
Abstract
The kinetics and mechanism of base hydrolysis of tris(3-(2-pyridyl)-5,6-bis(4-phenyl sulphonic acid)-1,2,4-triazine)iron(II), \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) have been studied in aqueous, sodium dodecyl sulphate (SDS) and cetyltrimethyl ammonium bromide (CTAB) media at 25, 35 and 45 °C under pseudo-first-order conditions, i.e. \(\left[ {\text{OH}^{ - } } \right]\) ≫ \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\). The reaction is first order each in \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) and hydroxide ion. The rate increases with increasing ionic strength in aqueous and SDS media, whereas this parameter has little effect in CTAB. In SDS medium, the rate-determining step involves the reaction between \(\left[ {\text{OH}^{ - } } \right]\) and \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\), whereas in CTAB medium, it involves reaction between a neutral ion pair, {\({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\)·4CTA+} and \(\left[ {\text{OH}^{ - } } \right]\) ions. The specific rate constants and thermodynamic parameters (E a, ΔH #, ΔS # and ΔG #35°C ) have been evaluated in all three media. The near equal values of ΔG #35°C obtained in aqueous and SDS media suggest that these reactions occur essentially by the same mechanism. Slightly lower ΔG #35°C values in CTAB medium can be attributed to a higher concentration of reactants in the Stern layer. The reaction is inhibited in SDS medium but catalysed in CTAB. The former can be attributed to the anionic surfactant creating more repellent space between the reactants. Catalysis in CTAB medium is ascribed to electrophilic and hydrophilic interactions between hydroxide ion/substrate with the cationic Stern layer, resulting in increased local concentrations of both reactants.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In recent years, base hydrolysis of low-spin iron(II)-diimine complexes and their derivatives in aqueous solution, water–methanol mixtures, micellar, mixed micellar and reverse micellar media, has received the attention of several workers [1,2,3,4,5,6]. Smith and Case synthesized a group of ligands by introducing various substituents on the diimine moiety; 3-((2-pyridyl)-5,6-diphenyl)-1,2,4-triazine (PDT), 3-(4-(4-phenyl-2-pyridyl)-5,6-di-phenyl-1,2,4-triazine (PPDT) and 2,4-bis (5,6-bis(4-phenyl)-1,2,4-triazin-3-yl) pyridine (BDTP) are some among them. To overcome the difficulties of insolubility of these ligands in water, they also prepared sulphonated derivatives, 3-(2-pyridyl)-5,6-bis(4-phenyl sulphonic acid)-1,2,4-triazine disodium salt (PDTS), 3-(4-(4-phenyl sulphonic acid)-2-pyridyl)-5,6-bis(4-phenyl sulphonic acid)-1,2,4-triazine (PPDTS) and (2,4-bis(5,6-bis(4-phenyl sulphonic acid)-1,2,4-triazin-3-yl) pyridine tetrasodium salt (BDTPS). These triazines as well as their sulphonated derivatives have adequate ligand strength to force spin pairing in iron(II) and form the kinetically inert \(t_{2g}^{6}\) configuration in 1:3 mol ratio [7,8,9,10,11,12]. These triazines are also of considerable interest in the study of kinetics of substitution reactions due to their high molar absorptivities. Studies on the acid fission, base hydrolysis and cyanide ion attack of iron(II)-diimine complexes having ionisable substituents revealed that the anionic sulphonate-substituted complexes react faster than their unsubstituted analogues [13,14,15,16,17]. Moreover the electron-withdrawing effect of \({\text{SO}}_{3}^{ - }\) reduces electron density at the iron(II) centre and so facilitates hydroxide ion attack. We have previously reported on the base hydrolysis of tris(3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine)iron(II), \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\) in aqueous, SDS and CTAB media [18]. This work was aimed at the study of the effect of the base hydrolysis of iron(II) complexes with the bidentate 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine ligand in aqueous, sodium dodecyl sulphate (SDS) and cetyltrimethyl ammonium bromide (CTAB) media. In this paper, the effect of sulphonate group on the base hydrolysis of the analogous complex, \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\), is reported.
Experimental
Reagents and instrumentation
PDTS was obtained from GFS Chemicals Inc., USA, and used without purification. A standard PDTS solution of 1.0 × 10−2 mol dm−3 was prepared by dissolving the required quantity in water. A 1.0 × 10−2 mol dm−3 solution of iron(II) was prepared by dissolving the requisite quantity of ammonium iron(II) sulphate hexahydrate (BDH, AnalaR) in 1.0 × 10−2 mol dm−3 H2SO4· \({\text{Fe}}\text{(}{\text{PDTS}}\text{)}_{\text{3}}^{{\text{4} - }}\) was prepared by mixing iron(II) and PDTS solutions in 1:3 molar ratio [19]. All other solutions were prepared as previously described [18]. A Varian Cary 100 Bio UV–visible spectrophotometer equipped with a Varian Peltier temperature controller accurate to within ±0.05 °C was used to perform kinetic runs for slow reactions and record the absorption spectra of reactants and products. Kinetic measurements of fast reactions were made with an Applied Photophysics SX20 stopped-flow spectrophotometer coupled with an online data acquisition system. The temperature of the sample chamber was controlled to within ± 0.1 °C (Scheme 1).

Structures of tris(3-(2-pyridyl)-5,6-bis(4-phenyl sulphonic acid)-1,2,4-triazine disodium salt)ferrate(II), \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\)
Kinetic procedure
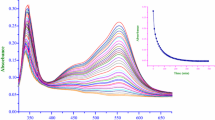
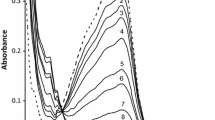
The base hydrolysis of \(\text{Fe(PDTS)}_{\text{3}}^{{\text{4} - }}\) in aqueous and SDS is relatively slow and hence its kinetics were followed by conventional UV–visible spectrophotometry. The spectral changes were recorded over 350–700 nm. The progress of the reaction was monitored at λ max = 562 nm, where the maximum change in absorbance was observed, as a function of time. The reaction conditions and procedure employed were as described earlier [18]. The effect of temperature on the reaction was studied by performing kinetic runs at 25, 35 and 45 °C. Figure 1a shows the UV–Vis spectral changes observed for the base hydrolysis of \(\text{Fe(PDTS)}_{\text{3}}^{{\text{4} - }}\) in aqueous media, and the inset shows a typical kinetic trace at 562 nm. The absorbance–time data at the selected wavelength were best fit to single exponential, using Origin 7.5® graphical analysis software [20]. The observed pseudo-first-order rate constant, k obs, values were calculated using nonlinear least-square fit of the data to equation [21];
where A o , A t and A∞ represent the absorbance of the reaction mixture initially, at time, t and at the end of the reaction, respectively. All the runs were performed in duplicate. The rate constants were reproducible to within ±4%. The k obs values for the base hydrolysis of \(\text{Fe(PDTS)}_{\text{3}}^{{\text{4} - }}\) in aqueous and SDS media at 35 °C are shown in Table 1.

Visible absorption changes of \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) with time in aqueous medium as acquired by UV–Vis spectrophotometry. Inset is a typical kinetic trace at λ max = 562 nm (a) and a typical kinetic trace acquired on a stopped-flow reaction analyser showing a perfect fit to a single exponential at λ max = 562 nm (b). \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) = 2.0 × 10−5 mol dm−3, \(\text{[OH}^{ - } \text{]}\) = 5.0 × 10−2 mol dm−3, μ = 0.2 mol dm−3, temperature = 35 °C
The base hydrolysis of \(\text{Fe(PDTS)}_{\text{3}}^{{\text{4} - }}\) in the cationic CTAB medium is relatively very fast; hence, its kinetics were studied by stopped-flow spectrometry using an Applied Photophysics SX20 stopped-flow spectrophotometer. This apparatus consists of two syringes, which are filled with the reactants through separate valves from the reservoir syringes, containing the individual reactants. The drive syringes were thermostatted by means of a water bath. The reactants were charged into the reaction chamber by a compressed gas-driven piston (800 kPa) to allow ultra-rapid mixing (within 10−3 s). The absorbance–time kinetic trace was recorded at the set wavelength and the pseudo-first-order rate constants were evaluated [22, 23] (k obs values at 35 °C are given in Table 1). A typical single exponential kinetic trace acquired from these experiments is shown in Fig. 1b.
Results and discussion
Base hydrolysis in aqueous medium
The rate constants for the title reaction are linearly dependent on \([ {\text{OH}}^{ - } ]\), showing first-order dependence on hydroxide. A plot obtained in aqueous medium is presented in Fig. 2, and those for SDS and CTAB media are given in supplementary data S1. The rate increases with increasing ionic strength, indicating that the rate-limiting step involves ions of like charges, i.e. \([ {\text{OH}}^{ - } ]\) and \(\text{Fe(PDTS)}_{\text{3}}^{{\text{4} - }}\). Thus, in the rate-limiting step \(\text{Fe(PDTS)}_{\text{3}}^{{\text{4} - }}\) reacts with \([ {\text{OH}}^{ - } ]\) to give an intermediate [Fe(PDTS)2(PDTS-η1)(OH)]5− which furthers reacts with another \({\text{OH}}^{ - }\) to give [Fe(PDTS)2(OH)2]4−. Subsequently this intermediate decomposes to Fe(OH)2 and two PDTS ions. Under aerobic conditions, Fe(OH)2 rapidly oxidizes to colourless Fe(OH)3 [18]. A detailed stepwise mechanism for the base hydrolysis is given in supplementary data S2. The rate law is described by the equation k obs = k \([ {\text{OH}}^{ - } ]\), with the rate data given in Table 1. Hereafter, the rate constant k obs in aqueous medium is referred to as k w. The specific rate constants k have been evaluated from the slopes of plots of k obs versus \([ {\text{OH}}^{ - } ]\). By measuring k at different temperatures, the activation parameters (E a, ΔH #, ΔS # and ΔG #35°C ) were computed using the Arrhenius and Eyring equations [24]. The specific rate constants and activation parameters data are presented in Table 2.

Effect of hydroxide ion on the base hydrolysis of \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) in aqueous media
Blandamer et al. [25] studied the reaction of tris(ferrozine)iron(II) abbreviated as \({\text{Fe}}({\text{ppsa}})_{3}^{4 - }\)(where ppsa = PDTS) with hydroxide in the concentration range from 0.67 to 1.33 mol dm−3 in both water and aqueous methanol. They postulated a ligand-substituted intermediate on the basis of changes in the spectrum of \({\text{Fe}}({\text{ppsa}})_{3}^{4 - }\). On addition of hydroxide ion, the magnitude of the charge transfer band of \({\text{Fe}}({\text{ppsa}})_{3}^{4 - }\) at 562 nm was observed to decrease markedly, accompanied by the emergence of a new band at 635 nm. They also found that the intensities of both peaks decreased with time. However, the overlay spectra obtained in the present study (in which \([ {\text{OH}}^{ - } ]\) = 0.01–0.20 mol dm−3) do not show any such peak at 635 nm (see Fig. 1). The lack of an observable intermediate in the present study is likely due to the lower hydroxide concentrations employed. The substrate reacts directly with \([ {\text{OH}}^{ - } ]\) in the rate-limiting step to give [Fe(PDTS)2(PDTS-η 1)(OH)]5− (see Supplementary data S2).
Base hydrolysis in micellar media
Micelle formation occurs at the critical micelle concentration (CMC), and reactions are frequently studied at the CMC to understand the effect of micelles on the reaction [26, 27]. The CMC values of CTAB-\({\text{OH}}^{ - }\) and SDS-\({\text{OH}}^{ - }\) mixtures are reported to be 1.5 × 10−3 and 1.0 × 10−3 mol dm−3, respectively [18]. Hence, the kinetic runs in CTAB and SDS media were carried out at their respective CMC values, maintaining the same conditions as employed in aqueous medium. The results suggest that the pseudo-first-order rate constants, k ψ, increase with increasing hydroxide concentration (see Table 1) and follow the same trend as in aqueous medium, suggesting that the reactions in micellar media occur by a similar mechanism to that proposed for aqueous medium. Hence, the rate equation can be expressed as k ψ = k \([ {\text{OH}}^{ - } ]\). In SDS medium, the rate increases with increase in ionic strength, whereas in CTAB medium, there is little effect on the rate (see Table 1). Plots of k ψ versus \([ {\text{OH}}^{ - } ]\) are given in Fig. 2 and Supplementary data S1.
The specific rate constants, k, have been evaluated from the slopes of the plots of k obs versus \([ {\text{OH}}^{ - } ]\). From these specific rate constants E a, ΔH #, ΔS # and ΔG #308K for the reaction in CTAB and SDS media were computed using the Eyring equation [28] (Table 2). Eyring plots [29] for base hydrolysis of \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) in aqueous, SDS and CTAB media are given Fig. 3.

Eyring plots for the base hydrolysis of \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) in aqueous and different surfactant media
The results indicate that the reaction is inhibited in anionic surfactant (SDS), but catalysed by cationic surfactant (CTAB). Inhibition is only to the extent of about 50% of the rate in aqueous medium, whereas catalysis is of the order of 104. The effect of ionic strength is interesting, with the rate increasing with ionic strength in both aqueous and SDS media, whereas it has little effect in CTAB medium. We infer that in aqueous and SDS media, the rate-determining step involves ions of like charges, i.e. \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) and \(\text{OH}^{ - }\). In SDS solutions both substrate and hydroxide ions are repelled by dodecyl sulphate anion (DS−), resulting in a decrease in rate. In CTAB medium, the rate-determining step involves an ion and a neutral species. Due to electrostatic interactions, the \({\text{Fe}}(\text{PDTS})_{3}^{4 - }\) molecules will be ion-paired with cetyltrimethyl ammonium cations, CTA+ to form {\({\text{Fe}}(\text{PDTS})_{3}^{4 - }\)·4CTA+}. These neutral species react with OH−; hence, there is no effect of ionic strength on the reaction rate. The E a values in Table 2 also suggest inhibition in SDS but catalysis in CTAB media. The large negative value of ΔS # and low positive ΔH # values indicate favourable conditions for base hydrolysis [30]. The data in CTAB medium indicate the formation of a more ordered transition state than in the aqueous and SDS media. Further the high values of free energy of activation ΔG # and enthalpy ΔH # suggest that the transition state is highly solvated. Comparable ΔG # values observed in aqueous and SDS media clearly indicate that these reactions occur by an equivalent mechanism in either media.
The reactants \(\text{Fe}({\text{PDTS}})_{3}^{4 - }\) and \({\text{OH}}^{ - }\), together with the medium (dodecyl sulphate anion, \({\text{DS}}^{ - }\)) are all anionic, resulting in electrostatic repulsion of \({\text{OH}}^{ - }\) and \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) by \({\text{DS}}^{ - }\) aggregates. In other words, SDS creates more anionic space between the reactants which slows down the reaction. Catalysis to the extent of 104 times was noticed in CTAB compared to aqueous medium. Electrostatic and hydrophobic interactions between hydroxide and cetyltrimethyl ammonium cation, CTA+, lead to concentration of \({\text{OH}}^{ - }\) in the Stern layer [18]. Likewise the substrate (\(Fe(\text{PDTS})_{3}^{4 - }\)) is a negatively charged species; hence, both reactants are concentrated in the Stern layer of CTAB. This facilitates the reaction between \(Fe(\text{PDTS})_{3}^{4 - }\) and \({\text{OH}}^{ - }\) ions, hence the significant catalysis in this medium. The low ΔG # values in CTAB medium are consistent with this interpretation.
Effect of SDS/CTAB on the rate
As discussed above, SDS inhibits the base hydrolysis of \(Fe(\text{PDTS})_{3}^{4 - }\), whereas CTAB catalyses it. Hence, the effect of SDS/CTAB mixtures on the rate of reaction was studied at different [SDS] (1.0 × 10−5–1.0 × 10−2 mol dm−3)/[CTAB] (zero to 1.0 × 10−3 mol dm−3) keeping \([ {\text{OH}}^{ - } ]\) (5.0 × 10−2 mol dm−3) and µ (0.2 mol dm−3) constant. The k ψ-[SDS] and k ψ-[CTAB] profiles can be explained by using the Berezin’s pseudo-phase [27, 31] and the Menger–Portnoy [32] models, respectively. Similar results obtained for the base hydrolysis of \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\) in these media indicate that the same reaction trend is followed by both \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\) and its sulphonated analogue in SDS and CTAB media. The data for these experiments, together with plots of k ψ versus [SDS]/[CTAB], are given in Supplementary data S3–S11.
Comparison with base hydrolysis of \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\)
It is expected that under similar conditions, the rate of base hydrolysis of anionic \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) should be lower than that of its cationic unsulphonated analogue, \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\). However, the present experimental results are contrary to this expectation. This may be ascribed to weakening of the iron–nitrogen bonds, due to the electron-withdrawing sulphonate groups. According to Burgess, sulphonation generally results in lower stability with respect to ligand substitution for iron(II) complexes [33]. For example, substituting the electron-withdrawing sulphonate group into the coordinated 1,10-phenanthroline ligands of \({\text{Fe}}({\text{phen}})_{3}^{2 + }\) weakens the iron–nitrogen bond significantly and increases its rate of dissociation to a small but significant extent. For dissociation of \({\text{Fe}}({\text{phen}})_{3}^{2 + }\), the ligand dissociation rate constant (k 1d) is 7.3 × 10−5 s−1 [17, 34, 35] as against 11.0 × 10−5 s−1 for \({\text{Fe}}({\text{SO}}_{3} {\text{phen}})_{3}^{ - }\) [33] in aqueous solution at 298.2 K. Similarly the strongly electron-withdrawing nitro group increases the k 1d value for \({\text{Fe}}({\text{NO}}_{3} {\text{phen}})_{3}^{ - }\) to 49 × 10−5 s−1 under the same conditions [36]. Burgess and coworkers [13] reported that sulphonate substitution of the 3-(2-pyridyl)-5,6-bis(2-furyl)-1,2,4-triazine (fertri) ligand has a similar effect. The electron-withdrawing properties of sulphonate substituents affect both σ-and π-bonding between iron and the nitrogen donors in substituted triazine ligands. The σ-bonding involves electron donation from the ligands to the metal centre through the coordinated nitrogen atoms, whereas sulphonation enhances π-back donation of electrons from the metal to the ligand. This latter effect appears to be dominant in the present case.
Rate inhibition in SDS and catalysis in CTAB medium are common features observed for the base hydrolysis of both \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) and \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\). In the case of \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\), the decrease in rate in SDS medium is to the extent of 6–8 times depending on the conditions. \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\) concentrates in the Stern layer because of electrostatic interactions, thus partitioning between aqueous and micellar phases leading to lowering of [\({\text{Fe}}({\text{PDT}})_{3}^{2 + }\)] in the aqueous phase. \({\text{OH}}^{ - }\) is repelled by the Stern layer and thus remains only in the aqueous phase, reacting only with the partitioned \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\) which results in the observed inhibition. In the case of \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\), both the substrate and \({\text{OH}}^{ - }\) are repelled from the Stern layer and hence both reactants remain in the aqueous phase. The inhibition (~50%) noticed may be due to medium effects, i.e. \({\text{DS}}^{ - }\) anions cause more anionic space between substrate and hydroxide ions resulting in a decrease in interactions between these two reactants.
For the base hydrolysis of \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\) in CTAB medium, the PDT ligands in the substrate will orient around the Stern layer due to their high electron density. The \([ {\text{OH}}^{ - } ]\) in the Stern layer will be high because of electrostatic interactions between hydroxide and CTA+. This brings hydroxide ions and \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\) together in the Stern layer, leading to the observed catalysis enhancement of only ~2 times in this cationic micelle. In the base hydrolysis of \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\), both the substrate and \({\text{OH}}^{ - }\) are anionic and concentrate in the cationic Stern layer due to electrostatic and hydrophobic interactions. This results in a marked catalysis of 104 times.
Conclusions
The rate of base hydrolysis of \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) is higher than that of its unsulphonated analogue, \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\) in aqueous medium, contrary to the expectation that it should be slower on electrostatic considerations. This can be explained by weakening of the iron–nitrogen bond due to the presence of electron-withdrawing sulphonate groups. Base hydrolysis of \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\) and its sulphonated analogue is inhibited in SDS medium, but catalysed in CTAB. In SDS medium, the k ψ-[surfactant] profile can be explained by the Berezin’s pseudo-phase model and by the Menger–Portnoy model in CTAB phase, indicating that the same reaction trend is followed in both SDS and CTAB media. The decrease of rate in SDS medium for the iron(II)-PDT complex can be explained by partitioning of \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\) between the aqueous and micellar phases. The inhibition is ~50% in the case of \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\) as both the substrate and hydroxide ions remain in the aqueous phase, and this decrease can be ascribed to medium effects in the presence of SDS. In CTAB medium, the \({\text{OH}}^{ - }\) ion concentrates in the cationic Stern layer. In the case of base hydrolysis of \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\), PDT ligands in the substrate orient around the Stern layer due to their high electron density and thus hydroxide ions and \({\text{Fe}}({\text{PDT}})_{3}^{2 + }\) are concentrated in the Stern layer, leading to moderate catalysis, whereas in the case of \({\text{Fe}}({\text{PDTS}})_{3}^{4 - }\), the anionic substrate concentrates highly in the Stern layer, leading to marked catalysis of the order of 104 times.
References
Kundu A, Dasmandal S, Rudra S, Mahapatra A (2015) J Mol Liq 209:99–103
Abdel-Rahman RM, Makki MS, Ali TE, Ibrahim MA (2015) J Heterocycl Chem 52:1595–1607
Kundu A, Dasmandal S, Majumdar T, Mahapatra A (2014) Colloids Surf A Phys Chem Eng Asp 452:148–153
Mandal HK, Majumdar T, Mahapatra A (2011) Int J Chem Kinet 43:579–589
Gharib A, Ezz-Eldin A, Gosal N, Burgess J (2001) Croat Chem Acta 74:545–558
Mandal HK, Dasmandal S, Mahapatra A (2016) J Disp Sci Technol 37:555–564
Gibbs CR (1976) Anal Chem 48:1197–1201
Stookey LL (1970) Anal Chem 42:779–781
Ratnam S, Anipindi NR (2012) Trans Met Chem 37:453–462
Bellam R, Anipindi NR (2014) Trans Met Chem 39:311–326
Bellam R, Jaganyi D (2017) Int J Chem Kinet 49:182–196
Bellam R, Sivamadhavi S, Ramakrishna S, Mambanda A, Jaganyi D, Anipindi NR (2017) J Coord Chem 70:1893–1909
Burgess J, Hubbard CD, Miyares PH, Cole TL, Dasgupta TP, Leebert S (2005) Trans Met Chem 30:957–963
Baxendale J, George P (1950) Trans Faraday Soc 46:736–744
Baxendale J, George P (1950) Trans Faraday Soc 46:55–63
Krumholz P (1949) Nature 163:724–725
Lee T, Kolthoff I, Leussing D (1948) J Am Chem Soc 70:3596–3600
Bellam R, Raju GG, Anipindi NR, Jaganyi D (2016) Trans Met Chem 41:271–278
Bellam R, Anipindi NR (2012) Trans Met Chem 37:489–495
Origin 7.5™ SRO, v7.5714 (B5714) (2003) Origin Lab Corporation, Northampton One, Roundhouse Plaza, Northampton, MA, 01060 USA
Atwood JD (1997) Inorganic and organometallic reaction mechanisms. Whiley-VCH Inc, NY, pp 43–61
Laidler K (2003) Physical chemistry. Houghton Mifflin Company, Boston
Connors KA (1990) Chemical kinetics: the study of reaction rates in solution. VCH, New York
Evans MG, Polanyi M (1935) Trans Faraday Soc 31:875–894
Blandamer MJ, Burgess J, Wellings P (1979) Trans Met Chem 4:95–97
Desando M, Reeves L (1986) Can J Chem 64:1817–1822
Berezin IV, Martinek K, Yatsimirskii AK (1973) Russ Chem Rev 42:787–802
Eyring H (1935) J Chem Phys 3:107–115
Atwood JD (1997) Inorganic and organometallic reaction mechanisms, 2nd edn. Wiley, New York, pp 43–61
Gangwar S, Rafiquee M (2007) Int J Chem Kinet 39:638–644
Berezin IV, Martinek K, Yatsimirsky A (1973) Usp Khim 42:1729–1756
Menger FM, Portnoy CE (1967) J Am Chem Soc 89:4698–4703
Burgess J (1967) J Chem Soc A Inorg Phys Theor. doi:10.1039/J19670000431
Moroi Y, Braun AM, Graetzel G (1979) J Am Chem Soc 101:567–572
Holyer R, Hubbard C, Kettle S, Wilkins R (1966) Inorg Chem 5:622–625
Burgess J, Prince R (1963) J Am Chem Soc (Resumed) 12:5752–5758
Acknowledgements
The authors are gratefully indebted to the University Grants Commission, New Delhi, India, and University of KwaZulu-Natal, South Africa, for financial support to Rajesh Bellam.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bellam, R., Anipindi, N.R. & Jaganyi, D. Base hydrolysis of tris(3-(2-pyridyl)-5,6-bis(4-phenyl sulphonic acid)-1,2,4-triazine)iron(II) in aqueous, SDS and CTAB media: kinetic and mechanistic study. Transit Met Chem 42, 719–725 (2017). https://doi.org/10.1007/s11243-017-0179-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-017-0179-z