
Abstract
Three isostructural, Iron(III), Manganese(III) and Chromium complexes, with a tetradentate Schiff base ligand (H2L2) from o-phenylenediamine and o-vanillin having the general formulae, [M(H2L2)(C2H3O2)(H2O)], where M = Cr, Mn, Fe) have been prepared. The complexes were then subsequently characterized by physico-chemical and spectroscopic methods and further investigated for their magnetic nature. All the complexes are neutral mononuclear species and crystallize in a triclinic space group P-1(2). Single-crystal diffraction studies reveal that the ligand, H2L2, coordinates to the central metal ion in a tetradentate fashion through both the azomethine nitrogen and the deprotonated phenolic oxygen generating neutral complexes. One of the acetate counter anions and a water molecule is involved in the coordination, generating a distorted octahedral geometry in all the complexes. Temperature dependence magnetic susceptibility was measured in a squid magnetometer in the temperature range 300–1.8 K and reveals that all of the three complexes show antiferromagnetic behavior in the temperature range studied. The experimental susceptibility data fitting, by considering the intermolecular interactions, enabled to estimate the magnetic anisotropy D and lead to the parameters (D = + 0.14 cm−1 and zJ = − 0.35 cm−1) for FeIII and (D = − 3.95 cm−1; E = + 0.54 cm−1; zJ = − 0.35 cm−1) for MnIII ions. This study is aiming and boosting to 'open up' our interest to focus more on mononuclear complexes with SMM character with first-row transition metals, which will be more interesting as far as the application side is concerned.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Molecular magnetism became an interesting field of research after the discovery of impressive properties and potential applications of d and/or f-metal molecular magnets [1, 2]. Due to this, chemists, chemical engineers, physicists, and material scientists closely collaborate and try to design, synthesize, characterize and model the magnetic properties of molecule-based materials of the Schiff bases [3,4,5,6]. Antiferromagnetic behavior is among the most energetically preferred magnetic configuration. Antiferromagnets are magnetic materials with zero net magnetization, which is due to the ordered moments being antiparallel between the adjacent atomic sites, and are not affected by applied external magnetic fields.
For a long time, these materials were thought to be interesting from the theoretical viewpoint only. But, nowadays, they are interested in the application viewpoint due to their possible use in spintronic technologies, where spin transport is the foundation of their functionalities [7,8,9,10] and they are used in information storage and manipulation [11, 12]. Even they can undergo antiferromagnetic switching up to two orders of magnitude faster than ferromagnetic switching in spintronic devices [13]. These materials are ranging from insulators to superconductors in their application and also play an important role in spintronic sensor and memory devices [14].
Another class of magnetic materials is single-molecule magnets (SMMs). These are materials that show characteristic magnetic properties, such as slow relaxation at a molecular level. Since the magnetization is retained even in the absence of an applied field, SMMs can be utilized in information storage at the molecular level. They can also be applied in spintronic devices as antiferromagnetic materials [15,16,17]. In the prior history, for this utilization, SMMs of polynuclear complexes possessing a high spin ground state with magnetic anisotropy were needed. This is because these materials can obtain a leading energy barrier to the reorientation of their magnetization [18,19,20]. But, due to structural complexity in polynuclear complexes, prediction of the magnetic anisotropy for enhancing the energy barrier is becoming complicated. The same problem was observed to study the stability in solution for explaining the overall magnetic property of these complexes [21,22,23,24]. Due to this, the design of SMMs is still a big challenge concerning the fundamental understanding of the origin of magnetic anisotropy and dynamic relaxation. Magnetic anisotropy achieved by strict regulation of geometry is the most important factor for high-performance mononuclear SMMs [25], which is dependent on axial anisotropy (D), along with spin ground state (S) [26]. Nowadays, the studies on magnetic properties of mononuclear complexes are growing fast and striking that few of the compounds reported to date are mononuclear magnets in zero fields [18,19,20, 27]. Designing the ligand field, which can preserve strict axial symmetry around the metal ion, is one of the best approaches to increasing the zero-field in d-metal mononuclear magnets. To do so, the design and synthesis of new ligands and the proper use of existing ones will remain to the fore [28]. In mononuclear molecular magnetism, the transition metals should be in low-coordinate ions of + 2 and + 3 oxidation states and extremely Lewis acidic, so that they can readily bind nucleophiles (ligands) and can directly bound atoms weakly by compensating for this charge [29]. The majority of mononuclear 3d molecular magnets are based on the half-integer spin of the metal ion. This is due to its ability to display slow relaxation of magnetization in a range of coordination environments [30]. For these molecules, magnetic anisotropy is widely used to control the mechanism for the relaxation of magnetizations.
In the past, several mononuclear Mn(II), Fe(II), Cr(III), and Fe(III) molecular magnets were reported [20, 29, 31,32,33]. Some of them are SMMs [18, 34,35,36,37] and others are even spin-crossover (SCO) compounds with N2O4 coordination environments [38]. Also, octahedral pseudo-dimeric molecular magnets are very common and show specific properties in their structural features [39].
On account of these, herein we report our investigations on structural and magnetic properties of three iso-structural CrIII, MnIII, and FeIII mononuclear complexes with a tetradentate Schiff base ligand having N2O4 coordination with hydrogen-bonded dimers.
Experimental
Materials and methods
The chemicals, o-phenylenediamine, o-vanillin, Mn(C2H3O2)2·4H2O, FeCl3·6H2O, and CrCl3·6H2O, were used from Sigma-Aldrich. Commercially available, diethyl ether, methanol, chloroform, hydrogen peroxide, n-hexane, DMSO-d6, and ethanol were purchased. All chemicals and solvents were used without further purification.
Preparation of the ligand and complexes
Preparation of the ligand (H2L2)
o-Phenylenediamine (OPDA) (8 mmol, 1.112 g) was dissolved in 10 mL MeOH and stirred for 20 min. o-Vanillin (8 mmol, 1.216 g) dissolved in 10 mL MeOH was added gradually to the above solution and stirred again for additional 20 min. Three drops of H2O2 were added to oxidize the aldehyde to carboxylic acid to enhance cyclization. The resultant mixture was left under reflux for 6 h with constant stirring and cooled (Fig. 1). Yellowish orange precipitate formed on cooling was filtered and washed thoroughly by diethyl ether and dried under vacuum. The existence of both H2L1 and H2L2 was seen by MALDI-TOF analysis having a molecular ion peak m/z at 241.09 (M + H) with molecular formula C14H12N2O2 for H2L1and 377 (M + H) with the molecular formula (C22H20O4N2) for H2L2, respectively (Fig. S6). The mixture was further purified by column chromatography in silica gel (eluent CH2Cl2: n-hexane 100:10) leading to pure compound H2L2 (1.427 g, 53%.) IR cm−1 (KBr) 3418(s), 1472(s), 1576(m), (Fig. S2) (3), 1H-NMR (DMSO-d6; 500 MHz)δ (ppm): 13.2(d, 2H), 10.7(s, 1H), 10.2(s, 1H), 9.0 (s, 1H), 8.9(d, 2H), 7.7(t, 3H), 6.9(d, 2H), 6.6(d, 2H), 3.3(s, 6H) (Fig. S1); 13C-NMR (DMSO-d6; 500 MHz) δ (ppm) 162,149, 141, 133, 123, 119, 116, 112 (Fig. S2); MALDI-TOF spectrum 377 m/z (M + H), H2L2 (C22H20O4N2) (Fig. S6 and S7). CHN calc. (%) for (H2L2: C22H20N2O4): C, 70.21: H, 5.3; N, 7.44. Found (%): C, 71.02; H, 5.03; N, 7.11.

The synthesis scheme of the ligand H2L2
1.2.3 Preparation of [Mn(H2L2)(C2H3O2)(H2O)]·2CH3OH (1)
A hot methanolic solution of (10 ml) (60 °C) of Mn(C2H3O2)2·4H2O (0.490 g, 2 mmol) was added to a stirred solution of Schiff base ligand H2L2 (0.754 g, 2 mmol) in 10 mL methanolic solution. The mixture was stirred under reflux for 6 h at 120 °C. The resulting solution was left overnight with constant stirring at room temperature. It was then filtered and left undisturbed. The yellow needle-like crystals those suitable for single-crystal X-ray diffraction obtained by slow evaporation after 20 days with a yield of 0.624 g, 75%) CHN calc. (%) for (Mn(H2L2)(C2H3O2)(H2O)·2CH3OH (1) C, 61.25, H, 4.64, N, 6.49, Mn, 12.76 Found (%) C, 60.22, H, 4.41, N, 6, Mn, 12.05, IR cm−1 (KBr) (3431, s), (2924, w), (1581, s), (1435, s), (1193, s), (973, m), (737, s) (Fig. S3).
2.2.2 Preparation of [Cr(H2L2)(C2H3O2)(H2O)]·2CH3OH (2)
A hot methanolic solution (60 °C) of CrCl3·6H2O (0.474 g, 3 mmol) was added to a stirred solution of the Schiff base ligand H2L2 (1.131 g, 3 mmol) in 10 mL methanol in presence of sodium acetate (1.23 g, 15 mmol). The resulting mixture was stirred under reflux for 6 h at 140 °C. Then, it was kept overnight with constant stirring at room temperature. It was then filtered and left undisturbed for crystallization. The yellow crystal that is suitable for single-crystal X-ray diffraction obtained after a month by slow evaporation with a yield (0.624 g, 57%). CHN calc. (%) for (Cr(H2L2)(C2H3O2)(H2O)·2CH3OH (2) C, 61.68, H, 4.67, N, 6.54, Cr, 12.12 Found (%) C, 60.87, H, 4.97, N, 6.02, Cr, 12.44, IR cm−1 (KBr); 3258(s), 2086(s), 1604(m), 1484(s), 1459(m), 1239 (s), 742(s) (Fig. S4).
2.2.2 Preparation of [Fe(H2L2)(C2H3O2)(H2O)]·2CH3OH(3)
A hot 5 mL methanolic solution (60 °C) of FeCl3·6H2O (0.474 g, 3 mmol) was added to a stirred solution of the Schiff base ligand H2L2 (1.131 g, 3 mmol) in 10 mL methanol in the presence of sodium acetate (0.82 g, 10 mmol). The mixture was stirred under reflux for 6 h at 140 °C. The resulting solution was left overnight with constant stirring at room temperature. It was then filtered and left undisturbed for crystallization. The yellow crystal suitable for single-crystal X-ray diffraction was obtained after 15 days by slow evaporation with a yield of 0.624 g, 69%) CHN calc. (%) for (Fe(H2L2)(C2H3O2)(H2O)·2CH3OH(3) C, 61.11, H, 4.62, N, 6.48, Fe, 12.96, Found (%) C, 62.33, H, 4.41, N, 6.02, Fe, 12.01, IR cm−1 (KBr); 3428 (s), 2922(w), 2085(s), 1604(m), 1479(s), 733(s)(Fig. S5).
Physical measurements
Infrared (FTIR) spectra for ligand and complexes were recorded on a Perkin Elmer 100FTIR spectrometer. 1H NMR and 13C-NMR spectra for a ligand were obtained by using a Bruker Advance II 400 MHz spectrometer in DMSO-d6. The mass spectrum for a ligand was collected on a Bruker Microflex LT MALDI-TOF–MS spectrometer. The single-crystal X-ray data for complexes were collected on an STOE IPDS2T diffractometer equipped with a variable-temperature nitrogen cold stream using monochromatic MoK\α radiation (λ = 0.71073 Å). The magnetic susceptibility measurements were performed on crystal samples on a Quantum Design MPMS XL-7 SQUID magnetometer between 1.8 and 300 K with polycrystalline samples of 22 mg, 20 mg, and 23 mg for Cr, Mn and Fe, respectively.
X-ray crystallography
X-ray diffraction data of single crystals for the three complexes up to a maximum of 50° in the 2θ scan mode were collected in single-crystal X-ray at 180 K (Table 1). Crystal parameters and refinement results for three compounds are summarized in Table 1. The structures were solved with the olex2.solve structure solution program. The obtained models were refined with olex2.refine, refinement package using Gauss–Newton minimization. In all three systems, all non-hydrogen atoms were refined anisotropically.
SQUID magnetometer measurements
Magnetic susceptibility measurements were made on microcrystalline samples of the complexes 1–3 from 1.8–300 K using the SQUID MPMS-XL magnetometer restricted to the field BDC = 7 T and T = 1.8–400 K. In measuring DC magnetic susceptibility experiments the small field BDC = 0.1 T was used.
has been applied in taking the temperature dependence of the static magnetic susceptibility between T = 1.8–300 K. Analyses were performed on crushed polycrystalline samples of 20 mg, 23 mg and 22 mg for compounds 1, 2 and 3, respectively. These data were corrected for the underlying diamagnetism.
Result and discussion
The structural description of complexes 1–3
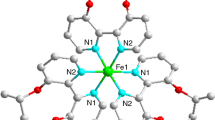
Selected bond lengths and bond angles for the complexes (1–3) are presented in Table 2, Table S1, S2, S3, and S4. The comparative bond parameters for three complexes are presented in Table 3. The atom-labeled mononuclear complexes 1–3 are presented in (Fig. 2). The molecular formula determined at 180 K, full space group descriptions and unit cell parameters are depicted in Table 1. In three of the complexes, the crystal lattice contains two slightly different metal centers identified as M1A and M1B, where M = MnIII, CrIII and FeIII (Figs. 2, S14 and S17). The structural view of the complexes along a and b axis is shown in Fig. S9 for 1, Fig. S10a and b for 2, and Fig. S14a and b for 3. The complexes are composed of a deprotonated Schiff base ligand, a water ligand, and a deprotonated acetate ligand as well. The equatorial plane contains two imine nitrogen atoms, N1 and N2, and two phenoxido oxygen atoms, O5 and O7, from deprotonated Schiff base ligand H2L2.

a The representative crystal structures of 1, 2 and 3; b Hydrogen bonding with 50% probability (crystal structures and hydrogen bonding of 1 and 2 are presented in Figs. S16 and S17)
The axial positions are occupied by a water ligand and a deprotonated acetate ligand. The long Mn–O bond distances, (Mn-O1 = 2.305(2) and Mn-O2 = 2.153(3), observed in 1 for axial coordinating ligands are indicative of a high valent Mn(III) oxidation state (Tables 2 and S1 [39, 40]. The observed slight variation in bond distances among the metal centers of the dimers (M = MnIII, CrIII and FeIII), which is (Mn1A-N1A = 1.991 Å to Mn1B-N1B = 1.988 Å for 1, (Cr1A-N1A = 1.985(3) Å to Cr1B-N1B = 1.986(4) Å, for 2 and Fe1A–N1A = 1.990(2) to Fe1B–N1B = 1.9924(18) for 3 is indicative for the distortion of the octahedral geometry in two centers of three complexes.
The characteristic bond distances and bond angles for 3 appear very similar to 1 and 2, except for the shorter bond distances of 2.1506(18) for Fe1A-O2A in 3, which is 2.153(3) Å for Mn1A-O2A in 1 and 2.152(3) Å for Cr1A-O2A in 2. In the second hydrogen-bonded dimer, it is 2.1409(17) Å for Fe1B-O2B, which is 2.143(3) Å for Mn1B-O2B and 2.144(2) Å for Cr1B-O2B (Table 2). These observed similarities among the three structures can also be supported by the elongation of a bond distance of Mn1A-O1A (water) = 2.305(3) Å in the Mn1A center to 2.360(3) Å (Mn1B-O1B (water)) in Mn1B and a bond distance of Cr1A-O1A (water) = 2.305(3) Å in Cr1A center to 2.358(3) Å in Cr1B-O1B (water). These can be further supported by the observed compression in bond distance of 1.894(3) Å in Mn1A-O5A (hydroxyl) to 1.887(2)Å in Mn1B-O5B (hydroxyl) (Table.S1) for 1. Similarly, 1.894(3) Å in Cr1A-O5A (hydroxyl) to 1.885(2)Å in Cr1B O5B(hydroxyl) (Table S2) for 2 and 1.8915(15)Å in (Fe1A-O5A (hydroxyl) to 1.8922(18) Å in (Fe1B-O5B (hydroxyl) for 3 (Table S3).
The average Cr–O distance of 2.305(2)Å for Cr1A–O1A and 2.152(3)Å for Cr1A–O2A is longer than the average Cr–N distance of 1.987(2)Å for Cr1A–N1A and 1.994(2)Å for Cr1A–N2A (Table 2). This is due to the nitrophilic character of the Lewis acidic Cr3+unit [41, 42].
The observed slight difference in bond distance of 1.989(3)Å (Mn1A-N2A) to 1.995(3)Å (Mn1B-N2B) and 1.988(3)Å (Mn1A-N1A) to 1.995(3)Å (Mn1B-N1B) for 1 shows that even though the two imines are from the same ligand, they are coming from different sides (from water and acetate). This is due to the difference in steric hindrance for two axial groups (less hindered oxygen from water ligand and more hindered oxygen from acetate ligand in our compounds). This scenario avoided steric hindrance and maintained the stability of the compounds. Also, a very small variation in increasing bond length from (Mn1A–N2A) to (Mn1B–N2B) in the range of 1.989(3) Å –1.995(3)Å = 0.006 Å and from 1.988(3)Å (Mn1A–N1A) to 1.995(3)Å (Mn1B–N2B) is observed. This is indicative for, even though two axial groups (water ligand oxygen and acetate ligand oxygen) are different on the steric bulk, their effect on the bond distance is insignificant and both bond distances are comparable. On the other hand, the pronounced bond angle variation from 85.91(10)° O2A (acetate oxygen)-Mn1A-N2A) to 83.59(10)° (O1A(water oxygen)-Mn1A-N2A) is due to the bulk acetate group compared to water, which prefers to be away from imine nitrogen and hydroxyl oxygen, so that to maximize the stability of the compound. These scenarios complete the tasks of the bonds to be compressed and elongated to a high extent. These all properties hold true for complexes 2 and 3 except with slight variation in bond distance and bond angle. This slight variation is due to difference in metal centers, proving the iso-structural properties of the three complexes.
The observed H-bonding in three compounds played a significant role in governing the architecture of the structures giving hydrogen-bonded dimers. This was expected because the H-bonding and π–π stacking have an important role in structure modification [18, 19, 39, 43,44,45]. The dominant H-bonding in the complexes is coming in between the coordinated H2O, non-coordinated methoxy and hydroxy oxygen of the o-vanillin entity and the solvent molecules. The longest hydrogen bonding in 1 is observed between the hydrogen atoms of the water molecules interacting with neighboring coordinated hydroxy oxygen of the ligand present in one of the hydrogen-bonded dimers with the bond distances of 2.899 Å. Similarly, the shortest hydrogen bonding is observed between the hydrogen atoms of the solvent molecule interacting with the neighboring oxygen atom of the coordinated acetate group which is present in the second dimer with a bond distance ranging from 2.88 to 2.94 Å (Fig. 2b).
In complex 2, the longest hydrogen bonding is observed between the hydrogen atoms of the water ligand interacting with neighboring non-coordinated methoxy oxygen of the ligand with the bond distances of 2.896 Å. Similarly, the shortest hydrogen bonding is observed between the hydrogen atoms of the solvent molecule interacting with the neighboring oxygen atom of the coordinated acetate ligand of the dimer with a bond distance of 2.88 Å (Fig. S13). In complex 3, a similar hydrogen bond formulation happened as complex 1, except with the longest and shortest bond distances of 2.898 Å and 2.88 Å, respectively (Fig. S16). The presence of solvent in the structure lattice did not show any disorder in the crystal structure, probably due to their coordination in crystal lattice only. As seen in 1, the short C–C interplanar distances of two rings of 3.646 Å observed in 3 reveal the presence of pronounced π–π stacking in the molecule (Fig. 3).

The representative π–π stacking of aromatic groups with approximately parallel molecular planes in 2 (complexes 1 and 3 are presented in Fig. S18)
Magnetic properties
Magnetic susceptibility measurements have been performed for compounds 1, 2 and 3, from 300 to 1.8 K at 0.1 T. From the χmT vs T curves, it has been concluded that the complexes show dominant antiferromagnetic interaction in the temperature studied (Fig. 4).

The χmT versus T plot for compounds 1, 2 and 3 between 1.8 K and 300 K
For the FeIII complex (3), with a spin state of S = 5/2, at 300 K, the χmT value is 4.48 cm3 K mol−1, which is less than the expected value of five unpaired electrons, which may be due to weak antiferromagnetic interaction between the monomeric metal centers. The χmT product decreases with temperature and 1.15cm3K mol−1 at 1.8 K, which indicates the presence of intramolecular antiferromagnetic interactions between metal centers [46, 47].
Somewhat the same magnetic behavior has been observed for the Manganese analog, (1). At room temperature, the χmT value observed for MnIII is 2.99cm3K mol−1 which is close to the value of non-interacting high-spin d4 MnIII ion with four unpaired electrons of χmT = 3.0 cm3K mol−1 [17, 18]. As the temperature decreases, the χmT product decreases gradually and reaches a minimum value of 1.03 cm3K mol−1 at 1.8 K, which is indicative of intramolecular antiferromagnetic interaction between the metal centers [48, 49].
Similarly, for chromium complex (2), χmT versus T trends have been investigated in the temperature range of 300–1.8 K at 0.1 T. The χmT value of 1.71 cm3K mol−1 at 300 K is less than the expected calculated value for three unpaired electrons of CrIII, indicating the weak antiferromagnetically interacting chromium centers. The χmT value remains almost constant until 50 K, and then, it decreases abruptly to reach a value of 0.93 cm3 K mol−1 at 1.8 K suggesting that a dominant intramolecular antiferromagnetic interaction between the hydrogen-bonded dimer metal centers [27].
The experimental susceptibility data for the complexes were fitted simultaneously including intermolecular interactions to estimate the magnetic anisotropy D and lead to the following parameters: for FeIII ion the CASSCF data (D = + 0.14 cm−1) and the fitting including intermolecular interactions result in the best fit with (D = + 0.14 cm−1 and zJ = − 0.35 cm−1). Similarly for the MnIII ion, the CASSCF data (D = − 3.95 cm−1, E = ± 0.54 cm−1) and the fitting including intermolecular interactions result in the best fit with (D = − 3.95 cm−1; E = ± 0.54 cm−1; zJ = − 0.35 cm−1).
Conclusion
In summing up, three isostructural complexes of Fe(III), Mn(III) and Cr(III) with the Schiff base ligand from o-phenylenediamine and o-vanillin have been synthesized, characterized, and further investigated for their magnetic behavior. Both the azomethine nitrogen and the hydroxyl groups in the deprotonated fashion are coordinated to the central metal in all the complexes. In addition, one of the acetate groups and a water molecule is also involved in coordination to generate a distorted octahedral geometry around the central metal. Hydrogen-bonded dimers close packed structural arrangement is observable in the unit cell of all the species with a considerable amount of π–π interaction between the molecules. Magnetic behaviors of the complexes were investigated in the temperature range 300–1.8 K. All of the three complexes show antiferromagnetic behavior in the temperature range studied.
References
Maniaki D, Pilichos E, Perlepes SP (2018) Coordination clusters of 3d-metals that behave as single-molecule magnets (SMMs): synthetic routes and strategies. Front Chem 6:461
Madhu NT, Tang JK, Hewitt IJ, Clérac R, Wernsdorfer W, Van Slageren J, Christopher EA, Powell AK (2005) What makes a single molecule magnet? Polyhedron 24:2864–2869. https://doi.org/10.1016/j.poly.2005.03.015
Feng X, Mathonière C, Jeon IR, Rouzières M, Ozarowski A, Aubrey ML, Gonzalez MI, Clérac R, Long JR (2013) Tristability in a light-actuated single-molecule magnet. J Am Chem Soc 135:15880–15884. https://doi.org/10.1021/ja407332y
Bałanda M, Fitta M (2019) Molecular magnets. Crystals 9(3):132
Khan A, Fuhr O, Akhtar MN, Lan Y, Thomas M, Powell AK (2020) Synthesis, characterization and magnetic st. J Coord Chem 73:1045–1054. https://doi.org/10.1080/00958972.2020.1759044
Madhu NT, Radhakrishnan PK (2000) Cobalt(II) complexes of 1,2-(diimino-4′-antipyrinyl) ethane and 4-N-(4′-antipyrylmethylidene) aminoantipyrine. Trans Metal Chem 25(3):287–292
Gomonay O, Jungwirth T, Sinova J (2017) Concepts of antiferromagnetic spintronics. Phys Status Solidi RRL. https://doi.org/10.1002/pssr.201700022
Jungwirth T, Sinova J, Manchon A, Marti X, Wunderlich J, Felser C (2018) The multiple directions of antiferromagnetic spintronics. Nat Phys 14:200–203. https://doi.org/10.1038/s41567-018-0063-6
Rezende SM, Azevedo A, Rodríguez-Suárez RL (2019) Introduction to antiferromagnetic magnons. J Appl Phys. https://doi.org/10.1063/1.5109132
Bai H, Zhou X, Zhou Y, Chen X, You Y, Pan F, Song C (2020) Functional antiferromagnets for potential applications on high-density storage and high frequency. J Appl Phys. https://doi.org/10.1063/5.0029799
Gebrezgiabher M, Bayeh Y, Gebretsadik T, Gebreslassie G, Elemo F, Thomas M, Linert W (2020) Lanthanide-based single-molecule magnets derived from Schiff base ligands of salicylaldehyde derivatives. Inorganics 8(12):66
Senthil Kumar K, Bayeh Y, Gebretsadik T, Elemo F, Gebrezgiabher M, Thomas M, Ruben M (2019) Spin-crossover in iron(II)-Schiff base complexes. Dalton Trans 48:15321–15337. https://doi.org/10.1039/c9dt02085c
Siddiqui SA, Sklenar J, Kang K, Gilbert MJ, Schleife A, Mason N, Hoffmann A (2020) Metallic antiferromagnets. J Appl Phys. https://doi.org/10.1063/5.0009445
Jungwirth T, Marti X, Wadley P, Wunderlich J (2016) Antiferromagnetic spintronics. Nat Nanotechnol 11:231–241. https://doi.org/10.1038/nnano.2016.18
Campbell VE, Tonelli M, Cimatti I, Moussy JB, Tortech L, Dappe YJ, Rivière E, Guillot R, Delprat S, Mattana R, Seneor P, Ohresser P, Choueikani F, Otero E, Koprowiak F, Chilkuri VG, Suaud N, Guihéry N, Galtayries A, Miserque F, Arrio MA, Sainctavit P, Mallah T (2016) Engineering the magnetic coupling and anisotropy at the molecule-magnetic surface interface in molecular spintronic devices. Nat Commun. https://doi.org/10.1038/ncomms13646
Coronado E (2020) Molecular magnetism: from chemical design to spin control in molecules, materials and devices. Nat Rev Mater 5:87–104. https://doi.org/10.1038/s41578-019-0146-8
Shao D, Wang XY (2020) Development of single-molecule magnets. Chin J Chem 38:1005–1018. https://doi.org/10.1002/cjoc.202000090
Novitchi G, Jiang S, Shova S, Rida F, Hlavička I, Orlita M, Wernsdorfer W, Hamze R, Martins C, Suaud N, Guihéry N, Barra AL, Train C (2017) From positive to negative zero-field splitting in a series of strongly magnetically anisotropic mononuclear metal complexes. Inorg Chem 56:14809–14822. https://doi.org/10.1021/acs.inorgchem.7b01861
Jin XX, Chen XX, Xiang J, Chen YZ, Jia LH, Wang BW, Cheng SC, Zhou X, Leung CF, Gao S (2018) Slow magnetic relaxation in a series of mononuclear 8-coordinate Fe(II) and Co(II) complexes. Inorg Chem 57:3761–3774. https://doi.org/10.1021/acs.inorgchem.7b03071
Dolai M, Mondal A, Liu JL, Ali M (2017) Three novel mononuclear Mn(III)-based magnetic materials with square pyramidal: versus octahedral geometries. New J Chem 41:10890–10898. https://doi.org/10.1039/c7nj02919e
Luo QC, Zheng YZ (2021) Methods and models of theoretical calculation for single-molecule magnets. Magnetochemistry. https://doi.org/10.3390/magnetochemistry7080107
Zadrozny JM, Telser J, Long JR (2013) Slow magnetic relaxation in the tetrahedral cobalt(II) complexes [Co(EPh)4]2-(EO, S, Se). Polyhedron 64:209–217. https://doi.org/10.1016/j.poly.2013.04.008
El-Khatib F, Cahier B, Shao F, López-Jordà M, Guillot R, Rivière E, Hafez H, Saad Z, Girerd JJ, Guihéry N, Mallah T (2017) Design and magnetic properties of a mononuclear Co(II) single molecule magnet and its antiferromagnetically coupled binuclear derivative. Inorg Chem 56:4601–4608. https://doi.org/10.1021/acs.inorgchem.7b00205
Gomez-Coca S, Cremades E, Aliaga-Alcalde N, Ruiz E (2013) Mononuclear single-molecule magnets: tailoring the magnetic anisotropy of first-row transition metal complexes. J Am Chem Soc 135:7010–7018. https://doi.org/10.1021/ja4015138
Lan W, Hao X, Dou Y, Zhen Zhou Lu, Yang HL, DL, Yunhui Dong LK and DZ, (2019) Investigation of the magnetic anisotropy in a series of trigonal bipyramidal Mn(II) complexes. Polymers 11:1585
Hay MA, Sarkar A, Marriott KER, Wilson C, Rajaraman G, Murrie M (2019) Investigation of the magnetic anisotropy in a series of trigonal bipyramidal Mn(II) complexes. Dalton Trans 48:15480–15486. https://doi.org/10.1039/c9dt02187f
Oliveira WXC, Pereira CLM, Pinheiro CB, Cano J, Lloret F, Julve M (2015) Relatively strong intramolecular antiferromagnetic coupling in a neutral CrIII2Nb2 heterobimetallic molecular square. Chem Commun 51:11806–11809. https://doi.org/10.1039/c5cc04285b
Perlepe PS, Maniaki D, Pilichos E, Katsoulakou E, Perlepes SP (2020) Smart ligands for efficient 3d, 4d and 5d-metal single-molecule magnets and single-ion magnets. Inorganics 8(6):39
Moseley IP, Lin CY, Zee DZ, Zadrozny JM (2020) Synthesis and magnetic characterization of a dinuclear complex of low-coordinate iron(II). Polyhedron 175:114171. https://doi.org/10.1016/j.poly.2019.114171
Hay MA, Sarkar A, Craig GA, Bhaskaran L, Nehrkorn J, Ozerov M, Marriott KER, Wilson C, Rajaraman G, Hill S, Murri M (2019) In-depth investigation of large axial magnetic anisotropy in monometallic 3d complexes using frequency domain magnetic resonance and: Ab initio methods: a study of trigonal bipyramidal Co(II). Chem Sci 10:6354–6361. https://doi.org/10.1039/c9sc00987f
Yu H, Nan Q (2019) Synthesis and crystal structures of manganese(III) complexes derived from bis-Schiff bases with antibacterial activity. Trans Metal Chem. https://doi.org/10.1007/s11243-018-00296-x
Čižmár E, Šoltésová D, Kazheva ON, Alexandrov GG, Kravchenko AV, KosenkoID SIB, Bregadze VI, Fedorchenko AV, Starodub VA, Buravov LI, Feher DOA, A, (2018) Large magnetic anisotropy of chromium(III) ions in a bis (ethylenedithio ) tetrathiafulvalenium salt of chromium bis(dicarbollide), (ET)2[3,3′-Cr(1,2-C2B9H11)2]. Trans Metal Chem 43(7):647–655. https://doi.org/10.1007/s11243-018-0253-1
Li X, Yang J, Zhou Y, Li B, Wang L (2018) Synthesis, characterization and magnetic properties of cobalt(II) and manganese(II) metal-organic frameworks assembled from 4,6-bis(imidazole-1-yl)isophthalic acid ligands. Trans Metal Chem 43(6):473–478. https://doi.org/10.1007/s11243-018-0247-z
Su J, Yin L, Ouyang Z, Wang Z, Zheng W (2020) 1,2-Diaza-4-phospholide complexes of chromium(II): dipotassium organochromates behaving as single-molecule magnets. Dalton Trans 49:6945–6949. https://doi.org/10.1039/d0dt00878h
Mossin S, Tran BL, Adhikari D, Ink M, Heinemann FW, Sutter J, Szilagyi RK, Meyer K, Mindiola DJ (2012) A mononuclear Fe(III) single-molecule magnet with a 3/2↔5/2 spin crossover. J Am Chem Soc 134:13651–13661. https://doi.org/10.1021/ja302660k
Realista S, Fitzpatrick AJ, Santos G, Ferreira LP, Barroso S, Pereira LCJ, Bandeira NAG, Neugebauer P, Hrubý J, Morgan GG, Slageren JV, Calhorda MJ, Martinho PN (2016) A Mn(III) single ion magnet with tridentate Schiff-base ligands. Dalton Trans 45:12301–12307. https://doi.org/10.1039/c6dt02538b
Cremades E, Ruiz E (2011) Mononuclear Fe II Single-Molecule Magnets : A Theoretical Approach. Inorg Chem 50:4016–4020
Saha R, Biswas S, Steele IM, Dey K, Mostafa G (2011) A supramolecular spin crossover Fe(iii) complex and its Cr(iii) isomer: stabilization of water-chloride cluster with in the supramolecular host. Dalton Trans 40:3166–3175. https://doi.org/10.1039/c0dt01256d
Tang J, Costa JS, Smulders S, Molnár G, Bousseksou A, Teat SJ, Li Y, Van Albada GA, Gamez P, Reedijk J (2009) Two-step spin-transition iron(III) compound with a wide [high spin-low spin] plateau. Inorg Chem 48:2128–2135. https://doi.org/10.1021/ic801973x
Kühne IA, Gavin LC, Harris M, Gildea B, Müller-Bunz H, Stein M, Morgan GG (2021) Mn(III) complexes with nitro-substituted ligands—spin states with a twist. J Appl Phys. https://doi.org/10.1063/5.0050276
Molcˇanov Kresˇimir, Biserka Kojic´-Prodic, (2019) Towards understanding p-stacking interactions between non-aromatic rings. IUCrJ 6:156–166. https://doi.org/10.1107/S2052252519000186
Chen Z, Naidu R, Subramanian A (2001) Separation of chromium(III) and chromium(VI) by capillary electrophoresis using 2,6-pyridinedicarboxylic acid as a pre-column complexation agent. J Chromatogr 92:219–227
Güveli Ş, Özdemir N, Bal-Demirci T, Soylu MS, Ülküseven B (2019) Hydrogen-bonded and π-stacked nickel(II) thiosemicarbazone complexes: synthesis, spectral and structural studies. Trans Metal Chem 44:115–123. https://doi.org/10.1007/s11243-018-0275-8
Wheeler SE (2013) Understanding substituent effects in noncovalent interactions involving aromatic rings. Acc Chem Res 46:1029–1038. https://doi.org/10.1021/ar300109n
Oshita H, Suzuki T, Kawashima K, Abe H, Tani F, Mori S, Yajima T, Shimazaki Y (2019) The effect of π-π stacking interaction of the indole ring with the coordinated phenoxyl radical in a nickel(II)-salen type complex. Comparison with the corresponding Cu(II) complex. Dalton Trans 48:12060–12069. https://doi.org/10.1039/c9dt01887e
Basak T, Gómez-García CJ, Gomila RM, Frontera A, Chattopadhyay S (2021) Hydrogen bond mediated intermolecular magnetic coupling in mononuclear high spin iron (III ) Schiff base complexes: synthesis, structure and magnetic study with theoretical insight. RSC Adv 11:3315–3323. https://doi.org/10.1039/D0RA09425K
Lan W, Wang X, Yang L, Si W, Zhuang S, Liu H, Liu Q, Zhang D (2019) Cyanide-bridged polynuclear heterobimetallic complexes: synthesis, crystal structures, and magnetic properties. Trans Metal Chem 44:383–389. https://doi.org/10.1007/s11243-019-00304-8
Cheng W, Zhang L, Ni Z, Kou H (2013) Synthesis, crystal structure, and magnetic properties of a Mn(III)-radical complex. Bull Korean Chem Soc 34:2195–2198
Kar P, Biswas R, Drew MGB, Ida Y, Ghosh A (2011) Structure and magnetic properties of an unprecedented syn-anti l-nitrito-1kO:2kO’ bridged Mn(III)-salen complex and its isoelectronic and isostructural formate analogue. Dalton Trans 40:3295–3304. https://doi.org/10.1039/c0dt01521k
Acknowledgements
This study was supported by Addis Ababa Science and Technology University (AASTU), Addis Ababa, Ethiopia. We are also thankful to AASTU, for a Ph.D. studentship for one of us (F.E.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors are declaring that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Elemo, F., Sani, T. & Thomas, M. Antiferromagnetically coupled iso-structural CrIII, MnIII and FeIII complexes of a tetradentate Schiff base ligand derived from o-phenylenediamine. Transit Met Chem 47, 265–274 (2022). https://doi.org/10.1007/s11243-022-00510-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-022-00510-x