
Abstract
The intramolecular [3 + 2] cycloaddition (32CA) reactions of allenic nitrones have been studied within the molecular electron density theory (MEDT) at the MPWB1K/6-311G(d,p) computational level. These zwitter-ionic type 32CA reactions show high activation free energies between 22.2 and 34.9 kcal mol−1 in ethanol consistent with their predicted non-polar character and follow one-step mechanism with highly asynchronous transition states. Interestingly, when the nitrone and the allene moieties are separated by two methylene units, the [3 + 2] addition is energetically feasible along the C5-C6 terminal double bond of the allene, while the presence of four methylene units change the cyclization selectivity towards the internal C4-C5 double bond of the allene. This is in complete agreement with the experimental outcomes. The molecular mechanism study in terms of bonding evolution theory (BET) shows varied electron density changes along these two reaction paths. Finally, the topological analysis of AIM (atoms-in-molecules) reveals the presence of non-covalent interactions at the interatomic bonding regions of the transition states, which agrees well with the electron localization function analysis and the forming C–C and C-O bond distances.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Allenes present an intriguing group of reacting counterparts in [3 + 2] cycloaddition (32CA) reactions owing to the presence of two cumulative unsaturations [1]. Although the simplest allene shows limited reactivity in the 32CA reactions with C-phenyl-N-methyl nitrone 1, incorporation of electron-deficient substituents (such as cyano (2), carbomethoxy (3), phenylsulfonyl (4), methoxy (5), fluoro (6)) overcomes the unreactive nature, affording high yield of isoxazolidines under milder reaction conditions with site selectivity towards the generation of CA4-5 and CA5-6 addition (Scheme 1) [2,3,4,5,6,7]. Nitrone-allene 32CA reactions show well-established applications in the total synthesis of alkaloids and natural products [8], and also exhibit interesting selectivity and mechanistic implications. Recently, Lee et al. have reported the mechanism and selectivity of the intermolecular 32CA reactions of nitrones with activated allenes [9].

Intermolecular 32CA reactions of nitrones and allenes
An interesting alternative to explore the reactivity of unactivated allenes was designed by Lebel and Banucci in 1970 [10] from the intramolecular [3 + 2] cycloaddition (IM32CA) reactions of allenic nitrones, and was also reported by Padwa et al. [11] in 1993 to proceed smoothly affording reasonably good yields. The IM32CA reactions consist of both nitrone and the allenic function suitably placed in the same molecule and exhibit interesting site selectivity for the two allenic double bonds depending on the substrate. For instance, the IM32CA reaction of exocyclic nitrone 7 affords isoxazolidine 8 by addition along the terminal C5-C6 double bond, while the allenic nitrone 9 involves addition along the internal C4-C5 double bond and affords the bridged bicyclic isoxazolidine 10 (Scheme 2).

IM32CA reactions of allenic nitrones 7 and 8
The IM32CA reaction of the allenic nitrone 11 generated from 5,6-heptadien-2-one and N-methylhydroxylamine hydrochloride in ethanol afforded unsaturated bicyclic isoxazolidine 14 with complete site selectivity for addition along the C5-C6 terminal double bond [10] (Scheme 3). The cyclization of the homologue nitrone 12 under similar reaction conditions afforded the bicyclic adducts 15 by addition along the terminal C5-C6 double bond, and 16 by addition along the terminal C4-C5 double bond, while the latter undergoes acid-catalyzed ethanol addition to the exocyclic double bond to afford the ethers 17 and 18 (Scheme 3). The homologue nitrone 13 (n = 4) proceeded with exclusive site selectivity for addition along the internal C4-C5 double bond of the allenic function, leading to the bicyclic adduct 19 (Scheme 3). These experimental findings imply that the separation of the nitrone and the allenic functions plays the decisive role in the mode of cyclization. Although the generation of preferred adducts have been advocated qualitatively by considering the strain factor in some cases to eliminate the possibility of the competing site selectivity, yet the correlation of molecular reactivity with the electron density changes along the two feasible cumulative unsaturations are worth investigating to outline the plausible mechanism and accordingly analyze the observed selectivity of these IM32CA reactions.

IM32CA reactions of allenic nitrones 11, 12 and 13
The molecular electron density theory [12] (MEDT) proposed by Domingo in 2016 studies the role of electron density changes in the molecular reactivity and has emerged as an appealing alternative to the FMO theory for analysis of organic reactions. Several aspects of [3 + 2] cycloaddition (32CA) reactions [13, 14] have been successfully studied within the MEDT framework, namely the strain promotion [15, 16], reactivity [17, 18], catalysis [19, 20], substituent effects [21, 22], regio- [23, 24], stereo [25, 26] and chemoselectivity [27, 28]. Very recently, we have reported the MEDT studies for the IM32CA reactions of nitrones [29] at the MPWB1K/6-311G(d,p) level of theory, recommended as a precise computational model for the analysis of 32CA reactions.
This MEDT report is presented in five sections: (1) first, the topological analysis of the electron localization function [30, 31] (ELF) at the ground state (GS) of the reagents is performed to correlate the electronic structure and the molecular reactivity; (2) second, the electronic behaviour at the GS of the reagents is analyzed on the basis of the global reactivity indices defined with the conceptual density functional theory [32, 33] (CDFT); (3) then, the potential energy surface (PES) along the feasible reactions paths is studied to locate the stationary points and analyze the energy profile with the evaluation of global electron density transfer [34] (GEDT) at the TSs to assess the polar character; (4) the mechanistic implications are studied in terms of the bonding evolution theory [35] (BET) to analyze the changes in electron density along the preferred reaction paths; (5) finally, the electronic structure at the TSs is analyzed from the ELF study, while the interatomic interactions are characterized from the AIM [36, 37] (atoms-in-molecules) parameters and subsequent NCI-Plot [38] visualization.
Computational methods
Optimization of the reagents, TSs and the products was done using Berny analytical gradient optimization method [39] at the MPWB1K/6-311G(d,p) level of theory. The stationary points were characterized as minima by the absence of imaginary frequency, while the TSs by one imaginary frequency along each reaction path. Solvent effects in ethanol were considered by full optimization at the same computational level using polarized continuum model within the self-consistent reaction field (SCRF) framework [40,41,42]. The relative enthalpies, entropies and free energies were calculated at 298 K and 1 atm in ethanol. The intrinsic reaction coordinate [43] (IRC) calculations using the second-order Gonzales-Schlegel integration method [44, 45] were performed to verify the reaction path connecting the reactants and the products. The GEDT at the TSs was determined by GEDT (f) = \(\sum\limits_{q \in f} q\), where q is the NBO-derived charge [46, 47] at the considered reacting framework. The reactivity indices defined within the CDFT, namely the electronic chemical potential μ [48], global hardness η [49], global electrophilicity ω [50, 51] and nucleophilicity N [52], were calculated according to reference [32].
All calculations were performed using Gaussian 03 suite of programs [53]. The electron localization function [30, 31] (ELF) ((high-quality grid with a spacing of 0.06 Bohr) and AIM [36, 37] parameters were calculated using the Multiwfn software [54]. The ELF localization domains were visualized using the UCSF Chimera software [55] at an isovalue 0.86 and Visual Molecular Dynamics (VMD 1.9.3.) was used to visualize the NCI isosurfaces [56].
Results and discussion
ELF topological analysis at the ground state (GS) of the reactants
The ELF [30, 31] allows characterizing the electronic structures at the GS of the reagents and accordingly establishing the correlation with molecular reactivity. The three atom components (TACs) participating in 32CA reactions can be classified as pseudodiradical, pseudo(mono)radical, carbenoid and zwitter-ionic, respectively, within the MEDT framework [12,13,14]. The pseudodiradical TACs [57] are associated with the presence of two pseudoradical centres (monosynaptic basin integrating less than 1 e), and show highest reactivity, while the pseudo(mono)radical [58] and carbenoid TACs [59] show relatively lower reactivity and are associated respectively with the presence of a pseudoradical and a carbenoid (monosynaptic basin integrating approximately 2 e) centre. The zwitter-ionic TACs [23] show the least reactivity in 32CA reactions and do not show the presence of pseudoradical and carbenoid centres. The most significant ELF valence basin populations, attraction positions and the ELF localization domains at the GS of the allenic nitrones 11, 12 and 13 are represented in Fig. 1. The ELF of 11, 12 and 13 show the presence of monosynaptic V(O1) and V′(O1) basins integrating 5.96–5.97 e associated with the non-bonding electron density at O1 oxygen. The N2-C3 and N2-O1 bonding regions integrate at 3.92–3.94 e and 1.43–1.44 e respectively associated with the N2-C3 double bond and N2-O1 single bond. The allenic moiety shows the presence of disynaptic V(C4,C5) and V′(C4,C5) basins integrating 3.68–3.75 e associated with the underpopulated C4-C5 double bond and the disynaptic V(C5,C6) and V′(C5,C6) basins integrating 3.70–3.77 e associated with the underpopulated C5-C6 double bond. Thus, the absence of pseudoradical and carbenoid centre in the allenic nitrones 11, 12 and 13 allows their classification as the zwitter-ionic TACs associated with high energy barrier.

MPWB1K/6-311G(d,p) calculated total electron density (isovalue = 0.1) and ELF localization domains (isovalue = 0.81) of gas phase allenic nitrones 11, 12 and 13 along with the most significant ELF basin populations. Protonated basins are shown in blue, monosynaptic basins in red, disynaptic basins in green and the core basins in black colour. ELF attractor positions are shown in pink colour
After establishing the electronic structure of the TACs, the proposed Lewis-like structures on the basis of ELF study is represented in Fig. 2 along with the NBO-derived charges. O1 of the nitrone moiety is negatively charged by −0.60 e, while C3 is positively charged by + 0.25 e, with the negligible charge 0.08 e at N2. In the allenic part, the terminal C6 carbon is negatively charged by −0.47 e, while C4 shows the negative charge of −0.28 e and −0.29 e owing to the alkyl chain substitution.

Proposed Lewis-like structures together with the natural atomic charges in average number of electrons, e, of the allenic nitrones 11, 12 and 13. Negative, negligible and positive charges are shown in red, green and blue colours, respectively
Analysis of the CDFT indices
The reactivity indices defined with the CDFT [32, 33] have been employed in numerous studies [12,13,14,15,16,17,18,19,20,21,22] to analyze the electronic behaviour at the GS of the reagents. The electronic chemical potential µ [48], chemical hardness η [49], global electrophilicity ω [50, 51] and global nucleophilicity N [52] at the GS of the allenic nitrones 11, 12 and 13 are calculated at the B3LYP/6-31G(d) level of theory (Table 1) to characterize the reagents within the standard electrophilicity and nucleophilicity scales defined at the same computational level [51]. The electronic chemical potentials µ of the allenic nitrones are between −2.75 eV and −2.83 eV, showing minimal increase from 11 to 13 with the increase in the alkyl chain length separation between the nitrone and the allene moieties. The nitrones are classified as marginal electrophiles (ω < 0.80 eV) within the electrophilicity scale and strong nucleophiles (N > 3.00 eV) within the standard nucleophilicity scale.
Exploring the potential energy surface along the IM32CA reactions of the allenic nitrones 11–13
Herein, the two feasible reaction channels associated with the IM32CA reactions of allenic nitrones 11–13 involving the addition of the nitrone moiety to the terminal C5-C6 double bond and to the internal C4-C5 double bond have been studied (Scheme 4).

Studied reaction paths for the IM32CA reactions of allenic nitrones
Search for the stationary points along these reaction paths allowed locating the reagents, only one TS and the bicyclic adduct in each case, suggesting one-step mechanism. The TSs and adducts associated with the addition along the C5-C6 and C4-C5 double bonds are respectively prefixed as TS1 and TS2, followed by 2, 3 and 4 respectively indicating the number of CH2 groups separating the nitrone and the allenic function. Thus, TS1-2, TS1-3 and TS1-4 are the TSs associated with the addition of the nitrone moiety of the reagents 11, 12 and 13 along the C5-C6 double bond respectively leading to the bicyclic adducts 14, 15 and 20, while TS2-2, TS2-3 and TS2-4 are associated with the addition of nitrone moiety of the reagents 11, 12 and 13 along the C4-C5 double bond respectively leading to the bicyclic adducts 21, 16 and 19. The relative energies, enthalpies, entropies and free energies in ethanol of the TSs and adducts are given in Table 2. The total thermodynamic data are gathered in the Supplementary Information. The studied IM32CA reactions are exergonic, with reaction free energies between −17.3 and −35.1 kcal mol−1, suggesting kinetic control and show activation free energies between 22.2 and 34.9 kcal mol−1, consistent with the zw- type character. Some appealing conclusions can be derived from the relative free energies. (1) For the IM32CA reaction of nitrone 11, the activation free energy of TS1-2 associated with addition along the C5-C6 bond is lowered by 5.9 kcal mol−1 relative to that of TS2-2 associated with the addition along the C4-C5 bond. This is in complete agreement with the experimental results [10] showing exclusive formation of the unsaturated bicyclic isoxazolidine 14. (2) The activation free energy of TS1-2 is lowered than that of TS1-3 and TS1-4 by 4.3 and 12.7 kcal mol−1 suggesting that the addition of the nitrone moiety to the terminal C5-C6 double bond of the allenic function is relatively more feasible when the nitrone and the allene groups are separated by two methylene units. (3) For the IM32CA reaction of nitrone 13, the activation free energy of TS2-4 associated with addition along the C4-C5 bond is lowered by 7.7 kcal mol−1 relative to that of TS1-4 associated with the addition along the C5-C6 bond. This is in complete agreement with the experimental results showing exclusive formation of the bicyclic isoxazolidine 19 [10]. (3) These unimolecular IM32CA reactions show negative relative entropies of the TSs between −9.2 and −20.4 kcal mol−1 and that of the adducts are between −12.7 and −25.6 kcal mol−1. Note that the relative entropies of 19 and 20 differ by 0.1 kcal mol−1, that of 15 and 16 differ by 2.2 kcal mol−1 and that of 14 and 21 differ by 2.8 kcal mol−1 suggesting the influence of ring size on the entropy differences of the two possible adducts. Inclusion of thermodynamic correction decreases the activation enthalpies by 0.6–1.3 kcal mol−1 relative to the activation energies, while the reaction enthalpies are increased by 0.4–1.9 kcal mol−1 relative to the reaction energies.
The MPWB1K/6-311G(d,p) optimized geometries of the TSs in ethanol are shown in Fig. 3, with the bond distances between the four interacting atomic centres, GEDT and the imaginary frequencies in gas phase and ethanol are given in Table 3. The distances between the C–C interacting centres are greater than 2.0 Å, and that between the C-O interacting centres are greater than 1.9 Å suggesting that the formation of new C–C and C-O covalent bond formation has not been started at the TSs, considering the formation of C–C bonds at 1.9–2.0 Å and the C-O bonds at 1.7–1.8 Å [13]. This is in complete agreement with the ELF and AIM studies (“ELF and AIM topological analyses of the electron density at the TSs associated with the IM32CA reactions” section). Note that TS2-2, TS2-3 and TS2-4 associated with the addition along the C4-C5 double bond are more asynchronous and relatively more advanced compared to TS1-2, TS1-3 and TS1-4 associated with the addition along the C5-C6 double bond.

MPWB1K/6-311G(d,p) optimized geometries of the TSs in ethanol
The GEDT [34] allows evaluating the flux of electron density at the TSs and hence the polar character. GEDT values above 0.2 e are associated with polar reactions, while those below 0.1 e are the non-polar ones. Accordingly, the calculated GEDT values at the TSs are given in Table 4. In both gas phase and ethanol, the GEDT values are between 0.01 and 0.06 e, indicating non-polar character allowing the classification of these zw- type 32CA reactions as the null electron density transfer (NEDF) [60] type. The predicted non-polar character is consistent with the calculated high activation parameters associated mainly with the rupture of the C–C double bond revealed from the ELF topological analysis along the reaction paths (“Revealing the molecular mechanism and flux of electron density along the reaction paths associated with the IM32CA reactions of the allenic nitrones 11 and 13” section).
Revealing the molecular mechanism and flux of electron density along the reaction paths associated with the IM32CA reactions of the allenic nitrones 11 and 13
The bonding evolution theory (BET) [35] is a quantum chemical methodology to establish the molecular mechanism of a chemical reaction by studying the nature of electronic rearrangement along a reaction path. The bonding changes are analyzed topologically and energetically within the MEDT perspective, allowing a complete understanding of the bonding changes and the origin of the energy profile. Herein, the molecular mechanism of the preferred reaction paths for the IM32CA reaction of the allenic nitrones 11 and 13, leading to the adducts 14 and 19, is studied. The complete BET studies are given in the Supplementary material. In this section, we explain the appealing conclusions arising from these BET studies in a chemical fashion.
(1) The molecular mechanism associated with the IM32CA reactions of the allenic nitrones 11 and 13 are represented in Schemes 5 and 6 respectively. The IM32CA reaction of 11 can be topologically characterized by eight differentiated phases while that of 13 by seven topological phases. (2) For the IM32CA reaction of 11, the starting structure of the phases is denoted as S0-I, S1-I, S2-I, S3-I, S4-I, S5-I, S6-I and S7-I, and for that of 13 is represented as S0-II, S1-II, S2-II, TS2-4, S3-II, S4-II and S5-II. (3) For the IM32CA reaction of 11, S1-I is associated with the creation of non-bonding electron density at N2 nitrogen with energy cost (EC) of 15.4 kcal·mol−1, S2-I is associated with the rupture of C5-C6 double bond with energy cost (EC) of 16.2 kcal·mol−1, S3-I is associated with the formation of pseudoradical centre at C3 with energy cost (EC) of 16.6 kcal·mol−1 and TS1-2 with the EC of 16.9 kcal·mol−1 belongs to Phase-III. Therefore, the activation energy associated with this IM32CA reaction is related to the formation of non-bonding electron density at N2 nitrogen, rupture of C5-C6 double bond and the creation of pseudoradical centre at C3 carbon. The subsequent phases are related to the creation of pseudoradical centre at C5 (S4-I), formation of C3-C5 single bond (S5-I), creation of pseudoradical centre at C6 (S6-I) and finally the formation of O1-C6 single bond (S7-I). The ELF localization domains of these structures are given in Fig. 4. The formation of second O1-C6 bond begins when the first C3-C5 bond formation has been completed by 94%, suggesting two-stage one-step mechanism.

Simplified representation of the molecular mechanism of the reaction path associated with the IM32CA reaction of allenic nitrone 11 leading to adduct 14 arising from the topological analysis of the ELF

Simplified representation of the molecular mechanism of the reaction path associated with the IM32CA reaction of allenic nitrone 13 leading to adduct 19 arising from the topological analysis of the ELF

MPWB1K/6-311G(d,p) ELF localization domains of S4-I, S5-I, S6-I and S7-I associated with the formation of C3-C5 and O1-C6 bonds in the IM32CA reaction of allenic nitrone 11 (protonated basins are shown in blue, monosynaptic basins in red, disynaptic basins in green and the core basins in black colour (isovalue = 0.85). The ELF attractors are shown in pink colour
(4) For the IM32CA reaction of 13, S1-II is associated with the creation of non-bonding electron density at N2 nitrogen and rupture of the C4-C5 double bond with energy cost (EC) of 17.2 kcal·mol−1, TS2-4 is associated with the formation of pseudoradical centre at C4 with energy cost (EC) of 17.8 kcal·mol−1. Therefore, the activation energy associated with this IM32CA reaction is related to the formation of non-bonding electron density at N2 nitrogen, rupture of C4-C5 double bond and the creation of pseudoradical centre at C4 carbon. The subsequent phases are related to the creation of pseudoradical centre at C3 (S2-II), creation of pseudoradical centre at C5 (S3-II), formation of C3-C4 single bond (S4-II) and finally the formation of O1-C5 single bond (S5-II). The ELF localization domains of these structures are given in Fig. 5. The formation of second O1-C5 bond begins when the first C3-C4 bond formation has been completed by 77%, suggesting asynchronous one-step mechanism for this IM32CA reaction. (5) The minimal GEDT along the reaction path to reach the TSs establishes non-polar character of these IM32CA reactions consistent with the high activation energies.

MPWB1K/6-311G(d,p) ELF localization domains of S2-II, S3-II, S4-II and S5-II associated with the formation of C3-C4 and O1-C5 bonds in the IM32CA reaction of allenic nitrone 13 (protonated basins are shown in blue, monosynaptic basins in red, disynaptic basins in green and the core basins in black colour (isovalue = 0.85). The ELF attractors are shown in pink colour
ELF and AIM topological analyses of the electron density at the TSs associated with the IM32CA reactions
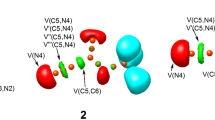
Finally, the ELF topological analysis at the TSs involved in the 32CA reactions was performed. The relevant valence basin populations are given in Table 4 and the ELF localization domains are shown in Fig. 6. TS1-2, TS1-3 and TS1-4 associated with the addition along the C5-C6 bond of the allenic nitrones 11, 12 and 13 present similar ELF topology, and that of TS2-2, TS2-3 and TS2-4 associated with the addition along the C4-C5 bond show similitude in the electronic structure. All six TSs show the presence of V(N2) monosynaptic basin integrating 1.19–1.32 e associated with the accumulation of non-bonding electron density at N2 nitrogen, which is not found in the allenic nitrones 11–13. The ELF of TS1-2, TS1-3 and TS1-4 shows the formation of V(C3) monosynaptic basin integrating 0.36–0.39 e associated with the presence of pseudoradical centre at C3, which is not found in TS2-2, TS2-3 and TS2-4. Note that to create the pseudoradical centre at C3 carbon and non-bonding electron density at N2 nitrogen, the N2-C3 bonding region at TS1-2, TS1-3 and TS1-4 experiences depopulation between 1.27 e and 1.41 e relative to the separated reagents, while this depopulation is between 0.98 e and 1.01 e at TS2-2, TS2-3 and TS2-4, since the non-bonding electron density at N2 nitrogen is only formed in these three TSs. The N2-O1 bonding region is also depopulated between 0.12 e and 0.15 e at TS1-2, TS1-3 and TS1-4 and between 0.15 e and 0.20 e at TS2-2, TS2-3 and TS2-4 to contribute for the accumulation of non-bonding electron density at the N2 nitrogen. Interestingly, TS1-4 shows the presence of V(C5) monosynaptic basin integrating 0.59 e deriving electron density from the C4-C5 bonding region, while no such change in electronic structure is observed in TS1-2 and TS1-3. On the other hand, TS2-2, TS2-3 and TS2-4 show the presence of V(C4) monosynaptic basin integrating 0.45 e, 0.37 e and 0.38 e respectively associated with the formation of pseudoradical centre at C4 carbon, the electron density of which comes from the depopulation of the C4-C5 bonding region along the reaction path. Note that the addition along the C5-C6 and C4-C5 bonds of the allenic nitrones show varied pattern of changes in electron density and accordingly follow different molecular mechanism. Finally, the absence of disynaptic basins associated with the formation of new covalent bonds reveal early nature of these TSs in each case in which the new covalent bonds have not been started.

MPWB1K/6-311G(d,p) ELF localization domains of the TSs associated with the IM32CA reactions of nitrones 11, 12 and 13 (protonated basins are shown in blue, monosynaptic basins in red, disynaptic basins in green and the core basins in black colour (isovalue = 0.85)
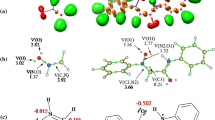
The topological analysis of the AIM was performed to characterize the interatomic interactions at the TSs. The AIM parameters, namely the total electron density ρ and the Laplacian of electron density \(\nabla^{2} \rho (r_{c} )\), at the bond critical points BCP1 and BCP2 associated respectively with the forming C–C and C-O bonds are given in Table 5. The total electron density ρ less than 0.1 au and the positive Laplacian of electron density indicate non-covalent interactions in each case, consistent with the ELF topological analysis and the forming bond distances greater than 2.0 Å. The NCI isosurfaces at TS1-2, TS1-3 and TS1-4 represented in Fig. 7 show green isosurfaces at the interatomic regions, indicative of weak non-covalent interactions.

NCI isosurfaces (isovalue = 0.5), in the range –0.04 < sign(λ2)ρ < 0.04 a.u., of TS1-2, TS1-3 and TS1-4
Conclusion
The zw- type intramolecular 32CA reactions of allenic nitrones 11, 12 and 13 experimentally reported by Lebel and Banucci [10] have been studied within MEDT at the MPWB1K/6-311G(d,p) computational level. The topological analysis of the ELF of the nitrones 11, 12 and 13 allows establishing their zwitter-ionic structure. Analysis of the global reactivity indices defined within CDFT classifies the nitrones as strong nucleophiles and marginal electrophiles. These IM32CA reactions follow non-concerted one-step mechanism with asynchronous TSs. In the allenic nitrone 11, the nitrone and allenic frameworks are separated by two methylene groups and the addition is energetically preferred along the C5-C6 double bond, while for the allenic nitrone 13, in which the nitrone and allenic frameworks are separated by four methylene groups, the addition takes place exclusively along the C4-C5 double bond of the allene. This study allows understanding the cyclization selectivity in intramolecular 32CA reactions of allenic nitrones. The molecular mechanism revealed from the bonding evolution theory predicts varied changes in the electron density along the reaction paths associated with the IM32CA reactions of the nitrones 11 and 13. However, in each case, the early TSs are located in which the formation of new C–C and C-O covalent bonds have not been started.
Data availability
All datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Pinho e Melo TM (2009) Allenes as dipolarophiles and 1,3-dipole precursors: synthesis of carbocyclic and heterocyclic compounds. Curr Org Chem 13:1406–1431. https://doi.org/10.2174/138527209789055090
Padwa A, Tomioka Y, Venkatramanan MK (1987) A study of the 5-exo methylene-isoxazolidine to 3-pyrrolidinone rearrangement. Tetrahedron Lett 28:755–758. https://doi.org/10.1016/S0040-4039(01)80981-3
Padwa A, Matzinger M, Tomioka Y, Venkatramanan MK (1988) Study of the thermal transformation of the 5-exo methyleneisoxazolidines to 3-pyrrolidinones. J Org Chem 53:955–963. https://doi.org/10.1021/jo00240a005
Padwa A, Kline DN, Koehler KF, Matzinger M, Venkatramanan MK (1987) Cycloaddition of nitrones with allenes. An example of steric control of regiochemistry. J Org Chem 52:3909–3917. https://doi.org/10.1021/jo00226a035
Padwa A, Carter SP, Chiacchio U, Kline DN (1986) Dipolar cycloaddition reaction of (phenylsulfonyl)propadiene with nitrones and alkylation studies of the cycloadducts. Tetrahedron Lett 27:2683–2686. https://doi.org/10.1016/S0040-4039(00)84616-X
Padwa A, Bullock WH, Kline DN, Perumattam J (1989) Heterocyclic synthesis via the reaction of nitrones and hydroxylamines with substituted allenes. J Org Chem 54:2862–2869. https://doi.org/10.1021/jo00273a018
Dolbier WR Jr, Wicks GE, Burkholder CR (1987) The cycloadditions of nitrones with fluoroallene. J Org Chem 52:2196–2201. https://doi.org/10.1021/jo00387a015
Alkayar ZTI, Coldham I (2019) Cascade cyclization and intramolecular nitrone dipolar cycloaddition and formal synthesis of 19-hydroxyibogamine. Org Biomol Chem 17:66–73. https://doi.org/10.1039/C8OB02839G
Lee W, Yuan M, Acha C, Onwu A, Gutierrez O (2019) Mechanism of nitrones and allenoates cascade reactions for the synthesis of dihydro[1,2-a] indoles. Org Biomol Chem 17:1767–1772. https://doi.org/10.1039/C8OB02346H
LeBel NA, Banucci E (1970) Intramolecular nitrone-allene cycloadditions. J Am Chem Soc 92:5278–5280. https://doi.org/10.1021/ja00720a080
Padwa A, Meske M, Ni Z (1993) Intramolecular [3+2]-cycloaddition of nitrones with allenes and alkynes. Tetrahedron Lett 34:5047–5050. https://doi.org/10.1016/S0040-4039(00)60672-X
Domingo LR (2016) Molecular electron density theory: a modern view of reactivity in organic chemistry. Molecules 21:1319. https://doi.org/10.3390/molecules21101319
Ríos-Gutiérrez M, Domingo LR (2019) Unravelling the mysteries of the [3+2] cycloaddition reactions. Eur J Org Chem 267–282 https://doi.org/10.1002/ejoc.201800916
Domingo LR, Acharjee N (2020) In: Ul-Haq Z, Wilson AK (ed) Molecular electron density theory: a new theoretical outlook on organic chemistry. Front Comput Chem 5:174–227. https://doi.org/10.2174/9789811457791120050007
Domingo LR, Acharjee N (2020) Unravelling the strain-promoted [3+2] cycloaddition reactions of phenyl azide with cycloalkynes from the molecular electron density theory perspective. New J Chem 44:13633–13643. https://doi.org/10.1039/D0NJ02711A
Domingo LR, Acharjee N (2020) Unveiling the high reactivity of strained dibenzocyclooctyne in [3+2] cycloaddition reactions with diazoalkanes through the molecular electron density theory. J Phys Org Chem 33:e4100. https://doi.org/10.1002/poc.4100
Domingo LR, Ríos-Gutiérrez M, Silvi B, Pérez P (2018) The mysticism of pericyclic reactions: a contemporary rationalisation of organic reactivity based on electron density analysis. Eur J Org Chem 1107–1120 https://doi.org/10.1002/ejoc.201701350
Domingo LR, Acharjee N, Mohammad Salim HA (2020) Understanding the reactivity of trimethylsilyldiazoalkanes participatingin [3+2]cycloaddition reactions towards diethylfumarate with a molecular electron density theory perspective. Organics 1:3–18. https://doi.org/10.3390/org1010002
Domingo LR, Ríos-Gutiérrez M, Acharjee N (2022) A molecular electron density theory study of the Lewis acid catalyzed [3+2] cycloaddition reactions of nitrones with nucleophilic ethylenes. Eur J Org Chem e202101417 https://doi.org/10.1002/ejoc.202101417
Domingo LR, Ríos-Gutiérrez M, Pérez P (2018) A molecular electron density theory study of the role of the copper metalation of azomethine ylides in [3 + 2] cycloaddition reactions. J Org Chem 83:10959–10973. https://doi.org/10.1021/acs.joc.8b01605
Domingo LR, Acharjee N (2021) Unveiling the substituent effects in the stereochemistry of [3+2] cycloaddition reactions of aryl- and alkyldiazomethylphosphonates with norbornadiene within a MEDT perspective. ChemistrySelect 6:10722–10733. https://doi.org/10.1002/slct.202102942
Domingo LR, Acharjee N (2018) [3+2] Cycloaddition reaction of C-phenyl-N- methyl nitrone to acyclic-olefin-bearing-electron-donating substituent: a molecular electron density theory study. ChemistrySelect 3:8373–8380. https://doi.org/10.1002/slct.201801528
Domingo LR, Ríos-Gutiérrez M, Pérez P (2018) A molecular electron density theory study of the reactivity and selectivities in [3 + 2] cycloaddition reactions of C, N-dialkyl nitrones with ethylene derivatives. J Org Chem 83:2182–2197. https://doi.org/10.1021/acs.joc.7b03093
Domingo LR, Ríos-Gutiérrez M, Adjieufack AI, Ndassa IM, Nouhou CN, Mbadcam JK (2018) Molecular electron density theory study of fused regioselectivity in the intramolecular [3+2] cycloaddition reaction of nitrones. ChemistrySelect 3:5412–5420. https://doi.org/10.1002/slct.201800224
Acharjee N, Mohammad Salim HA, Chakraborty M, Rao MP, Ganesh M (2021) Unveiling the high regioselectivity and stereoselectivity within the synthesis of spirooxindolenitropyrrolidine: a molecular electron density theory perspective. J Phys Org Chem 34:e4189. https://doi.org/10.1002/poc.4189
Acharjee N (2020) Unravelling the regio- and stereoselective synthesis of bicyclic N, O- nucleoside analogues within the molecular electron density theory perspective. Struct Chem (Springer) 31:2147–2160
Domingo LR, Acharjee N (2021) Unveiling the chemo- and regioselectivity of the [3+2] cycloaddition reaction between 4-chlorobenzonitrile oxide and β-aminocinnamonitrile with a MEDT perspective. ChemistrySelect 6:4521–4532. https://doi.org/10.1002/slct.202100978
Domingo LR, Ríos-Gutiérrez M, Acharjee N (2019) A molecular electron density theory study of the chemoselectivity, regioselectivity, and diastereofacial selectivity in the synthesis of an anticancer spiroisoxazoline derived from α-santonin. Molecules 24:832. https://doi.org/10.3390/molecules24050832
Acharjee N, Mondal A, Chakraborty M (2022) Unveiling the intramolecular [3 + 2] cycloaddition reactions of C, N-disubstituted nitrones from the molecular electron density theory perspective. New J Chem 46:7721–7733. https://doi.org/10.1039/d2nj00888b
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92:5397. https://doi.org/10.1063/1.458517
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371:683–686. https://doi.org/10.1038/371683a0
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21:748. https://doi.org/10.3390/molecules21060748
Geerlings P, Proft FD, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103:1793–1874. https://doi.org/10.1021/cr990029p
Domingo LR (2014) A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4:32415–32428. https://doi.org/10.1039/C4RA04280H
Krokidis X, Noury S, Silvi B (1997) Characterization of elementary chemical processes by catastrophe theory. J Phys Chem A 101:7277–7282. https://doi.org/10.1021/jp9711508
Bader RFW (1994) Atoms in molecules: a quantum theory. Oxford University Press, Oxford, New York
Bader RFW, Essén H (1984) The characterization of atomic interactions. J Chem Phys 80:1943. https://doi.org/10.1063/1.446956
García JC, Johnson ER, Keinan S, Chaudret R, Piquemal JP, Beratan DN, Yang W (2011) NCIPLOT: a program for plotting noncovalent interaction regions. J Chem Theory Comput 7:625–632. https://doi.org/10.1021/ct100641a
Hehre WJ, Radom L, PvR S, Pople J (1986) In: AB INITIO molecular orbital theory. Wiley-Interscience, New York
Tomasi J, Persico M (1994) Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem Rev 94:2027–2094. https://doi.org/10.1021/cr00031a013
Cancès E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107:3032–3041. https://doi.org/10.1063/1.474659
Barone V, Cossi M, Tomasi J (1998) Monte-Carlo model for the hydrogenation of alkenes on metal catalyst. J Comput Chem 19:404–417. https://doi.org/10.1002/(SICI)1096-987X(199803)19:4%3c404::AID-JCC3%3e3.0.CO;2-W
Fukui K (1970) Formulation of the reaction coordinate. J Phys Chem 74:4161–4163. https://doi.org/10.1021/j100717a029
González C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527. https://doi.org/10.1021/j100377a021
González C, Schlegel HB (1991) Improved algorithms for reaction path following: Higher-order implicit algorithms. J Chem Phys 95:5853–5860. https://doi.org/10.1063/1.461606
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735–746. https://doi.org/10.1063/1.449486
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926. https://doi.org/10.1021/cr00088a005
Parr RG, Yang W (1989) In: Density-functional theory of atoms and molecules. Oxford University Press, New York
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516. https://doi.org/10.1021/ja00364a005
Parr RG, Szentpály LV, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922–1924. https://doi.org/10.1021/ja983494x
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 58:4417–4423. https://doi.org/10.1016/S0040-4020(02)00410-6
Domingo LR, Pérez P (2011) The nucleophilicity N index in organic chemistry. Org Biomol Chem 9:7168–7175. https://doi.org/10.1039/C1OB05856H
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery Jr JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision D.01. Gaussian, Inc., Wallingford CT
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592. https://doi.org/10.1002/jcc.22885
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Molec Graphics 14:33–38. https://doi.org/10.1016/0263-7855(96)00018-5
Domingo LR, Chamorro E, Perez P (2010) Understanding the high reactivity of the azomethine ylides in [3+2] cycloaddition reactions. Lett Org Chem 7:432–439. https://doi.org/10.2174/157017810791824900
Domingo LR, Ríos-Gutiérrez M (2017) A molecular electron density theory study of the reactivity of azomethine imine in [3+2] cycloaddition reactions. Molecules 22:750. https://doi.org/10.3390/molecules22050750
Ríos-Gutiérrez M, Domingo LR (2019) The carbenoid-type reactivity of simplest nitrile imine from a molecular electron density theory perspective. Tetrahedron 75:1961–1967. https://doi.org/10.1016/j.tet.2019.02.014
Domingo LR, Ríos-Gutiérrez M, Perez P (2020) A molecular electron density theory study of the participation of tetrazines in aza-Diels–Alder reactions. RSC Adv 10:15694–15405. https://doi.org/10.1039/D0RA01548B
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Data collection and analysis were performed by Barsali Banerjee under the supervision of Nivedita Acharjee. The draft of the manuscript was written and subsequently reviewed by Barsali Banerjee, Nivedita Acharjee and Debnath Palit. All authors read, reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Banerjee, B., Acharjee, N. & Palit, D. Revealing the cyclization selectivity in intramolecular [3 + 2] cycloaddition reactions of allenic nitrones from the molecular electron density theory perspective. Struct Chem 35, 209–221 (2024). https://doi.org/10.1007/s11224-023-02175-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-023-02175-3