expression vector library (Fig. 4c). Finally, a more efficient and balanced pathway could be obtained according to the titer of the target product (Fig. 4d, e). Therefore, the PLMC technology significantly reduces the redundant and duplicated efforts for matching the control elements with target genes and constructing the various plasmids in the combination and optimization of multi-module pathway (Zhang et al. 2015a; Zhang et al. 2015b).

Schematic diagram of the promoter library-based module combination (PLMC) technology. a The primers designed for PLMC technology. b Construction of varied modules. c Combination of multi-modules via Golden Gate assembly to generate a plasmid library. P promoter, T terminator, B BasI restriction sites. d, e Comprehensive evaluation of the various clones. The best candidate was selected according to the titer of the target production
Module optimization to improve threonine production in C. glutamicum
The threonine-producing C. glutamicum strain was constructed as described in Fig. S1. Threonine dehydratase encoded by ilvA and tdcB were firstly deleted to eliminate the degradation of threonine. In order to remove the feedback inhibition by threonine, the aspartate kinase and homoserine dehydrogenase were introduced the point mutation, LysCT311I and HomG378E, respectively. Homoserine acetyltransferase (MetX) was also deleted to remove the competitive pathway of precursor homoserine. Finally, the initial strain rLHdAXB produced 2.1 g/L of threonine, whereas this strain still accumulated 3.2 g/L of homoserine and 2.3 g/L of lysine as the major by-products. The low titer was largely attributed to the unbalanced pathway fluxes, and overmuch metabolic flux was wasted to generate by-products.
In order to improve the threonine production, the threonine biosynthesis pathway was reconstructed and optimized using the PLMC technology. To facilitate the construction of genetic modules, three different strength promoters, L6, M6, and H6, were randomly selected from three strength groups to regulate the expression of genes (Fig. 3c). In C. glutamicum, the first enzyme of threonine biosynthesis pathway, LysCR (LysC threonine-feedback-resistant mutant), has been demonstrated as the key node driving the carbon flux into the threonine biosynthesis pathway (Dong et al. 2011). Hence, the key enzyme, LysCR, was expressed using high-strength promoter to improve the metabolic flux of the pathway (Fig. 5a). A mathematical model describing the combinatorial approach was applied to simplify the optimization of four genetic modules as described previously (Zhang et al. 2015a) (Fig. 5b). The various module combinations were scaled down from the theoretical 81 to 9, and assembled with the PLMC technology (Fig. 5c).

Combination and optimization of multi-module for threonine biosynthesis. a The structure and components of the threonine biosynthetic modules. The arrows in different colors indicate different expression strengths of the genes. Red “T” symbol represents the terminator. b The mathematic model of combinatorial approach for multi-module optimization. Three levels of promoter strength (H6, M6, and L6) were selected to control the expression of target genes. c Assembly of the threonine biosynthesis pathway modules. P promoter, T terminator
The fermentation profile analysis of the candidate strains was carried out with the initial strain rLHdAXB harboring the empty plasmid pXMJ22 as the control. The results confirmed the presence of clear variations on both threonine production and growth among different module combinations (Fig. 6a). In particular, the threonine titers of strains A, B, D, and H reached 8.9, 10.1, 12.8, and 11.3 g/L, respectively, which was distinctly higher than that of the control stain. Importantly, strain D exhibited the highest threonine titer among all of the strains, an almost 6.1-fold increase compared with the control strain, indicating that the module LHAMHHBMCL was the best combination for threonine biosynthesis in C. glutamicum. Moreover, the titer of the by-product lysine was low in strain D (1.8 g/L), relative to other strains (Fig. 6b). Besides, the variation of the by-product lysine was correlated to the expression of Hom. It may be because the third enzyme of the threonine pathway, Hom, is located at the metabolic node that competes for a common substrate with the dihydrodipicolinatesynthase of the lysine pathway (Fig. S1). Therefore, the activity of Hom for each strain was also determined to demonstrate the hypothesis. As shown in Fig. 6c, the activity of Hom under the control of high-strength promoters exhibited almost 11-fold higher than that under the control of low-strength promoters, supporting the opinion that the high-level expression of Hom was more propitious to drive fluxes toward threonine biosynthesis. In addition, the biochemical stability of Hom-overexpression strains was determined by evaluating the activities of Hom after 20 generations of passage. There was no significant difference between the original and subcultured strains (Fig. S6), revealing that the overexpression of hom with low-copy vector generated in this study was effective and stable as described previously (Reinscheid et al. 1994). Nevertheless, the by-product homoserine of the strain D was still found even though it was much lower in comparison with the control strain (Fig. 6b). This is mainly because the activity of ThrB is competitively inhibited by threonine but is relieved with the increase of homoserine (Dong et al. 2011). Therefore, the optimized combination of ThrB expression and an appropriate concentration of homoserine was required for threonine accumulation. By contrast, the high expression of Asd and ThrC led to only a marginal increase of threonine accumulation, which was consistent with the previous reports (Dong et al. 2011; Eikmanns et al. 1991; Zhang et al. 2015c).

Fermentation profile analysis of C. glutamicum strains carrying the different genetic module combinations. a Threonine production (orange column) and cell growth (black circle) of the candidate strains. Strain rLHdAXB harboring pXMJ22 was used as the control. b The titer of by-products lysine and homoserine. c The activity of homoserine dehydrogenase (Hom) in various tested strains. All data and standard errors were derived from three independent biological replicates
Discussion
One important goal of synthetic biology is the development of well-characterized, standardized, and reusable elements that can be more easily and rapidly assembled into an industrial organism with the desired properties (Xu et al. 2012). A number of elements have been established, designed, collected, and registered in several databases, such as the iGEM Registry of Standard Biological Parts (http://parts.igem.org) and the AddGene database (www.addgene.org). However, the lack of well-characterized genetic control elements is still a limiting factor for the design of platform strains and the assembly of desired pathways in C. glutamicum (Li and Borodina 2015). In view of its industrial importance, the paucity of control biobricks for the engineering of C. glutamicum is particularly pressing. In the recent years, several research groups have developed some promoter control elements to regulate gene expression in C. glutamicum (Yim et al. 2013; Rytter et al. 2014). Yim et al isolated and characterized multiple potential promoters of various strengths from fully synthetic random sequences, including the strongest promoter H36 ever reported in the literature. Rytter et al established synthetic promoter libraries using the β-galactosidase reporter to assess promoter strength. However, PCR amplification bias of fully random templates generally reduces the diversity of the promoter library (Kanagawa 2003), and the single-stage screening procedure of promoter library also makes the assessment of promoter strength insufficiency and imprecision. In order to develop a comprehensive promoter library, the core sequence of an overall length of 80 bp was designed to contain − 35 NNGNCN motif, − 10 NNTANANT motif, and a common RBS AAAGGA motif (Fig. 1a). A total of about 5 × 107 clones were screened by a three-step, high-throughput screening method. Fortunately, the promoter library exhibited an efficient regulatory capacity with a wide range. In particular, the β-galactosidase activity of H10 strain was 1.3 and 1.5 times higher than those of H36 and P7, respectively (Fig. 3d). To the best of our knowledge, H10 promoter is the strongest promoter so far identified in C. glutamicum (Pátek et al. 2003; Patek and Nesvera 2011; Yim et al. 2013). According to the statistics of nucleotide usage frequency in high-strength promoters, a high-strength RBS sequence was also predicted as AAAGGAAGKRY (Fig. S4), which could also be used for tuning gene expression at the translational level. However, the space distances between the − 10 and − 35 regions of the sorted promoters were 10–16 bp (Fig. S3), which was not consistent with the designed 17 ± 1-bp space as described previously (Patek et al. 2013). The most likely reason is that the available software for promoter analysis was exploited with the general bacterial but not C. glutamicum-specific, which probably made the prediction imperfect (Yim et al. 2013). To sum up, the promoter library exhibited advantages of wide-regulated range with satisfied accuracy and simple constitute, making it be efficiently applied to tune the expression of target genes for metabolic engineering in C. glutamicum.
For engineering of cell factories, the unregulated expression of several related pathway genes, whether native or heterologous, can cause a large metabolic burden due to the futile expression of a large number of enzymes or the accumulation of toxic metabolic intermediates (Biggs et al. 2014). Many advanced strategies have been designed to optimize the carbon flux toward the target product through systematic investigation of pathway input variables (Biggs et al. 2014; Keasling 2012). Xu et al. has combined the plasmid library with RBS elements to tune gene expression for optimization of fatty acid production (Xu et al. 2013). Moreover, the ePathbrick method was developed to achieve a diversified combination library by iteratively integrating multiple regulatory biobricks into the ePathBrick vector with compatible restriction enzyme (Xu et al. 2012). However, their strategies are implemented through several rounds of plasmid construction, which are time-consuming and labor-intensive. Therefore, there is an urgent need for convenient and practical tools that enable multivariate modular metabolic engineering. In this study, the PLMC technology was developed, based on the Golden Gate assembly, to streamline the process of reconstruction and optimization of pathway modules in C. glutamicum (Fig. 4). Golden Gate assembly is an efficient DNA construction method based on type II restriction enzymes, which can cleave the target DNA fragment outside the recognition site to form 3–4-bp cohesive ends for fragment ligation (Engler et al. 2014; Peccoud et al. 2011). In the assembly process, the irreversible ligation enormously enhances the efficiency of assembly compared with the classical restriction-ligation methods (Werner et al. 2012). With the rational design of the sticky-end nucleotides, the Golden Gate assembly method is able to effectively generate a multi-gene shuffled library spanning more than 30 kb (Engler et al. 2014; Peccoud et al. 2011), which suggests that it is ideally suitable for multi-gene assembly. Combined the Golden Gate assembly and the practical promoter elements, the PLMC technology significantly reduces the time and workload required for the optimization of multi-gene pathways in C. glutamicum.
To date, many efforts have been made to improve threonine production via overexpression of key genes, elimination of feedback inhibition, weakening of the competing branches, and reduction of intracellular consumption (Dong et al. 2011). Nevertheless, the imbalance of metabolic fluxes and accumulation of toxic intermediates remain significant engineering issues that need to be solved. In an innovative approach, a DNA scaffold system is designed to adjust the spatial distribution of pathway enzymes, which improves threonine productivity more than 3-fold, and decreases the accumulation of the toxic intermediate homoserine about 15-fold in E. coli (Lee et al. 2013). However, there are few reports about the engineering of C. glutamicum for threonine production, even though the workhorse has numerous intrinsic advantages. In this study, an efficient threonine cell factory was constructed and optimized using the PLMC technology, and it displayed the highest titer of threonine in C. glutamicum to date (Fig. 6a). Besides the first enzyme LysC, both Hom and ThrB nodes were also identified as the important control valves of threonine biosynthesis through the analysis of fermentation profiles, consistent with the results found in E. coli (Zhang et al. 2015c). In order to construct a more cost-effective threonine producer, reliving the feedback inhibition of ThrB by threonine will need to be solved by protein engineering. However, overexpression of ThrC had little effect on threonine production (Fig. 6a), which was consistent with a previous report in C. glutamicum (Dong et al. 2011), even if it could improve the productivity in E. coli (Lee et al. 2013). Although the titer of threonine was clearly lower compared with that in E. coli, C. glutamicum is still a promising candidate for the industrial production of threonine, and its productivity can be further improved by future metabolic engineering.
To sum up, a convenient and effective promoter element library was successfully constructed with broad range of strength in C. glutamicum. Based on the sorted promoter elements, a rapid and efficient module combination technology was developed to fine-tune the expression of genes in multi-gene pathway. Using the PLMC technology, the threonine biosynthesis pathway was reconstructed and optimized. The titer of threonine was significantly improved, demonstrating that this is an efficient method to construct and optimize pathways in metabolic engineering. Additionally, the PLMC technology would be more efficient by combing with a high-throughput screening method of target products. Fortunately, with the development of synthetic biology, more and more high-throughput screening technologies for several valuable chemicals have been established based on biosensors, riboswitches, and visible light (Mahr et al. 2015; Sang-Woo and Min-Kyu 2015). Therefore, there is reason to believe that the PLCM technology will be widely applied to optimize cell factories and improve the production of target chemicals via metabolic engineering in the future.
References
Ajikumar PK, Xiao WH, Tyo KEJ, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G (2010) Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science 330(6000):70–74. https://doi.org/10.1126/science.1191652
Becker J, Zelder O, Häfner S, Schröder H, Wittmann C (2011) From zero to hero—design-based systems metabolic engineering of Corynebacterium glutamicum for L-lysine production. Metab Eng 13(2):159–168. https://doi.org/10.1016/j.ymben.2011.01.003.
Biggs BW, De Paepe B, Santos CN, De Mey M, Kumaran Ajikumar P (2014) Multivariate modular metabolic engineering for pathway and strain optimization. Curr Opin Biotechnol 29:156–162. https://doi.org/10.1016/j.copbio.2014.05.005
Reinscheid DJ, Kronemeyer W, Eggeling L, Eikmanns BJ, Sahm H (1994) Stable expression of hom-1-thrB in Corynebacterium glutamicum and its effect on the carbon flux to threonine and related amino acids. Appl Environ Microbiol 60:126–132
Dong X, Zhao Y, Zhao J, Wang X (2016) Characterization of aspartate kinase and homoserine dehydrogenase from Corynebacterium glutamicum IWJ001 and systematic investigation of L-isoleucine biosynthesis. J Ind Microbiol Biotechnol 43(6):873–885. https://doi.org/10.1007/s10295-016-1763-5
Dong XY, Quinn PJ, Wang XY (2011) Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for the production of L-threonine. Biotechnol Adv 29(1):11–23. https://doi.org/10.1016/j.biotechadv.2010.07.009
Du J, Yuan YB, Si T, Lian JZ, Zhao HM (2012) Customized optimization of metabolic pathways by combinatorial transcriptional engineering. Nucleic Acids Res 40(18):e142. https://doi.org/10.1093/nar/gks549
Eikmanns BJ, Metzger M, Reinscheid D, Kircher M, Sahm H (1991) Amplification of three threonine biosynthesis genes in Corynebacterium glutamicum and its influence on carbon flux in different strains. Appl Microbiol Biotechnol 34(5):617–622. https://doi.org/10.1007/BF00167910
Engler C, Youles M, Gruetzner R, Ehnert TM, Werner S, Jones JD, Patron NJ, Marillonnet S (2014) A golden gate modular cloning toolbox for plants. ACS Synth Biol 3(11):839–843. https://doi.org/10.1021/sb4001504
Feng L, Zhang Y, Fu J, Mao Y, Chen T, Zhao X, Wang Z (2016) Metabolic engineering of Corynebacterium glutamicum for efficient production of 5-aminolevulinic acid. Biotechnol Bioeng 113(6):1284–1293. https://doi.org/10.1002/bit.25886
Islam ZU, Klein M, Aßkamp MR, ASR Ø, Nevoigt E (2017) A modular metabolic engineering approach for the production of 1,2-propanediol from glycerol by Saccharomyces cerevisiae. Metab Eng doi: https://doi.org/10.1016/j.ymben.2017.10.002
Israelsen H, Madsen SM, Vrang A, Hansen EB, Johansen E (1995) Cloning and partial characterization of regulated promoters from Lactococcus lactis Tn917-lacZ integrants with the new promoter probe vector, pAK80. Appl Environ Microbiol 61(7):2540–2547
Jeschek M, Gerngross D, Panke S (2017) Combinatorial pathway optimization for streamlined metabolic engineering. Curr Opin Biotechnol 47:142–151. https://doi.org/10.1016/j.copbio.2017.06.014
Juminaga D, Baidoo EE, Reddingjohanson AM, Batth TS, Burd H, Mukhopadhyay A, Petzold CJ, Keasling JD (2012) Modular engineering of L-tyrosine production in Escherichia coli. Appl Environ Microbiol 78(1):89–98. https://doi.org/10.1128/AEM.06017-11
Kanagawa T (2003) Bias and artifacts in multitemplate polymerase chain reactions (PCR). J Biosci Bioeng 96(4):317–323. https://doi.org/10.1016/S1389-1723(03)90130-7
Keasling JD (2012) Synthetic biology and the development of tools for metabolic engineering. Metab Eng 14(3):189–195. https://doi.org/10.1016/j.ymben.2012.01.004
Kim HI, Kim JH, Park YJ (2016) Transcriptome and gene ontology (GO) enrichment analysis reveals genes involved in biotin metabolism that affect l-lysine production in Corynebacterium glutamicum. Int J Mol Sci 17(3):353. https://doi.org/10.3390/ijms17030353
Kirchner O, Tauch A (2003) Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J Biotechnol 104(1–3):287–299. https://doi.org/10.1016/s0168-1656(03)00148-2
Lee JH, Jung SC, Bui LM, Kang KH, Song JJ, Kim SC (2013) Improved production of L-threonine in Escherichia coli by use of a DNA scaffold system. Appl Environ Microbiol 79(3):774–782. https://doi.org/10.1128/aem.02578-12
Li M, Borodina I (2015) Application of synthetic biology for production of chemicals in yeast Saccharomyces cerevisiae. FEMS Yeast Res 15(1):1–12. https://doi.org/10.1111/1567-1364.12213
Liu Y, Li Q, Zheng P, Zhang Z, Liu Y, Sun C, Cao G, Zhou W, Wang X, Zhang D, Zhang T, Sun J, Ma Y (2015) Developing a high-throughput screening method for threonine overproduction based on an artificial promoter. Microb Cell Factories 14:121. https://doi.org/10.1186/s12934-015-0311-8
Lo TM, Teo WS, Ling H, Chen B, Kang A, Chang MW (2013) Microbial engineering strategies to improve cell viability for biochemical production. Biotechnol Adv 31(6):903–914. https://doi.org/10.1016/j.biotechadv.2013.02.001
Mahr R, Gatgens C, Gatgens J, Polen T, Kalinowski J, Frunzke J (2015) Biosensor-driven adaptive laboratory evolution of L-valine production in Corynebacterium glutamicum. Metab Eng 32:184–194. https://doi.org/10.1016/j.ymben.2015.09.017
Mao Y, Fu J, Tao R, Huang C, Wang Z, Tang YJ, Chen T, Zhao X (2017) Systematic metabolic engineering of Corynebacterium glutamicum for industrial-level production of optically pure D-(−)-acetoin. Green Chem 19:5691–5702. https://doi.org/10.1039/C7GC02753B
Nowroozi FF, Baidoo EE, Ermakov S, Redding-Johanson AM, Batth TS, Petzold CJ, Keasling JD (2014) Metabolic pathway optimization using ribosome binding site variants and combinatorial gene assembly. Appl Microbiol Biotechnol 98(4):1567–1581. https://doi.org/10.1007/s00253-013-5361-4
Okibe N, Suzuki N, Inui M, Yukawa H (2010) Antisense-RNA-mediated plasmid copy number control in pCG1-family plasmids, pCGR2 and pCG1, in Corynebacterium glutamicum. Microbiology 156(Pt 12):3609–3623. https://doi.org/10.1099/mic.0.043745-0
Okibe N, Suzuki N, Inui M, Yukawa H (2011) Efficient markerless gene replacement in Corynebacterium glutamicum using a new temperature-sensitive plasmid. J Microbiol Methods 85(2):155–163. https://doi.org/10.1016/j.mimet.2011.02.012
Pátek M, Nešvera J, Guyonvarch A, Reyes O, Leblon G (2003) Promoters of Corynebacterium glutamicum. J Biotechnol 104(1–3):311–323. https://doi.org/10.1016/s0168-1656(03)00155-x
Patek M, Holatko J, Busche T, Kalinowski J, Nesvera J (2013) Corynebacterium glutamicum promoters: a practical approach. Microb Biotechnol 6(2):103–117. https://doi.org/10.1111/1751-7915.12019
Patek M, Nesvera J (2011) Sigma factors and promoters in Corynebacterium glutamicum. J Biotechnol 154(2–3):101–113. https://doi.org/10.1016/j.jbiotec.2011.01.017
Peccoud J, Weber E, Engler C, Gruetzner R, Werner S, Marillonnet S (2011) A modular cloning system for standardized assembly of multigene constructs. PLoS One 6(2):e16765. https://doi.org/10.1371/journal.pone.0016765
Rytter JV, Helmark S, Chen J, Lezyk MJ, Solem C, Jensen PR (2014) Synthetic promoter libraries for Corynebacterium glutamicum. Appl Microbiol Biotechnol 98(6):2617–2623. https://doi.org/10.1007/s00253-013-5481-x
Sambrook J, Fritsch EF, Maniatis T (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, New York
Sang-Woo L, Min-Kyu O (2015) A synthetic suicide riboswitch for the high-throughput screening of metabolite production in Saccharomyces cerevisiae. Metab Eng 28:143–150. https://doi.org/10.1016/j.ymben.2015.01.004
Tauch A, Kirchner O, Loffler B, Gotker S, Puhler A, Kalinowski J (2002) Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr Microbiol 45(5):362–367. https://doi.org/10.1007/s00284-002-3728-3
Werner S, Engler C, Weber E, Gruetzner R, Marillonnet S (2012) Fast track assembly of multigene constructs using golden gate cloning and the MoClo system. Bioeng Bugs 3(1):38–43. https://doi.org/10.4161/bbug.3.1.18223
Xu P, Gu Q, Wang WY, Wong L, Bower AGW, Collins CH, Koffas MAG (2013) Modular optimization of multi-gene pathways for fatty acids production in E. coli. Nat Commun 4:1409. https://doi.org/10.1038/ncomms2425
Xu P, Vansiri A, Bhan N, Koffas MA (2012) ePathBrick: a synthetic biology platform for engineering metabolic pathways in E. coli. ACS Synth Biol 1(7):256–266. https://doi.org/10.1021/sb300016b
Yang TT, Cheng L, Kain SR (1996) Optimized codon usage and chromophore mutations provide enhanced sensitivity with the green fluorescent protein. Nucleic Acids Res 24(22):4592–4593. https://doi.org/10.1093/nar/24.22.4592
Yim SS, An SJ, Kang M, Lee J, Jeong KJ (2013) Isolation of fully synthetic promoters for high-level gene expression in Corynebacterium glutamicum. Biotechnol Bioeng 110(11):2959–2969. https://doi.org/10.1002/bit.24954
Zhang B, Zhou N, Liu YM, Liu C, Lou CB, Jiang CY, Liu SJ (2015a) Ribosome binding site libraries and pathway modules for shikimic acid synthesis with Corynebacterium glutamicum. Microb Cell Factories 14:14. https://doi.org/10.1186/s12934-015-0254-0
Zhang C, Zhang J, Kang Z, Du G, Chen J (2015b) Rational engineering of multiple module pathways for the production of L-phenylalanine in Corynebacterium glutamicum. J Ind Microbiol Biotechnol 42(5):787–797. https://doi.org/10.1007/s10295-015-1593-x
Zhang Y, Cai J, Shang X, Wang B, Liu S, Chai X, Tan T, Zhang Y, Wen T (2017) A new genome-scale metabolic model of Corynebacterium glutamicum and its application. Biotechnol Biofuels 10:169. https://doi.org/10.1186/s13068-017-0856-3
Zhang Y, Meng Q, Ma H, Liu Y, Cao G, Zhang X, Zheng P, Sun J, Zhang D, Jiang W, Ma Y (2015c) Determination of key enzymes for threonine synthesis through in vitro metabolic pathway analysis. Microb Cell Factories 14:86. https://doi.org/10.1186/s12934-015-0275-8
Acknowledgements
We are grateful to Prof. Masayuki Inui (Research Institute of Innovative Technology for the Earth, Japan) for generously providing strains and plasmids. This study was supported by the National Natural Science Foundation of China (Nos. 31500044 and 31601460), the Natural Science Foundation of Tianjin (Nos. 17JCQNJC09600 and 17JCYBJC24000), and “Hundred Talents Program” of the Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 1220 kb)
Rights and permissions
About this article

Cite this article
Wei, L., Xu, N., Wang, Y. et al. Promoter library-based module combination (PLMC) technology for optimization of threonine biosynthesis in Corynebacterium glutamicum. Appl Microbiol Biotechnol 102, 4117–4130 (2018). https://doi.org/10.1007/s00253-018-8911-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-8911-y