
Abstract
The kinetics of base hydrolysis of tris(3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine)iron(II), \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) has been studied in aqueous, cetyltrimethyl ammonium bromide (CTAB) and sodium dodecyl sulphate (SDS) media at 25, 35 and 45 °C under pseudo-first-order conditions, i.e. \( [ {\text{OH}}^{ - } ]\gg [{\text{Fe(PDT)}}_{ 3}^{2 + } ] \). The reactions are first order in both of substrate \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) and hydroxide ion. The rates decrease with increasing ionic strength in aqueous and CTAB media, whereas SDS medium shows little ionic strength effect. The rate also increases with CTAB concentration but decreases with SDS. The specific rate constant, k and thermodynamic parameters (E a, ΔH #, ΔS # and ΔG #) have also been evaluated. The near equal values of ΔG # obtained in aqueous and CTAB media suggest that these reactions occur essentially by the same mechanism such that \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) reacts with OH− in the rate-determining step. The ionic strength effect in SDS medium suggests that the rate-determining step involves an ion and a neutral species. The results in this study are compared with those obtained for other iron(II)-bipyridine complexes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The formation and dissociation kinetics of the low-spin iron(II) diimine complexes, tris(1,10-phenanthroline)iron(II) [1] and tris(2,2′-bipyridyl)iron(II) [2, 3] has been of interest to researchers for many years. Initially, the kinetics of these reactions were studied in aqueous solutions; later studies were also extended to organic solvents [4–6], micellar [7] and reverse micellar media [8]. Of these, the nucleophilic attack by hydroxide ion, i.e. base hydrolysis at the iron(II) centre, has been the most extensively studied [9–11]. The ligand 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine (PDT) is capable of forming stable five-membered chelate rings with iron(II). Its iron(II) complex, \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) is structurally similar to \( {\text{Fe(bpy)}}_{ 3}^{2 + } \), with three planar bidentate N-ligands coordinated to the central iron atom in an octahedral geometry (Scheme 1). PDT has two phenyl rings which are known for their electron donating tendency. This results in increased electron density on the iron(II) centre. Burgess and Prince studied the base hydrolysis of iron(II) 4,4′- and 5,5′-dimethyl-substituted bipyridine complexes and observed decreased rates. This was attributed to the electron donating nature of the methyl groups. It is observed that \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) undergoes base hydrolysis in aqueous medium. This reaction is catalysed by cationic cetyltrimethyl ammonium bromide (CTAB) and inhibited by anionic sodium dodecyl sulphate (SDS). We have taken up this work to understand the reactivity of hydroxide ion with \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) and compare the results with iron(II) bipyridine, 4,4′- and 5,5′-dimethyl-substituted bipyridine complexes. The results of our studies are presented in this paper.

Structural formula of \( {\text{Fe(PDT)}}_{ 3}^{2 + } \)
Experimental
Reagents
PDT (GFS Chemicals Inc., USA) and SDS (Fluka) were used as such without recrystallization. A 1.0 × 10−2 mol dm−3 solution of PDT was prepared by dissolving the requisite quantity in methanol (Merck, GR) and stored in an amber coloured bottle. 1.0 × 10−2 mol dm−3 solutions of SDS and NaOH were prepared by dissolving the requisite quantities in Millipore water. All other solutions were prepared as described earlier [12]. A 2.0 × 10−4 mol dm−3 solution of \( {\text{Fe(PDT)}}_{ 3}^{{ 2 { + }}} \) was prepared by mixing ferrous ammonium sulphate and PDT in 1:3 ratio. The λ max and ε max values for this solution were 555 nm and 2.34 × 104 mol−1 dm3 cm−1, respectively, in agreement with the literature [13].
Kinetic procedure
Base hydrolysis of the magenta coloured \( {\text{Fe(PDT)}}_{ 3}^{{ 2 { + }}} \) gives a colourless solution of Fe(OH)3. The kinetic runs in aqueous and surfactant media were performed at 555 nm, i.e. the λ max of \( {\text{Fe(PDT)}}_{ 3}^{{ 2 { + }}} \) by the procedure described earlier [12], and the product has negligible absorbance at this wavelength. Kinetic runs were performed using a Varian Cary 100 Bio UV–Visible spectrophotometer thermostatted with a Varian peltier temperature controller to within ±0.05 °C. The data obtained from the UV–VS instrument were fitted by nonlinear least-squares to the absorbance-time data according to Eq. (1), using Origin 7.5® graphical analysis software. All kinetic traces were fitted with exponential functions to generate the pseudo-first-order rate constants, k obs (for convenience the k obs values in aqueous medium are referred to as k w and those in CTAB/SDS media as k ψ ).
where A o, A t and A ∞ represent the absorbance of the reaction mixture at zero time, at time t and at the end of the reaction, respectively. The kinetic runs were performed at three different temperatures (25, 35 and 45 °C). Half of the runs were performed in duplicate. The rate constants were reproducible to within ±8 %.
Results and discussion
Base hydrolysis in aqueous medium
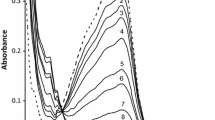
The kinetic runs were carried out under pseudo-first-order conditions with \( [ {\text{OH}}^{ - } ]\gg [{\text{Fe(PDT)}}_{ 3}^{2 + } ] \) (i.e., [OH−] was in 50-fold excess over \( [{\text{Fe(PDT)}}_{ 3}^{2 + } ] \)) at different initial concentrations of [OH−] in the range 1.0 × 10−2 − 20.0 × 10−2 mol dm−3, keeping the concentration of \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) at 2.0 × 10−5 mol dm−3 and ionic strength at 0.2 mol dm−3. The effects of \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) and ionic strength on the rate have also been studied. Figure 1 shows the spectral changes during the reaction; the inset is a kinetic trace of time dependence of the absorbance at the λ max of \( {\text{Fe(PDT)}}_{ 3}^{{ 2 { + }}} . \) The rate data (Table 1) show that the rate constants are linearly dependent on [OH−] (see Supplementary Fig. 1), indicating that the order with respect to hydroxide ion is unity. Furthermore, the rate of reaction is found to decrease with ionic strength, indicating that the rate-limiting step involves ions of opposite charge. The rate equation can be expressed as
or

UV–Vis spectral changes observed during the reaction; Inset is a typical kinetic trace at 555 nm. \( [{\text{Fe(PDT)}}_{ 3}^{2 + } ] = 2.0 \times 10^{ - 5} {\text{mol}}\;{\text{dm}}^{ - 3} ,\; [ {\text{OH}}^{ - } ]= 5.0 \times 10^{ - 2} \) mol dm−3, μ = 0.2 mol dm−3 and temperature = 25 °C
Hereafter the rate constant, k obs in aqueous medium is referred to as k w. The slope of a plot of k w versus [OH−] gives the specific rate constant, k (Table 2).
Hydroxide ion is capable of nucleophilic attack on Fe(II)-diimine complexes and gives an intermediate, [Fe(diimine)2(diimine-η1)(OH)]+ in which OH− is bonded to the metal centre and one dimimine has become monodentate. This ultimately gives Fe(OH)2 which is subsequently oxidised to Fe(OH)3 in the presence of O2. Similarly, base hydrolysis of \( {\text{Fe(phen)}}_{ 3}^{{ 2 { + }}} \) or \( {\text{Fe(bpy)}}_{ 3}^{{ 2 { + }}} \) gives Fe(OH)3 in the presence of dissolved oxygen. In the present studies, the solutions were optically clear up to ~80 % of the reaction. At the end of each run, a red colloidal precipitate was observed. Hence, we assume that \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) reacts with hydroxide to give \( \left[ {{\text{Fe(PDT)}}_{ 2} ( {\text{PDT-}}\upeta^{\text{1}} ) ( {\text{OH)}}} \right]^{ + } . \) This step is considered to be rate-limiting. \( \left[ {{\text{Fe(PDT)}}_{ 2} ( {\text{PDT-}}\upeta^{\text{1}} ) ( {\text{OH)}}} \right]^{ + } \) loses the monodentate PDT by subsequent nucleophilic attack of another OH− to give \( [ {\text{Fe(PDT)}}_{ 2} ( {\text{OH)}}_{2} ]_{{}}^{{}} \) which decomposes to Fe(OH)2 plus PDT. Under aerobic conditions, Fe(OH)2 rapidly oxidises to Fe(OH)3. The overall mechanism for the base hydrolysis of \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) is outlined in Scheme 2.

Mechanism for the base hydrolysis of \( {\text{Fe(PDT)}}_{ 3}^{2 + } \)
Base hydrolysis in micellar media
When a surfactant is dissolved in water, its molecules have a tendency to aggregate spontaneously into thermodynamically stable particles of colloidal dimensions, due to hydrophobic interactions [14, 15]. At lower concentrations, the surfactant molecules behave like ordinary solutes. After attaining a certain concentration, the individual surfactant molecules assemble into spherical aggregates known as micelles [16]. The minimum concentration for the onset of micellization is known as critical micelle concentration, CMC.
The electrical charge of the surfactant generally influences the rate of reaction. Hence, the title reaction has also been carried out in cationic CTAB and anionic SDS micellar media at their respective CMC values. These CMC values of CTAB-OH− and SDS-OH− mixtures were determined by conductometric methods at fixed [OH−] = 5.0 × 10−2 mol dm−3 varying the concentration of CTAB/SDS. The CMC is the point of intersection of two straight lines observed in plots of [CTAB]/[SDS] versus specific conductance. The CMC values obtained for CTAB-OH− and SDS-OH− mixtures were 1.5 × 10−3 and 1.0 × 10−3 mol dm−3, respectively. The results show that the pseudo-first-order rate constants in micellar media, k ψ , increase with increasing OH− concentration (see Table 1) and follow the same trend as observed in aqueous medium (Fig. 2), suggesting that the reactions in micellar media occur by a similar mechanism to that proposed for aqueous medium. Hence, the final rate equation can be expressed as

Effect of hydroxide ion on the base hydrolysis of \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) in (a) CTAB and (b) SDS media. \( \left[ {{\text{Fe(PDT)}}_{ 3}^{{ 2 { + }}} } \right] = 2.0 \times 10^{ - 5} \) mol dm−3, μ = 0.2 mol dm−3, [CTAB] = 1.5 × 10−3 mol dm−3, [SDS] = 1.0 × 10−3 mol dm−3
Comparison of the rate data in Table 1 shows that the rate is enhanced in CTAB medium, but significantly retarded in SDS. Representative plots of rate constants versus [OH−] at 35 °C in aqueous and micellar media are presented in Fig. 3.

Effect of hydroxide ion on the base hydrolysis of \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) in aqueous and different surfactant media at 35 °C. \( \left[ {{\text{Fe(PDT)}}_{ 3}^{{ 2 { + }}} } \right] = 2.0 \times 10^{ - 5} \) mol dm−3, μ = 0.2 mol dm−3, [CTAB] = 1.5 × 10−3 mol dm−3, [SDS] = 1.0 × 10−3 mol dm−3
The specific rate constants, k, have been evaluated from the slopes of the plots of k obs versus [OH−]. Using these specific rate constants, the activation parameters (E a, ΔH #, ΔS # and ΔG # 308K) for aqueous, CTAB and SDS media were computed using the Arrhenius and Eyring equations (Table 2).
The E a values in Table 2 suggest catalysis of the reaction in CTAB medium and inhibition in the presence of SDS. The large negative value of entropy and low-positive enthalpy of activation are consistent with base hydrolysis [17]. The large negative entropy value obtained in CTAB indicates that the transition state is more rigid and ordered than that in aqueous medium. Further the high values of free energy and enthalpy of activation suggest that the transition state is highly solvated. The very similar ΔG # values observed in aqueous and CTAB media clearly indicate that these reactions occur essentially by a similar mechanism in both cases. The inhibition noticed in SDS medium may be due to repulsion of OH− by anionic dodecyl sulphate, DS− aggregates. The rate of the reaction decreases with increasing ionic strength in aqueous and CTAB media, suggesting that the rate-determining step involves ions of opposite charge. However, in SDS medium the ionic strength has little effect on the reaction which indicates that the rate-determining step involves an ion and neutral species. Formation of a neutral ion pair between \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) and two DS− ions is likely, and this reacts with OH− in the rate-determining step. This explains the ionic strength effect and the slightly higher ΔG # value in SDS medium.
Effect of CTAB
The effect of CTAB on the reaction was also studied by varying the concentration of CTAB from zero to 1.0 × 10−3 mol dm−3 at [OH− = 5.0 × 10−2 mol dm−3 and µ = 0.2 mol dm−3 (see Supplementary Table 1). The k ψ versus [CTAB] profile shows that the rate of the reaction increases sharply with increasing [CTAB] and reaches a maximum at 1.5 × 10−3 mol dm−3 (CMC) and then decreases (Fig. 4). This is the characteristic feature of bimolecular micellar catalysis [18–20].

Profile of k ψ versus [CTAB] for base hydrolysis of \( \text{Fe(PDT)}_{\text{3}}^{{\text{2 + }}} \). \( \left[ {{\text{Fe(PDT)}}_{ 3}^{2 + } } \right] = 2.0 \times 10^{ - 5} \) mol dm−3, \( [ {\text{OH}}^{ - } ] = 5.0 \times 10^{ - 2} \) mol dm−3 and µ = 0.2 mol dm−3
These observations can be explained by a pseudo-phase kinetic model proposed by Menger and Portnoy [21]. According to this model, the total volume of surfactant or micelle is considered as a separate phase which is uniformly distributed in the aqueous phase. Base hydrolysis occurs in both bulk aqueous and micellar phases (Scheme 3).

Menger and Portnoy pseudo-phase kinetic model
The terms S, D n and K s in Scheme 3 denote substrate (\( {\text{Fe(PDT)}}_{ 3}^{2 + } \)), micellised surfactant (D n = [total surfactant] − CMC) and binding constant of substrate with micelle (K s = [S M]/[S W][D n]), respectively. The constants k M and k W are the first-order rate constants in the micellar and aqueous phases, respectively. According to this model, k ψ for the overall reaction is given by Eq. (5).
For k M > k W, the rate of the reaction increases with increase in [D n] and ultimately reaches the limiting value k M. Conversely, if k M < k W, an increase in [D n] produces a decrease in k ψ and k ψ tends to attain the limiting value k W. Equation (5) can be rearranged into its reciprocal form as
or
Bunton and Cerichlli [22] have pointed out that the treatment of Eq. (6), i.e. a plot of 1/(k ψ − k W) versus 1/[D n], is very sensitive to the values of CMC that may be affected by the reaction media. As no linear relationship between 1/(k ψ − k w) and 1/[D n] could be observed, Eq. (6) is modified (by neglecting k w) as
From the present experimental data, good straight line plots are obtained at all three temperatures employed when 1/k ψ is plotted against 1/[D n] (see Supplementary Fig. 2). The slope and intercept of these plots are equal to 1/k M K S and 1/k M, respectively. The resulting constants are given in Table 3.
Micellar effects of bimolecular reactions involving ionic reactants follow the Hartley principle [23], i.e. cationic reactions are accelerated by anionic micelles and inhibited by cationic micelles and vice versa. Counter-ions are effectively concentrated at surfaces of ionic micelles where they partially neutralise the head group charges. Polar substrates are located at micellar surfaces, so it is reasonable to assume that the reactions involving ions or polar molecules occur in the region occupied by the surfactant head groups, the so-called Stern layer [24]. Most micellar catalysed reactions are supposed to occur in the Stern layer [21, 25]. Micellar catalysis is often rationalised in terms of bringing together the reactants in the Stern layer surrounding the micellar surface.
The catalytic effect in the presence of CTAB is due to electrostatic and hydrophobic interactions between hydroxide anions and CTA+ cations. The positively charged cetyltrimethylammonium and substrate ions cannot easily approach each other in the Stern layer. However, the high electron density of the three PDT ligands in the substrate should lead to their orientation around the Stern layer, facilitating interaction between \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) and OH−. The two opposing effects, namely concentration effect and dilution effect, explain the profile of k ψ versus [CTAB] which shows a maximum at the CMC. At lower [CTAB], the number of ionic or polar reactant species going into the Stern layer increases, leading to enhancement of the rate. After saturation in the micellar phase, further increase in the concentration of CTAB favours an increase in the number of micelles, leading to a decrease in effective concentrations of both \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) and OH− ions around the Stern layer. This is probably one of the reasons for the decrease in rate at higher [CTAB].
Effect of SDS
The rate data in Table 1 show that SDS inhibits base hydrolysis of \( {\text{Fe(PDT)}}_{ 3}^{2 + } \). The effect of SDS on the rate of reaction was also studied by varying the concentration of SDS in the range from 1.0 × 10−5 to 1.0 × 10−2 mol dm−3, at fixed concentration of OH− (5.0 × 10−2 mol dm−3) and ionic strength (0.2 mol dm−3) (see Supplementary Table 2). The k ψ versus [SDS] profile shows that k ψ initially decreases with increasing [SDS], and then becomes nearly saturated at higher concentrations (Fig. 5).

Plot of k ψ versus [SDS] for base hydrolysis of \( \text{Fe(PDT)}_{\text{3}}^{{\text{2 + }}} \) in SDS medium. \( \left[ {{\text{Fe(PDT)}}_{ 3}^{2 + } } \right] = 2.0 \times 10^{ - 5} \) mol dm−3, [OH−] = 5.0 × 10−2 mol dm−3 and µ = 0.2 mol dm−3
The profile in Fig. 5 can be explained by using Berezin’s pseudo-phase model [25–27] (Scheme 4). According to this model, the micellar catalysis or inhibition is based on the assumption that a bimolecular interaction between the reactants may occur in both the micellar pseudo-phase and aqueous phase.

Berezin’s pseudo-phase model
In this scheme, S denotes substrate (\( {\text{Fe(PDT)}}_{ 3}^{2 + } \)), whilst the subscripts M and W denote a pseudo-phase associated with the surfactant and the bulk solvent, respectively. K S and K OH are the equilibrium constants for partition of substrate and hydroxide between the aqueous and micellar phases. The constants k M and k w are the rate constants for the micellar and aqueous phases, respectively. In such a two phase system, it has been shown that for dilute surfactant solutions, in which the volume fraction of the micellar phase is small, the quantitative expression for the pseudo-first-order rate constant for a bimolecular reaction occurring only in aqueous and micellar phase is given by
where D n = [total surfactant] − CMC, K S and K OH are the association constants of \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) and OH− with SDS and [SDS] = k M/V (where V is the molar volume of the micelle). Since the hydroxide ion is a charged species and the substrate is a large molecule, the hydrophobic and electrostatic interactions are significant and give high values of K S and K OH. Since [SDS] is small it may be possible that \( k_{\text{W}} \gg k_{\text{M}} K_{\text{S}} K_{\text{OH}} [D_{\text{n}} ] \) so that Eq. (9) takes the form
Further, since [D n] is very small and thus the terms containing [D n]2 may be neglected. The inverse of Eq. (10) then simplifies to
A plot of 1/k ψ versus D n gives a straight line with negative slope and positive intercept (see Supplementary Fig. 3). The value of k w has been evaluated from the intercept of this plot. Substituting this value into the slope of the plot gives the sum of the binding constants, (K S + K OH) which are presented in Table 4.
The rate inhibition in the presence of SDS is attributed to selective partitioning of \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) in the micellar region. According to this, \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) is preferentially bound to the micelle whereas OH− is repelled. Thus, the partition of \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) between different regions decreases the probability of interaction with OH−, resulting in a decrease in rate. Ionic strength has no effect on the reaction in SDS medium, indicating that the rate-determining step involves an ion and neutral molecule. Since OH− ion is one of the reactants, the other reactant namely the iron(II)-PDT complex should be in neutral form. This is possible only when \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) forms an ion pair with anionic dodecyl sulphate anion, \( {\text{DS}}^{ - } ,\{ {\text{Fe(PDT)}}_{ 3}^{2 + } \cdot 2{\text{DS}}^{ - } \}. \)
The base hydrolysis of Fe(II)-bipyridine, 4,4′- and 5,5′-dimethyl-substituted bipyridine and PDT all follow essentially the same mechanism. At 25 °C, the specific rate constant (×103) values for substitution of Fe(II)-bipyridine, 4,4′-dimethyl bipyridine, 5,5′-dimethyl bipyridine and PDT complexes by hydroxide are 7.0, 6.0, 5.67 [9] and 0.80 [this work] mol dm−3 s−1, respectively. Hence, the substitution of Fe(II)-PDT complex by hydroxide is extremely slow compared to analogous complexes. This can be explained as follows. The methyl and phenyl groups are weakly activating, i.e. these groups release electrons to the nitrogen, resulting in increased electron density on the iron atom. Comparing \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) with dimethyl-substituted \( {\text{Fe(bpy)}}_{ 3}^{2+}, \) the former has more electron density on the iron(II) centre due to the presence of triazine N-atoms. Hence, \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) is less able to react with hydroxide ion. Therefore, the order of the reactivity towards base hydrolysis is bipyridine › dimethyl bipyridine » PDT. The ΔH # values for the base hydrolysis of \( {\text{Fe(PDT)}}_{ 3}^{2 + } /{\text{Fe(bpy)}}_{ 3}^{2 + } \) in aqueous and SDS medium are 102/75 and 114/82 kJ mol−1, respectively [28]. These values also confirm the above order of reactivity.
Conclusions
The catalytic effect observed for the title reaction in the presence of CTAB is due to electrostatic and hydrophobic interactions between hydroxide ion and positively charged CTAB. The positively charged cetylammonium and substrate ions are not expected to come close in the Stern layer. However, the high electron density of three PDT molecules in \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) leads to their orientation around the Stern layer, facilitating the reaction between \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) and OH−. In SDS medium hydroxide ion cannot easily approach negatively charged DS− aggregates. The other reactant \( {\text{Fe(PDT)}}_{ 3}^{2 + } \) is preferentially bound to the Stern layer of the micelle and thus distributes itself between aqueous and SDS phases. This decreases the probability of interaction between the two reactants in the aqueous phase, resulting in a decrease in rate.
References
Lee T, Kolthoff I, Leussing D (1948) J Am Chem Soc 70:3596–3600
Baxendale J, George P (1950) Trans Faraday Soc 46:736–744
Baxendale J, George P (1950) Trans Faraday Soc 46:55–63
Margerum D (1957) J Am Chem Soc 79:2728–2733
Blandamer MJ, Burgess J (1979) Pure Appl Chem 51:2087–2092
Blandamer MJ, Burgess J (1980) Coor Chem Rev 31:93–121
Mandal HK, Majumdar T, Mahapatra A (2011) Int J Chem Kinet 43:579–589
Sarkar D, Khilar K, Begum G, SubbaRao P (2005) Colloids Surfaces A: Physico Eng Asp 268:73–77
Burgess J, Prince R (1965) J Chem Soc, pp. 6061–6066
Kundu A, Dasmandal S, Majumdar T, Mahapatra A (2014) Colloids Surfaces A: Physico Eng Asp 452:148–153
Blandamer MJ, Burgess J, Chambers JG, Haines RI, Marshall HE (1977) J Chem Soc Dalton Trans, pp. 165–170
Bellam R, Anipindi NR (2014) Trans Met Chem 39:311–326
Stephen W, Islam M (1993) Anal Chim Acta 274:335–346
Hartley G (1941) Transa Faraday Soc 37:130–133
Jada A, Lang J, Candau SJ, Zana R (1989) Colloid Surfaces 38:251–261
Menger FM (1979) Acc Chem Res 12:111–117
Gangwar S, Rafiquee M (2007) Int J Chem Kinet 39:638–644
Cordes E, Gitler C (1972) Prog Bioorg Chem 2:1–53
Bunton C (1979) Catal Rev Sci Eng 20:1–56
Rao PS, Rao GK, Ramakrishna K, Murty P (1991) React Kinet Catal Lett 43:209–216
Menger FM, Portnoy CE (1967) J Am Chem Soc 89:4698–4703
Bunton CA, Cerichelli G (1980) Inter J Chem Kinet 12:519–533
Bunton CA (1991) Surfactants in solution. Springer, New York, pp 17–40
Bacaloglu R, Bunton CA, Cerichelli G, Ortega F (1989) J Phy Chem 93:1490–1497
Berezin IV, Martinek K, Yatsimirskii AK (1973) Russ Chem Rev 42:787–802
Martinek K, Yatsimirsky A, Levashov A, Beresin I, Mittal K (1977) In: Mittal KL (ed) Plenum Press, New York
Berezin I, Martinek K, Yatsimirsky A (1973) Usp Khim 42:1729–1756
Kundu A, Dasmandal S, Majumdar T, Mahapatra A (2013) Colloid Surfaces A: Physico Eng Asp 419:216–222
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bellam, R., Raju, G.G., Anipindi, N.R. et al. Kinetics and mechanism of base hydrolysis of tris(3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine)iron(II) in aqueous and micellar media. Transition Met Chem 41, 271–278 (2016). https://doi.org/10.1007/s11243-015-0018-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-015-0018-z