
Abstract
The development of environmentally benign protocols to synthesize novel N-heterocycles is vital in the field of synthetic organic chemistry. We herein report a successful one-pot domino synthesis of novel 6,7 disubstituted 1H-pyrroles using substituted phenacyl bromide, barbituric acid/Meldrums acid, aromatic amines catalysed by 5 mol% Fluorescein in presence of visible light. This procedure is a useful and adaptable method for the synthesis of pyrroles since it is compatible with a wide range of sensitive functional groups, does not require column chromatography purification. During the reaction, Fluorescein may catalyse the formation of enamine leading to amino alcohol which subsequently undergoes dehydration to give 6,7 disubstituted 1H-pyrroles. All the synthesized derivatives were obtained in 90–95% yields and were characterized by 1H, 13C NMR and HRMS (ESI) analysis.
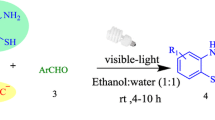
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Pyrrole and its derivatives belong to the class of biologically active scaffold and are well known to exhibit diverse and distinct physiological properties [1,2,3]. The electron-rich pyrrole ring is less basic and possesses a dipole moment of 1.5–1.9 D exactly contrary to other members of the same family such as thiofuran, furan, and azine. Pyrrole derivatives act as antihypertensive [4], antidepressant [5], analgesic [6], anti-inflammatory [7], anticancer [8, 9], fungicidal [10], insecticidal [11, 12] and plant growth regulators [13]. They are well known to inhibit HIV-1 integrase [14], CYP1 [15], DNA/RNA polymerase [16] and FMS kinase [17] enzymes present in several mammalian and microbial cells.
Substituted pyrroles behave as corrosion prohibitors [18], preservatives [19], precursors [20], form the building block for pigments such as chlorophyll and haemoglobin. They are present in the natural extracts of several alkaloids [21] and form the heart of vitamin B12, haematoidin, biliverdin, bacteriochlorin, porphobilinogen and proline. Several commercially available drugs such as calcimycin, ageliferin, aloracetam, isamoltane, obatoclax, ketorolac consist of pyrrole scaffold (Fig. 1).

Some representative drugs containing pyrrole ring
The synthesis of polyfunctional pyrrole rings and the study of their medicinal properties stand out as a zone of the immense scope of research in organic chemistry. Nevertheless, synthesizing pyrroles entailing multiple substituents and functional groups are challenging for chemists, and consequently, new methodologies [22,23,24,25,26,27,28,29,30,31,32] following the principles of green chemistry are in demand and highly profitable.
As a result, synthetic chemists have paid intense attention to the construction of such frameworks and several reactions involving the use of condensation [33], cascade [34], cyclization [35], decarboxylation [36], 1,3 dipolar addition [37], catalytic dehydrogenation [38] reactions are reported. However, they are associated with several liabilities such as longer reaction duration, use of catalyst, lesser yield, multiple steps and column chromatography method of purification.
Multicomponent reactions (MCRs), which combine three or more substrates to generate a highly complex and divergent [39] product in a single-step manner have experienced a rapid increase in applications in all domains of organic synthesis [40, 41]. They have obvious advantages over conventional multistep processes, including high reaction efficiency [42], atom economy [43], waste reduction, and energy savings [44]. These characteristics are in line with the green chemistry principles, making MCRs a suitable synthetic strategy in combinatorial chemistry and diversity-oriented synthesis [45]. Indeed, they have been frequently employed in the pharmaceutical industry to generate enormous libraries of small substrates that can be exploited to find lead compounds in a shorter period saving manpower. MCRs generally employ easily accessible substrates under cleaner and efficient energy transfer conditions to investigate the dimensions of the reaction to generate excellent product yields.
In this regard, visible light-mediated MCRs [46] have caught the attraction of synthetic organic chemists as they stimulate the synthesis of heterocycles under significantly moderate conditions generating the target molecules in excellent yields. Most of the photoinduced reactions work in presence of visible light [47], blue light [48], green light [49] and hence providing a cleaner and efficient pathway leading to the goal of sustainable synthesis. In a photochemical process, one of the starting materials is converted to a reactive intermediate which then subsequently interacts with other substrates generating product in a one-pot manner. The efficiency of the visible light catalyzed MCR can be further enhanced by the use of a suitable photocatalyst which can generate the radical intermediate in a shorter duration. Photocatalyzed MCRs have proven to be successful in the synthesis of polyhydroquinolines [50], 3-arylacetals, trisubstituted hydroxylamines [51], α-amino amides [52], secondary amine [53], indole [54], imidazole [55] and several others.
Contemplating these facts and in our continuous pursuit in synthesizing heterocycles [56] through cleaner reaction profiles following green chemistry principles [57] we herein successfully report a visible light-mediated, fluorescein catalyzed one-pot sustainable approach for the synthesis of twelve novel 6,7 disubstituted 1H-pyrroles in excellent yields (Fig. 2).

Synthesis of novel 6,7 disubstituted 1H-pyrroles
2 Materials and Methodology
2.1 Experimental Section
Reagents and solvents of commercial-grade were purchased from Sigma-Aldrich and TCI Chemicals (India) Pvt. Ltd. and were used without further purification except for aldehydes which were distilled before use. The progress of the reactions and the purity of products was assessed by TLC [analytical silica gel plates (Merck60 F254)]. Melting points were determined on a RAAGA, an Indian make apparatus. 1H NMR and 13C NMR spectra were recorded on Avance Bruker instrument operating at 400 MHz and 100 MHz in DMSO-d6 respectively. Chemical shifts are reported in δ ppm. HRMS data were obtained on a Varian IonSpec QFT-MS spectrometer with the technique of electrospray ionization.
2.2 General Procedure for the Synthesis of Novel 6,7 Disubstituted 1H-Pyrroles 4(a-l)
In a 50 mL round-bottomed flask, a mixture of substituted 2-bromo-1-(phenyl)ethanone (1 mmol), barbituric acid/Meldrum’s acid (1 mmol), aryl amine (1 mmol), EtOH (3 mL), Fluorescein (5% mol) along NBS (3% mol) was taken and stirred at room temperature using a magnetic stirrer for 30 min. The setup was irradiated using white light generated by a 22 W white LED. The reactions were monitored by thin-layer chromatography (TLC) using hexane: ethyl acetate (7:3) as eluent. After completion of the reaction (TLC), the reaction mixture was quenched with ice and the precipitate formed was filtered, washed, dried and recrystallized from ethanol to yield the pure product. The structures of all the products were confirmed by 1H NMR, 13C NMR and HRMS analysis.
2.3 Spectral Data
2.3.1 7-(4-Chlorophenyl)-6-(p-tolyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (4a)
Yellow solid, mp: 218 − 219 °C;
1H NMR (400 MHz, DMSO-d6): δ 10.38 (s, 1H, -NH), 9.29 (s, 1H, -NH), 7.86 − 7.22 (m, 8H, Ar–H), 6.53 (s, 1H, Ar–H), 2.56 (s, 3H, –CH3) ppm;
13C NMR (100 MHz, DMSO-d6): δ 162.2, 148.9, 142.8, 138.2, 136.2, 134.9, 133.6, 131.7, 127.9, 126.1, 124.4, 122.1, 106.0, 98.4, 23.5 ppm;
HRMS (ESI) calcd for C19H15ClN3O2 [M + H]+: 352.0853, found 352.0847.
2.3.2 7-(4-Nitrophenyl)-6-(p-tolyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (4b)
Daek Yellow solid, mp: 261 − 262 °C;
1H NMR (400 MHz, DMSO-d6): δ 10.71 (s, 1H, –NH), 9.72 (s, 1H, –NH), 8.27 − 7.25 (m, 8H, Ar–H), 6.73 (s, 1H, Ar–H), 2.27 (s, 3H, -CH3) ppm;
13C NMR (100 MHz, DMSO-d6): δ 163.7, 149.7, 147.5, 142.7, 138.7, 136.7, 134.8, 131.6, 126.4, 125.2, 123.7, 119.4, 105.5, 99.7, 23.1 ppm;
HRMS (ESI) calcd for C19H15N4O4 [M + H]+: 363.1093, found 363.1086.
2.3.3 7-(4-Chlorophenyl)-2,2-dimethyl-6-(p-tolyl)-[1,3]dioxino[4,5-b]pyrrol-4(7H)-one (4c)
Dark Yellow solid, mp: 233 − 234 °C;
1H NMR (400 MHz, DMSO-d6): δ 7.91 − 7.23 (m, 8H, Ar–H), 6.62 (s, 1H, Ar–H), 2.22 (s, 3H, -CH3), 1.46 (s, 3H, -CH3), 1.25 (s, 3H, -CH3) ppm;
13C NMR (100 MHz, DMSO-d6): δ 167.4, 143.6, 139.7, 138.2, 136.6, 132.1, 130.6, 128.8, 125.7, 123.7, 120.3, 110.5, 109.0, 106.2, 26.5, 23.0 ppm;
HRMS (ESI) calcd for C21H19ClNO3 [M + H]+: 368.1053, found 368.1047.
2.3.4 2,2-Dimethyl-7-(4-nitrophenyl)-6-(p-tolyl)-[1,3]dioxino[4,5-b]pyrrol-4(7H)-one (4d)
Yellow solid, mp: 220 − 221 °C;
1H NMR (400 MHz, DMSO-d6): 8.26 − 7.25 (m, 8H, Ar–H), 6.56 (s, 1H, Ar–H), 2.28 (s, 3H, –CH3), 1.47 (s, 3H, -CH3), 1.23 (s, 3H, -CH3) ppm;
13C NMR (100 MHz, DMSO-d6): δ 167.4, 146.5, 144.6, 142.8, 139.1, 137.3, 131.8, 125.9, 125.2, 123.9, 121.9, 111.6, 110.2, 106.1, 26.5, 24.2 ppm;
HRMS (ESI) calcd for C21H19N2O5 [M + H]+: 379.1294, found 379.1288.
2.3.5 7-(4-Chlorophenyl)-6-(4-methoxyphenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (4e)
Dark Yellow solid, mp: 226 − 227 °C;
1H NMR (400 MHz, DMSO-d6): δ 10.67 (s, 1H, –NH), 9.54 (s, 1H, –NH), 7.83 − 7.02 (m, 8H, Ar–H), 6.63 (s, 1H, Ar–H), 3.69 (s, 3H, -OCH3) ppm;14.
13C NMR (100 MHz, DMSO-d6): δ 161.4, 158.5, 149.7, 142.7, 137.6, 134.5, 133.6, 131.6, 129.1, 127.6, 121.9, 118.3, 105.6, 99.5, 57.0 ppm;
HRMS (ESI) calcd for C19H15ClN3O3 [M + H]+: 368.0802, found 368.0795.
2.3.6 6-(4-Methoxyphenyl)-7-(4-nitrophenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (4f)
Yellow solid, mp: 227 − 228 °C;
1H NMR (400 MHz, DMSO-d6): δ 10.22 (s, 1H, -NH), 9.16 (s, 1H, -NH), 8.26 − 7.03 (m, 8H, Ar–H), 6.65 (s, 1H, Ar–H), 3.79 (s, 3H, -OCH3) ppm; 14.
13C NMR (100 MHz, DMSO-d6): δ 161.6, 157.9, 149.7, 147.4, 142.8, 136.1, 132.8, 131.4, 130.3, 126.6, 120.6, 119.8, 104.5, 100.1, 56.6 ppm;
HRMS (ESI) calcd for C19H15N4O5 [M + H]+: 379.1042, found 379.1036.
2.3.7 7-(4-Chlorophenyl)-6-(4-methoxyphenyl)-2,2-dimethyl-[1,3]dioxino[4,5-b]pyrrol-4(7H)-one (4 g)
Yellow solid, mp: 255 − 256 °C;
1H NMR (400 MHz, DMSO-d6): δ 7.94 − 6.99 (m, 8H, Ar–H), 6.68 (s, 1H, Ar–H), 3.67 (s, 3H, –OCH3), 1.52 (s, 3H, -CH3), 1.28 (s, 3H, -CH3) ppm;
13C NMR (100 MHz, DMSO-d6): δ 167.1, 159.4, 142.7, 140.3, 136.1, 132.2, 131.4, 130.1, 129.0, 120.6, 118.4, 111.7, 110.4, 105.6, 39.4, 25.5, 23.2 ppm;
HRMS (ESI) calcd for C21H19ClNO4 [M + H]+: 384.1003, found 384.0096.
2.3.8 6-(4-Methoxyphenyl)-2,2-dimethyl-7-(4-nitrophenyl)-[1,3]dioxino[4,5-b]pyrrol-4(7H)-one (4 h)
Yellow solid, mp: 239 − 240 °C;
1H NMR (400 MHz, DMSO-d6): δ 8.20 − 7.04 (m, 8H, Ar–H), 6.67 (s, 1H, Ar–H), 3.78 (s, 3H, –OCH3), 1.46 (s, 3H, -CH3), 1.25 (s, 3H, -CH3) ppm;
13C NMR (100 MHz, DMSO-d6): δ 167.2, 158.5, 146.6, 144.8, 142.7, 136.9, 133.2, 130.4, 125.2, 121.6, 119.4, 112.5, 110.2, 107.3, 25.9, 23.1 ppm;
HRMS (ESI) calcd for C21H19N2O6 [M + H]+: 395.1243, found 395.1236.
2.3.9 6-(4-Bromophenyl)-7-(4-chlorophenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (4i)
Yellow solid, mp: 231 − 232 °C;
1H NMR (400 MHz, DMSO-d6): δ 10.95 (s, 1H, –NH), 9.52 (s, 1H, –NH), 7.92 − 7.37 (m, 8H, Ar–H), 6.67 (s, 1H, Ar–H) ppm;
13C NMR (100 MHz, DMSO-d6): δ 163.6, 149.3, 147.2, 143.4, 139.5, 131.2, 129.3, 127.5, 126.9, 125.6, 121.4, 120.3, 107.4, 98.5 ppm;
HRMS (ESI) calcd for C18H12BrClN3O2 [M + H]+: 415.9801, found 415.9794.
2.3.10 6-(4-Bromophenyl)-7-(4-nitrophenyl)-1H-pyrrolo[2,3-d]pyrimidine-2,4(3H,7H)-dione (4j)
Dark Yellow solid, mp: 249 − 250 °C;
1H NMR (400 MHz, DMSO-d6): δ 10.69 (s, 1H, –NH), 9.21 (s, 1H, –NH), 8.27 − 7.57 (m, 8H, Ar–H), 6.75 (s, 1H, Ar–H) ppm;
13C NMR (100 MHz, DMSO-d6): δ 161.6, 150.2, 142.5, 139.4, 137.4, 135.4, 133.5, 132.2, 127.9, 127.4, 122.0, 121.2, 106.9, 100.8 ppm;
HRMS (ESI) calcd for C18H12BrN4O4 [M + H]+: 427.0042, found 427.0036.
2.3.11 6-(4-Bromophenyl)-7-(4-chlorophenyl)-2,2-dimethyl-[1,3]dioxino[4,5-b]pyrrol-4(7H)-one (4 k)
Yellow solid, mp: 259 − 260 °C;
1H NMR (400 MHz, DMSO-d6): δ 8.84 − 7.49 (m, 8H, Ar–H), 6.68 (s, 1H, Ar–H), 1.55 (s, 3H, –CH3) 1.33 (s, 3H, –CH3) ppm;
13C NMR (100 MHz, DMSO-d6): δ 167.5, 145.3, 139.9, 139.1, 136.3, 132.8, 130.1, 129.4, 126.4, 121.3, 120.2, 111.4, 109.2, 105.4, 26.1 ppm;
HRMS (ESI) calcd for C20H16BrClNO3 [M + H]+: 432.0002, found 431.9995.
2.3.12 6-(4-Bromophenyl)-2,2-dimethyl-7-(4-nitrophenyl)-[1,3]dioxino[4,5-b]pyrrol-4(7H)-one (4 l)
Yellow solid, mp: 246 − 247 °C;
1H NMR (400 MHz, DMSO-d6): δ 7.97 − 7.34 (m, 8H, Ar–H), 6.52 (s, 1H, Ar–H), 1.54 (s, 3H, -CH3) 1.31 (s, 3H, -CH3) ppm;
13C NMR (100 MHz, DMSO-d6): δ 162.9, 150.1, 146.6, 142.7, 139.3, 136.4, 134.9, 131.6, 128.6, 125.3, 122.0, 120.2, 106.0, 99.4, 26.5 ppm;
HRMS (ESI) calcd for C20H16BrN2O5 [M + H]+: 443.0243, found 443.0240.
3 Results and Discussion
We initiated the investigation on optimizing the reaction conditions and diversification of the methodology by selecting 2-bromo-1-(p-tolyl)ethenone, barbituric acid, 4-chloroaniline in 1 mmol quantity and subjecting them to divergent catalysts, solvents and additives.
The substrates selected for the synthesis of 4a are initially exposed to visible light in presence of various solvents to conjecture their effects. The visible light-mediated model reaction was then performed in non-polar solvents such as n-pentane, benzene, diethyl ether, o-xylene and from Table 1, Entries 1 − 4, it was observed that the formation of 4a is not detected even after stirring for 600 min. The presence of polar aprotic solvents such as dioxane, acetone, acetonitrile and DMF generated a maximum of 20% yield of 4a (Table 1, Entries 5 − 8). To our amuse, the presence of polar solvents diethylene glycol, glycerol, acetic acid, H2O, MeOH progressively increased the yield upto 40% (Table 1, Entries 9 − 13). The reaction was then conducted in EtOH:H2O (1:1), and EtOH which generated a yield of 60% in 600 min (Table 1, Entries 14 − 15). Hence it was decided to use EtOH as a reaction medium for further studies.
To increase the yield of 4a we then exposed the substrates to varied photocatalysts such as tris(2-phenylpyridine)iridium(III) (Ir(ppy)3), dimanganese decacarbonyl [Mn2(CO)10], titanium dioxide (TiO2), red acid 52, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), [Acr+-Mes][ClO4−], eosin B, eosin Y and fluorescein in 10 mol% concentration and it was observed that the reaction yield significantly increased from trace to 90% (Table 1, Entries 16 − 23).
To ascertain the effect of visible light in the synthesis of 4a, the reaction was performed in 10 mol% Fluorescein in ethanol in absence of visible light, and only a trace quantity of yield was obtained (Table 1, Entry 25). Its seen that when the model reaction was specifically exposed to 30 W Blue LED, 24 W Green LED to uncover their effect, a maximum of 45% yield was obtained (Table 1, Entries 26 − 27).
To reduce the reaction duration, the effect of additives was then studied. Surprisingly, when the reaction was exposed to various additives such as AlCl3, ZnBr2, N-bromosuccinimide (NBS) in a 5 mol% quantity the reaction duration was considerably reduced to 30 min yielding 95% of 4a (Table 1, Entries 28 − 30). It was also noticed from the model reaction that the right quantity of fluorescein and NBS was 5 mol% and 3 mol% respectively (Table 1, Entry 32). Further reduction in the amount of fluorescein to 3% generated less yield (Table 1, Entry 33). The reaction was also performed in presence of NBS and in the absence of fluorescein as a catalyst to divulge the importance of Fluorescein. To our dismay, the reaction performed in the absence of fluorescein generated a yield of 15% reiterating the role of catalyst in the synthesis of 4a. Hence, visible light along with 5 mol% of fluorescein as a catalyst, 3 mol% NBS as an additive in EtOH was chosen as a valid condition in the synthesis of 6,7 disubstituted 1H-pyrroles. The scope of optimized conditions was later applied to the gram-scale synthesis of 4a and it proved to be rewarding as a yield of 95% was obtained within 60 min.
The optimized conditions of reaction were then enforced on a reaction constituting three substituted 2-bromo-1-phenylethanones, two 1,3 dicarbonyl compounds and anilines (Fig. 3) as starting materials and a series of corresponding twelve novel 6,7 disubstituted 1H-pyrroles were synthesized in excellent yield (Fig. 4). The designated protocol tolerates a variety of substituents which are electron-withdrawing or donating on both 2-bromo-1-phenylethanone and aniline.

Diversity of starting materials

Diversity of synthesized 6,7 disubstituted 1H-pyrroles
To confirm and establish the radical-mediated pathway, the designated model substrates were exposed to benzyl acrylate and 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) as radical scavengers to the optimized condition of the reaction. The presence of benzyl acrylate and TEMPO drastically reduced the yield of the expected product to 10% and 5% respectively (Fig. 5).

Effect of radical scavengers on the synthesis of 4a
The control experiments were further performed to decipher the order of the combination of the reactants. As observed from Fig. 6, the addition of 2-bromo-1-(p-tolyl)ethanone, barbituric acid generated an intermediate 5. The reaction also generated another intermediate 6 when barbituric acid, 4-chloroaniline were exposed to the standardized conditions. However, when 2-bromo-1-(p-tolyl)ethanone and 4-chloroaniline were taken as substrates no intermediates were generated. Thus it can be concluded that the reaction proceeds via the reaction of barbituric acid or intermediate 6 with 2-bromo-1-(p-tolyl)ethanone.

Control experiments
The probable mechanism based on the studies conducted is presented in Fig. 7. It is equitable to assume that NBS may initiate the formation of radical intermediate 1 from barbituric acid which reacts with 2-bromo-1-(p-tolyl)ethanone to generate 5. The condensation of 5 with 4-chloroaniline removes a molecule of water giving an imine which gets may get converted to a more stable enamine. The presence of Fluorescein may catalyse the formation of enamine which further leads to amino alcohol and its dehydration to give the target molecule 4a.

A plausible mechanism for the formation of 4a
The structures of all synthesized 6,7 disubstituted 1H-pyrroles were characterized using 1H NMR and 13C NMR, and HRMS (ESI) spectral techniques. The 1H NMR spectrum of molecule 4a shows two singlets at 10.38 and 9.29 ppm confirming the presence of -NH groups. The presence of eight protons of two aromatic rings is seen as multiplet in the range of 7.86 − 7.22 ppm. The proton of the pyrrole ring is seen as a characteristic peak at 6.53 ppm. The –CH3 group shows a singlet at 2.56 ppm. The 13C NMR spectrum shows a peak at 23.5 ppm which can be assigned to the carbon of –CH3 group. The other peaks in the range of 98.4–142.8 ppm are due to carbon atoms of the aromatic ring. The characteristic peak at 148.9 and 162.2 is due to carbonyl groups on carbon. The HRMS (ESI) spectrum showed a [M + H]+ peak at 352.0847 affirming the formation of 4a.
4 Conclusions
For the first time, a combination of multi-component approach and photocatalyst was applied for the synthesis of twelve 6,7 disubstituted 1H-pyrroles in excellent yield using 2-bromo-1-(phenyl)ethanone, barbituric acid/Meldrum’s acid, aromatic amines in EtOH with Fluorescein as catalyst, NBS as an additive. The protocol is novel, benign, is formidable in terms of reaction time, diversity of products and follows the positive aspects of MCRs following the principles of green chemistry.
References
Wang F, Sikma E, Duan Z, Sarma T, Lei C, Zhang Z, Humphrey SM, Sessler JL (2019) Shape-persistent pyrrole-based covalent organic cages: synthesis, structure and selective gas adsorption properties. Chem Commun 55:6185–6188
Jadoun S, Biswal L, Riaz U (2018) Tuning the optical properties of poly (o-phenylenediamine-co-pyrrole) via template mediated copolymerization. Des Monomers Polym 21:75–81
Ninis O, Kacimi R, Bouaamlat H, Abarkan M, Bouachrine M (2017) Theoretical studies of photovoltaic properties for design of new azo-pyrrole photo-sensitizer materials as dyes in solar cells. J Mater Environ Sci 8:2572–2578
Ahmad S, Alam O, Naim MJ, Shaquiquzzaman M, Alam MM, Iqbal M (2018) Pyrrole: an insight into recent pharmacological advances with structure activity relationship. Eur J Med Chem 157:527–561
Fatahala SS, Nofal S, Mahmoud E, Abd El-hameed RH (2019) Pyrrolopyrazoles: synthesis, evaluation and pharmacological screening as antidepressant agents. Med Chem 15:911–922
Battilocchio C, Poce G, Alfonso S, Porretta GC, Consalvi S, Sautebin L, Pace S, Rossi A, Ghelardini C, Mannelli LDC, Schenone S, Giordani A, Francesco LD, Patrignani P, Biava M (2013) A class of pyrrole derivatives endowed with analgesic/anti-inflammatory activity. Bioorganic Med Chem 21:3695–3701
Harrak Y, Rosell G, Daidone G, Plescia S, Schillaci D, Pujol MD (2007) Synthesis and biological activity of new anti-inflammatory compounds containing the 1, 4-benzodioxine and/or pyrrole system. Bioorganic Med Chem 15:4876–4890
Ghorab MM, Ragab FA, Heiba HI, Youssef HA, El-Gazzar MG (2010) Synthesis of novel pyrrole and pyrrolo [2 3-d] pyrimidine derivatives bearing sulfonamide moiety for evaluation as anticancer and radiosensitizing agents. Bioorganic Med Chem Lett 20:6316–6320
Pegklidou K, Papastavrou N, Gkizis P, Komiotis D, Balzarini J, Nicolaou I (2015) N-substituted pyrrole-based scaffolds as potential anticancer and antiviral lead structures. Med Chem 11:602–608
Yao TT, Xiao DX, Li ZS, Cheng JL, Fang SW, Du YJ, Zhao JH, Dong XW, Zhu GN (2017) Design, synthesis, and fungicidal evaluation of novel pyrazole-furan and pyrazole-pyrrole carboxamide as succinate dehydrogenase inhibitors. J Agric Food Chem 65:5397–5403
Ye Z, Shi L, Shao X, Xu X, Xu Z, Li Z (2013) Pyrrole-and dihydropyrrole-fused neonicotinoids: design, synthesis, and insecticidal evaluation. J Agric Food Chem 61:312–319
Boukouvala MC, Kavallieratos NG, Athanassiou CG, Hadjiarapoglou LP (2016) Insecticidal effect of two novel pyrrole derivatives against two major stored product insect species. Crop Prot 84:1–7
Rico R, Bermejo F (1995) Total synthesis of (−)-ampullicin and (+)-isoampullicin two growth regulators from Ampulliferina Sp no 27. Tetrahedron Lett 36:7889–7892
Bianco MDCAD, Marinho DILF, Hoelz LVB, Bastos MM, Boechat N (2021) Pyrroles as privileged scaffolds in the search for new potential HIV inhibitors. Pharmaceuticals 14:893
Williams IS, Joshi P, Gatchie L, Sharma M, Satti NK, Vishwakarma RA, Chaudhuri B, Bharate SB (2017) Synthesis and biological evaluation of pyrrole-based chalcones as CYP1 enzyme inhibitors for possible prevention of cancer and overcoming cisplatin resistance. Bioorganic Med Chem Lett 27:3683–3687
Wu C, Wang W, Fang L, Su W (2018) Programmable pyrrole-imidazole polyamides: a potent tool for DNA targeting. Chin Chem Lett 29:1105–1112
Peifer C, Selig R, Kinkel K, Ott D, Totzke F, Schächtele C, Heidenreich R, Röcken M, Schollmeyer D, Laufer S (2008) Design, synthesis, and biological evaluation of novel 3-aryl-4-(1 H-indole-3yl)-1, 5-dihydro-2 H-pyrrole-2-ones as vascular endothelial growth factor receptor (VEGF-R) inhibitors. J Med Chem 51:3814–3824
Zarrouk A, Hammouti B, Lakhlifi T, Traisnel M, Vezin H, Bentiss F (2015) New 1H-pyrrole-2, 5-dione derivatives as efficient organic inhibitors of carbon steel corrosion in hydrochloric acid medium: electrochemical XPS and DFT studies. Corros Sci 90:572–584
Ash M (2004) Handbook of preservatives. Synapse info resources.
Xu Y, Wang Z, Gan Z, Xi Q, Duan Z, Mathey F (2015) Versatile synthesis of phospholides from open-chain precursors application to annelated pyrrole–and silole–phosphole rings. Org Lett 17:1732–1734
Fan H, Peng J, Hamann MT, Hu JF (2008) Lamellarins and related pyrrole-derived alkaloids from marine organisms. Chem Rev 108:264–287
Vinayak A, Sudha M, Lalita SK (2017) Design synthesis and characterization of novel amine derivatives of 5-[5-(chloromethyl)-1, 3, 4-oxadiazol-2-yl]- 2-(4-fluorophenyl)-pyridine as a new class of anticancer agents Dhaka Uni. J Pharm Sci 16:11–19
Vinayak A, Sudha M, Jagadeesha AH, Lalita SK (2015) Design, synthesis, characterization and cancer cell growth-inhibitory properties of novel derivatives of 2-(4-fluoro-phenyl)-5-(5-aryl substituted-1, 3, 4-oxadiazol-2-yl) pyridine. Br J Pharm Res 7:34–43
Vinayak A, Sudha M, Lalita SK, Rao Prakash K (2014) Design, synthesis, characterization and anticancer properties of novel 2-chloro-N-(aryl substituted) acetamide derivatives of 5-[2-(4-methoxyphenyl) pyridin-3-yl]-1, 3, 4-oxadiazole-2-thiol. Int J Drug Dev Res 6:188–195
Vinayak A, Sudha M, Jaadeesha AH, Kulkarni P, Lalita KS, Rao PK (2014) Synthesis, characterization of some novel 1, 3, 4-oxadiazole compounds containing 8-hydroxy quinolone moiety as potential antibacterial and anticancer agents. Int J Pharm Res 4(4):180–185
Vinayak A (2014) Design, synthesis and cytotoxic evaluation of novel 2-(4-N, N-dimethyl) pyridine containing 1, 3, 4-oxadiazole moiety. Asian J Biomed Pharm Sci 4:1–5
Vinayak A, Sudha M, Lalita KS, Rao Prakash K (2014) Design, synthesis, characterization and anticancer properties of novel derivatives of 1-[2-(Aryl substituted)-5- (4’-methoxy-biphenyl-4-yl)-[1, 3, 4] oxadiazole-3-yl]- ethanone. Int J Pharm Sci Res 4:713–717
Vinayak A, Sudha M, Jagadeesha AH, Lalita KS, Rao K (2014) Synthesis, characterization and cytotoxic evaluation of novel derivatives of 1, 3, 4-oxadiazole containing 5-phenyl thiophene moiety. IOSR- JPBS 9:42–48
Vinayak A, Sudha M, Rao K, Lalita KS (2013) Synthesis, characterisation and anticancer acitivity of schiff base derivatives of 5-(2-phenoxypyridin-3-yl)-1, 3, 4-thiadiazol-2-amine. Int Res J Pharm 4:62–66
Vinayak A, Sudha M, Rao K, Lalita KS (2014) synthesis of n-{[5-(2,4-dichlorophenyl)- 1, 3, 4-oxadiazol- 2-yl] methyl} amine derivatives as anticancer precursors. Int J Med Chem Anal 4:231–235
Vinayak A, Anusha S, Santosh N, Basappa Y (2020) Synthesis, impedance, and current–voltage characteristics of strontium-manganese titanate hybrid nanoparticles. Macromol Symp 392:2000002
Vinayak A, Revaigh MG, Adarsha HJ (2020) Synthesis and fabrication of y-doped ZnO nanoparticles and their application as a gas sensor for the detection of ammonia. J Mater Eng Perform 29:4–5
Wang B, Gu Y, Luo C, Yang T, Yang L, Suo J (2004) Pyrrole synthesis in ionic liquids by Paal-Knorr condensation under mild conditions. Tetrahedron Lett 45:3417–3419
Li X, Chen M, Xie X, Sun N, Li S, Liu Y (2015) Synthesis of multiple-substituted pyrroles via gold (I)-catalyzed hydroamination/cyclization cascade. Organic Lett 17:2984–2987
Yamamoto H, Sasaki I, Mitsutake M, Karasudani A, Imagawa H, Nishizawa M (2011) An efficient pyrrole synthesis via silaphenylmercuric triflate catalyzed cyclization of homopropargyl azides. Synlett 2011:2815–2818
Kim Y, Kim J, Park SB (2009) Regioselective synthesis of tetrasubstituted pyrroles by 1, 3-dipolar cycloaddition and spontaneous decarboxylation. Organic Lett 11:17–20
St Cyr DJ, Arndtsen BA (2007) A new use of Wittig-type reagents as 1, 3-dipolar cycloaddition precursors and in pyrrole synthesis. J Am Chem Soc 129:12366–12367
Bonnaud B, Bigg DC (1994) Preparation of pyrroles by dehydrogenation of pyrrolidines with manganese dioxide. Synthesis 1994:465–467
Veltri L, Mancuso R, Altomare A, Gabriele B (2015) Divergent multicomponent tandem palladium-catalyzed aminocarbonylation-cyclization approaches to functionalized imidazothiazinones and imidazothiazoles. ChemCatChem 7:2206–2213
Cioc RC, Ruijter E, Orru RV (2014) Multicomponent reactions: advanced tools for sustainable organic synthesis. Green Chem 16:2958–2975
Das D (2016) Multicomponent reactions in organic synthesis using copper-based nanocatalysts. ChemistrySelect 1:1959–1980
van der Heijden G, Ruijter E, Orru RV (2013) Efficiency, diversity, and complexity with multicomponent reactions. Synlett 24:666–685
Marson CM (2012) Multicomponent and sequential organocatalytic reactions: diversity with atom-economy and enantiocontrol. Chem Soc Rev 41:7712–7722
Rahman I, Deka B, Thakuria R, Deb ML, Baruah PK (2020) l-Proline-catalyzed regioselective C1 arylation of tetrahydroisoquinolines through a multicomponent reaction under solvent-free conditions. Org Biomol Chem 18:6514–6518
Dandapani S, Marcaurelle LA (2010) Current strategies for diversity-oriented synthesis. Curr Opin Chem Biol 14:362–370
Dutta A, Patra SK, Khatua S, Nongkhlaw R (2021) Visible-light-mediated synthesis of 3, 4, 5-trisubstituted furan-2-one derivative via bifunctional based organo photocatalyst. New J Chem. https://doi.org/10.1039/D1NJ03238K
Zhang M, Chen MN, Li JM, Liu N, Zhang ZH (2019) Visible-light-initiated one-pot, three-component synthesis of 2-amino-4 h-pyran-3, 5-dicarbonitrile derivatives. ACS Combinatorial Sci 21:685–691
Chen J, Liu S, Lv X, Hong K, Lei J, Xu X, Hu W (2020) Blue light-promoted formal [4+ 1]-annulation of diazoacetates with o-aminoacetophenones: synthesis of polysubstituted indolines and computational study. J Org Chem 85:13920–13928
Jarrahi M, Tayebee R, Maleki B, Salimi A (2021) One-pot multicomponent green LED photoinduced synthesis of chromeno [4, 3-b] chromenes catalyzed by a new nanophotocatalyst histaminium tetrachlorozincate. RSC Adv 11:19723–19736
Harale RR, Shitre PV, Sathe BR, Shingare MS (2017) Visible light motivated synthesis of polyhydroquinoline derivatives using CdS nanowires. Res Chem Intermed 43:3237–3249
Cai BG, Li Q, Zhang Q, Li L, Xuan J (2021) Synthesis of trisubstituted hydroxylamines by visible light-promoted multicomponent reaction. Org Chem Front. https://doi.org/10.1039/D1QO01102B
Zhu J, Dai C, Ma M, Yue Y, Fan X (2021) Visible light-mediated cross-coupling of electrophiles: synthesis of α-amino amides from isocyanates and ketimines. Org Chem Front 8:1227–1232
Wang X, Zhu B, Dong J, Tian H, Liu Y, Song H, Wang Q (2021) Visible-light-mediated multicomponent reaction for secondary amine synthesis. Chem Commun 57:5028–5031
Wu CJ, Meng QY, Lei T, Zhong JJ, Liu WQ, Zhao LM, Li ZJ, Chen B, Tung CH, Wu LZ (2016) An oxidant-free strategy for indole synthesis via intramolecular C-C bond construction under visible light irradiation: cross-coupling hydrogen evolution reaction. ACS Catal 6:4635–4639
Patel G, Patel AR, Banerjee S (2020) Visible light-emitting diode light-driven one-pot four component synthesis of poly-functionalized imidazoles under catalyst-and solvent-free conditions. New J Chem 44:13295–13300
Tabassum S, Govindaraju S, Pasha MA (2016) Ultrasound mediated, green innovation for the synthesis of polysubstituted 1, 4-dihydropyridines. RSC Adv 6:29802–29810
Tabassum S, Govindaraju S, Pasha MA (2017) FeSO4⋅7H2O catalyzed rapid and efficient one-pot multicomponent synthesis of functionalized pyrazol-yl-pyrazolone methanes. ChemistrySelect 2:4054–4057
Acknowledgements
The authors thank SIF, IISc Bangalore for spectral analysis.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Govindaraju, S., Tabassum, S. Visible Light Mediated Organophotoredox-Catalyzed One-Pot Domino Synthesis of Novel 6,7 Disubstituted 1H-Pyrroles. Top Catal (2022). https://doi.org/10.1007/s11244-022-01580-y
Accepted:
Published:
DOI: https://doi.org/10.1007/s11244-022-01580-y