
Abstract
New N-substituted oxime derivatives (5b, 5d-g, 5i-k, and 5m) and known compounds (5a, 5c, 5h, 5g, 5l, and 5n-q) were obtained by reacting phenyl and butyl oxime chloride analogs with aniline, piperazine derivatives, piperidine, and diethylamine. Structures of all new compounds were determined by 1H NMR, 13C NMR, HRMS, and FTIR methods. All compounds were evaluated in vitro for their antiradical activity and hydrogen peroxide scavenging activity. Compounds 5g, 5h, 5i, 5j, 5k, 5l, 5m, and 5p have shown good antiradical activities with EC50 values < 1 (conc. antiradical (µM)/conc. DPPH (µM)). Similar results were obtained in the peroxide scavenging activity tests. These results have shown that piperidine and butyl substitution greatly increases the antiradical activity of the oximes. Compounds 5l and 5m were found to be the best radical scavengers. In addition, molecular docking studies were performed for compounds (5a-q) against CP450 (CYP1A2), NADPH oxidase, and Xanthine oxidase to predict their antioxidant capabilities through ROS-producing enzyme inhibitions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reactive oxygen species (ROS) such as superoxides (O2.−), hydrogen peroxide (H2O2), hydroxyl radical (.OH), and singlet oxygen (1O2) are unstable and highly reactive molecules that derive from molecular oxygen. ROS are biologically involved in cell signaling and cell defense against bacteria and xenobiotics [1]. ROS are produced by mitochondria during ATP production, peroxisomes, and as a result of enzyme activites such as NADPH oxidase, CP450, and Xanthine oxidase [2].
Increased levels of ROS in cells disrupt the redox homeostasis and cause oxidative stress and damage proteins, lipids, enzymes and DNA structure in body. This can cause diseases such as cancer, liver disease, neurodegenerative disorders, diabetes, and atherosclerosis [3]. Antioxidant molecules can maintain redox homeostasis in cells by showing antiradical activity and scavenging ROS or inhibiting ROS-producing enzymes such as superfamily of CP450, NADPH oxidase, Xanthine oxidase, Lipoxygenase, Cyclooxygenase, and Monoamine oxidase, thus hindering ROS production [4].
Amidoximes, oximes, and oxime esters have gained considerable attention in recent years due to many diverse biological activities such as antibacterial [5], antifungal [6], antioxidant [7,8,9], and anticancer [10]. Also these compounds are known as effective AChE reactivators [11]. In addition, piperazine and piperidine containing heterocyclic molecules are important compounds due to their wide biological activities such as antimicrobial [12], antiinflammatory [12], antioxidant [13, 14], and anticancer [15].
In literature, there are some works about antiradical activities of oxime compounds. Kala and coworkers obtained isatine oxime derivatives and they found DPPH reduction between 62.5 and 77.5% [8]. Also Harini and coworkers synthesized methyl-piperidinone oxime esters and found antiradical activity results with IC50 values between 11 and 165 µM [9]. Moreover, Özen and Taş obtained amido-carbonyl oxime derivatives and these compounds show antiradical activities with IC50 values between 39 and 104 µg/mL [10].
Considering the antioxidant capabilities of oxime derivatives in literature, N-substituted oxime derivatives (5a-q) were obtained to investigate these compounds’ antioxidant capabilities and effects of different substituents on antioxidant powers of these compounds. DPPH and Ruch methods were carried out to investigate in vitro antiradical activities. Also, in silico molecular docking studies were performed to investigate inhibition potentials and structure activity relationships against ROS-producing enzymes like CP450 (CYP1A2), NADPH oxidase, and Xanthine oxidase.
Results and discussion
Chemistry
The reactions of phenyl-substituted oxime chloride (3a) with amine derivatives (4a-f) are shown in Table 1. Reaction of 3a with aniline (4a) gave compound 5a in 43% yield (lit 30% [16]). Also, N-substituted oxime compound 5b (42%) was obtained by reaction of 3a with piperazine (4b). In addition, 5c (60%) (there is no information about yield in literature [17], 5d (69%), 5e (54%), and 5f (68%) were obtained by the reaction of 3a with piperazine derivatives 4c-f, respectively.
The reactions 3a-e with amine derivatives 4f-h are given in Table 2. Reaction of 3b with phenyl-piperazine (4f) gave 5g in 43% yield. N-substituted oxime derivative 5h (lit 80% [18]) was obtained in 67% yield from the reaction of 3a with 4g. Additionally, reactions of 2-hydroxyphenyl, 4-hydroxyphenyl, and 4-methoxyphenyl-substituted oxime chlorides (3c-e) with piperidine (4g) gave N-substituted oxime derivatives 5i (83%), 5j (70%), and 5k (67%) in high yields, respectively. Moreover, n-propyl-piperidine–substituted oxime derivative 5l (83%) (lit. 41% [19]) and n-propyl-diethylamine–substituted oxime derivative 5m (80%) were obtained from the reactions of 3b with 4g (83%) and 4h (80%), in high yields.
N-substituted oxime derivatives (5n-q) that are present in literature were also obtained to evaluate their antiradical and peroxide scavenging activities. Synthesis of 5n-q is presented in Scheme 1.

Synthesis of 5n-q
Antioxidant and peroxide scavenging activity results of N-substituted oxime derivatives (5a-q)
General reaction mechanism with an antiradical with DPPH is given in Scheme 2.

Reaction mechanism of DPPH with antiradical
According to this reaction mechanism, reaction of DPPH with the antiradical molecule (H-A) generates an electro-deficient radical A˙. The more stable the formed radical A˙, the higher the reactivity of the antiradical compound H-A against DPPH, thereby making H-A a potent antiradical compound. Inductive (electron donating) and mesomeric effects on these radicals increase its stability, and compounds in this work were designed by considering these effects. Antiradical activities of all synthesized compounds (5a-q) are shown in Table 3.
While compound 5p shows good antiradical activity and fast reaction rate with an EC50 value of 0.40, activity of 5a is radically low (EC50 = 10) with a slow reaction rate. However, electron-donating groups carrying N,N-diethyl (5n, EC50 = 3.71) and N-propyl (5q, EC50 = 3.71) oxime derivatives show much higher antiradical activity than 5a. Since piperazine carrying oxime derivative 5b has very low antiradical activity with an EC50 value of 31, morpholine-substituted 5o (EC50 = 9.51) shows higher activity compared to 5b. However interestingly, N-methyl piperazine carrying oxime derivative 5c has higher antiradical effect with an EC50 of 2.78. We think that this is due to electron-donating effect of methyl group on piperazine. Also, N-furoyl- (5d, EC50 = 7.81) and N-carbethoxy (5e, EC50 = 5.55)-substituted oxime derivatives having low antiradical powers show that electron-withdrawing groups on piperazine moiety lower the compounds’ antiradical capabilities. While phenyl and N-phenyl piperazine–substituted oxime derivative 5f shows high antiradical activity with an EC50 of 1.65, propyl and N-phenyl piperazine–substituted oxime derivative 5g shows higher antiradical activity (EC50 = 0.41). This result shows that inductive electron-donating effect caused by N-propyl group is more effective than mesomeric effect due to phenyl group on these oxime molecules. When comparing antiradical activity of morpholine- (5o, EC50 = 9.51), piperazine- (5b, EC50 = 31), and piperidine (5h, EC50 = 0.56)-substituted oxime compounds to see which of these cyclic groups are more effective on the antiradical efficiency, it can be seen that piperidine substitution is much more effective than morpholine and piperazine moieties.
In order to investigate the effect of phenyl substitiution on antiradical activity, phenyl-substituted oxime compounds such as o-OH (5i, EC50 = 0.96), p-OH (5j, EC50 = 0.38), and p-OMe (5k, EC50 = 0.49) were prepared. Among these compounds, best antiradical efficiency was observed on 5j with p-OH-phenyl-substituted oxime compound.
When these data are examined, it can be seen that antiradical activities of N-alkyl-substituted oximes ( 5g, 5h, 5n, 5q) and butyloxime (5g) are higher than the other compounds we obtained. For this reason, we designed piperidine-butyl–substituted oxime compound 5l and piperidine-N,N-diethyl–substituted oxime compound 5m to investigate antiradical activities of these compounds. As expected, 5l and 5m have the most antiradical power in this work with EC50 values of 0.26 and 0.28, respectively.
In the light of these informations, we think that N-alkyl substitution on oximes makes the formed oxygen radical more stable. This electron-deficient radical makes intramolecular interactions with the lone pair of electrons on N and therefore making radical more stable and more easy to form. Also electron-donating substituents on oximes further increase this stability (Scheme 3).

Intramolecular effects on the oxime radical to increase its stability
Also, no N-substitution carrying 2-butanoneoxime was evaluated for antiradical activity to compare it with N-substituted oxime derivatives and it shows low antiradical activity with an EC50 value of 35.10. This result further supports our theory that N-alkyl substitution increases the antiradical efficiency of oximes. Moreover, peroxide scavenging activity of N-substituted oxime derivatives (5a-q) and Trolox was investigated and results are presented in Table 4.
Compounds 5a, 5c, 5d, 5e, 5f, 5g, and 5k show higher peroxide scavenging activities than Trolox standard (Table 4). Peroxide scavenging activities of compounds 5b and 5l are lower than Trolox standard.
Molecular docking studies
In order to investigate binding energies and active site interactions of 5a-q with CP450 (CYP1A2), NADPH oxidase, and Xanthine oxidase, molecular docking studies were performed.
To validate the docking procedure, native ligands (α-naphtoflavone for CYP1A2, Adenosine-5'-Diphosphate for NADPH oxidase, and Hypoxhanthine for Xanthine oxidase) in crystallized enzyme structures were redocked into their original binding pockets. Generated docking conformations were superimposed into experimental conformations. Root mean square deviation (RMSD) values between docked conformation and experimental conformation were calculated. RMSD values must be less than 2.0 Å to consider the docking protocol valid [4]. Overlapping of the redocked conformations and native conformations is presented with RMSD values in Fig. 1.

Overlapping of the redocked conformations and experimental conformations
As can be seen in Fig. 1, all RMSD values obtained from redocking procedure are below 2.0 Å. This means the protocol used in this study is valid and can be carried out for the docking study of ligand compounds (5a-q). Nine docking poses were generated for each ligand in every docking calculation. Docking pose with highest docking score (binding free energy = ∆G = − Kcal/mol) was chosen to present. Binding energies of 5a-q and reference drugs (Fluvoxamine for CYP1A2, Dextrometorphan for NADPH oxidase, and Febuxostat for Xanthine oxidase) with CYP1A2, NADPH oxidase, and Xanthine oxidase are presented in Table 5.
By analyzing all docking scores, it can be seen that compounds 5a, 5d, and 5f show the highest three docking scores against all three receptors.
The docking scores against CP450 showed that 10 (5a, 5b, 5d, 5f-k, and 5o) out of 17 tested compounds have docking scores (between − 8.0 and − 10.1 kcal/mol) higher than reference drug Fluvoxamine (∆G = − 7.7 kcal/mol).
Docking scores against NADPH oxidase showed that 2 (5d = − 7.9 kcal/mol and 5f = − 7.9 kcal/mol) out of 17 compounds gave docking scores higer than reference drug Dextrometorphan (∆G = − 7.6 kcal/mol) but compound 5a showed lower binding score than Dextrometorphan (− 7.3 kcal/mol).
Docking score results of Xanthine oxidase showed that none of the 17 test compounds showed higher docking scores than reference drug Febuxostat (∆G = − 7.8 kcal/mol) but 5a, 5d, and 5f gave close docking scores between − 7.5 and − 7.7 kcal/mol.
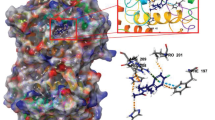
Interactions of three compounds with best docking scores against CP450 (5a, 5d, and 5f) are presented in Fig. 2.

Binding interactions of 5a, 5d, and 5f and Fluvoxamine with CP450
Target receptor CYP1A2 (PDB code: 2HI4) is a member of CP450 superfamily. According to literature and PDB records, important active site residues of CP450 are Thr118, Thr124, Phe125, Glu225, Phe226, Lys250, Arg251, Lys253, Phe260, Asn312 Ala317, Gly318, Thr319 Asp320, Thr321, Val322 Leu382, Thr385, Ile386, Leu497, and Thr498 [20, 21].
By investigating the interactions of Fluvoxamine with CYP1A2, it can be seen that phenyl group made π − σ interactions with ALA317 and LEU497 and π − π stackings with GLY316 and PHE226. Also, CF3 substitution on benzene ring interacts with LEU382. In addition, alkyl chain made π − alkyl interactions with PHE125 and PHE226.
By investigating the interactions of 5a, it can be seen that oxime moeity made hydrogen bonding with ALA317 and THR124. In addition, phenyl substitutions interact with PHE226, PHE260, GLY316, and LEU497 through π − π stackings.
By investigating the interactions of 5d, it can be seen that oxime moeity made hydrogen bonding with ASP313. Also, furan moeity made π − alkyl interactions with ALA317 and LEU382. In addition, piperazine ring made C-H bonding with THR124 and phenyl ring interacts with PHE226 and PHE260 through π − π stackings.
Lastly, phenyl rings of 5f made similar interactions with 5d. Two phenyl moeities made π − π stackings and π − alkyl interactions with GLY316, PHE226, PHE260, ALA317, and LEU382. Also oxime group made hydrogen bonding with ASP320.
Summarily, 5a, 5d, and 5f showed similar residue interactions with CYP1A2. It is clear that aromatic moeities of tested compounds interact with PHE226, PHE260, GLY316, ALA317, and LEU497. Reference drug Fluvoxamine showed similar interactions through its aromatic moeity. Also, oxime moeity on all test compounds made hydrogen bondings with THR124, ASP313, ALA317, and ASP320.
Interactions of three compounds with best docking scores against NADPH oxidase (5a, 5d, and 5f) are presented in Fig. 3.

Binding interactions of 5a, 5d, 5f, and Fluvoxamine with NADPH oxidase
According to literature and PDB site records, important active site residues of NADPH oxidase are GLY156, GLY158, TYR159, ILE160, ASP179, GLY180, HIS181, TYR188, LYS213, VAL214, ILE243, and GLY244 [4].
Six-membered cyclohexane ring of Dextrometorphan made π − alkyl interactions with TYR188, TYR159, and PRO298 and π − σ interaction with LEU299. Also, methoxy group on benzene interacts with TYR159 through π − alkyl interactions.
By looking at the interaction diagram of 5a, it can be seen that oxime and N–H moeity made hydrogen bondings with ASP179. In addition, two phenyl groups of 5a made π − alkyl interaction with LYS213 and π − π stacking and π − alky interaction with ILE243.
Oxime moeity of 5d made hydrogen bondings with GLY180 and HIS181. In addition, carbonyl group next to furan ring made another hydrogen bonding with LYS187. Benzene ring interacts with ILE243 and LYS213 through π − σ and π-alkyl interactions, respectively. Also, piperazine ring interacts with ASP179 through C-H bonding.
But investigating the interaction of 5f, it can be seen that oxime moeity made hydrogen bondings with HIS181. Also, two benzene rings interact with ILE243 and LYS213 through π − σ, π-π stacking, and π − alkyl interactions, respectively.
As a result, 5a, 5d, and 5f showed similar residue interactions with NADPH oxidase through their aromatic moeities. Aromatic groups of these compounds interact with LYS213 and ILE243. Also, like the interactions with CP450, oxime groups of 5a, 5d, and 5f made hydrogen bondings. The residues that were interacted with through hydrogen bondings are ASP179, GLY180, HIS181, and LYS187.
All these molecular docking predictions showed that compounds 5a, 5d, and 5f have potentials to be inhibitor compounds for CYP1A2 and NADPH oxidase.
Conclusion
In the presented work, N-substituted oxime derivatives (5a-q) were designed and evaluated in vitro for their antiradical capabilities and they have shown EC50 values between 0.26 and 31. Low EC50 values of piperidine-butyl– (5l, EC50 = 0.26) and diethylamine-butyl (5m, EC50 = 0.28)–substituted compounds indicate that amino and alkyl substitiuents on oximes highly increase antiradical activities.
Also, molecular docking studies of 5a-q against ROS-producing enzymes (CP450, NADPH oxidase, and Xanthine oxidase) have shown that compounds 5a, 5b, 5d, 5f-k, and 5o have higher docking scores against CP450 than reference drug Fluvoxamine. Also, compounds 5d and 5f have higher docking scores against NADPH oxidase than reference drug Dextrometorphan. These docking results are promising and molecules 5a, 5d, and 5f have potentials to be inhibitor molecules against CP450 and NADPH oxidase to hinder the ROS production of these enzymes. Obtaining new derivatives and evaluation of in vitro inhibition studies of these molecules against CP450 and NADPH oxidase is an ongoing research project of our group.
Experimental
Melting points were determined on a Gallenkamp capillary melting point apparatus. IR spectra (ATR) were obtained with a Bruker Tensor27 spectrophotometer in the 400–4000-cm−1 range with 2-cm−1 resolutions. 1H NMR and 13C NMR spectra were recorded on a Varian Mercury-400 high performance Digital FT-NMR and Varian Oxford NMR300 spectrometers. High Resolution Mass Time-of-Flight spectra (TOF) were measured on an Agilent 1200/6210 LC/MS spectrophotometer. UV absorbance was measured by Rigol Ultra-3000 UV–VIS Spectrophotometer. Thin layer chromatographies (TLC) were performed on Merck aluminum-packed silica gel plates. Purification of products was performed by column chromatography on silica gel (Merck silica gel 60, 40–60 µm) or preparative TLC on silica gel of Merck (PF254-366 nm). 2,2-diphenyl-1-picrylhydrazyl (DPPH) and all reagents and solvents were commercially purchased.
General synthesis procedure and spectroscopic data of N-substituted oxime derivatives (5a-q)
Firstly, oxime derivatives (2a-e) were obtained from the reaction of aldehydes with hydroxylamine.HCl by known methods [22]. All oxime chlorides (3a-e) were obtained from the reactions of aldoxime derivatives with N-chlorosuccinimide (NCS) [23] (Scheme 4).

General synthesis of oximes (2a-e) and oxime chlorides (3a-e)
N-substituted oxime derivatives (5a-q) were obtained with the procedure described below:
In a reaction vessel, corresponding N-substituted derivative (4a-k, 1.2 mmol) and Et3N (3.0 mmol) was homogenized in 5 mL of THF and stirred in ice bath. Solution of corresponding oxime derivative (3a-e, 1 mmol) in 2 mL of THF was added dropwise. After instillation, reaction vessel was removed from ice bath and allowed to stir overnight. Reaction was monitored with TLC. After completion, water was added and crude product was extracted with chloroform (3 × 20 mL). Combined organic phases were neutralized by water (3 × 20 mL), dried over anhydrous Na2SO4, and evaporated. Solid crude products were purified by crystallization from EtOAc, and oily substances were purified with column chromatography (Hexane/EtOAc = 1/1 as eluent). All obtained compounds were isolated as E/Z isomer mixtures but Z isomers are dominant. All spectral data provided belongs to Z isomers.
(Z)-N'-Hydroxy-N-phenylbenzimidamide (5a) [16]
Yield 43% (0.29 g) as yellow solid; m.p: 119–121 °C (lit m.p: 132–134 °C); 1H NMR (400 MHz, DMSO- d6), δ (ppm): 10.52 (1H, s, N–H), 8.26 (1H, s, O–H), 7.36–7.26 (5H, m, Ar–H), 7.02 (2H, t, J = 7.6 Hz, Ar–H), 6.74 (1H, t, J = 7.6 Hz, Ar–H), 6.62 (2H, d, J = 8.0 Hz, Ar–H).
(Z)-Phenyl(piperazin-1-yl)methanone oxime (5b)
Yield 42% (0.993 g) as yellow solid; m.p: 170–172 °C; IR (ATR) ʋmax 3299 (OH), 3200 (NH), 3057 (arom. C-H), 2863 (aliphatic C-H), 1632 (C = N), 1139 (C-N), 770, 698 (arom. C-H) cm−1; 1H NMR (400 MHz, DMSO- d6), δ (ppm): 9.29 (1H, s, O–H), 7.42–7.31 (5H, m, Ar–H), 3.27 (1H, s, N–H), 2.80 (4H, t, J = 4.8 Hz, -CH2-), 2.68 (4H, t, J = 4.8 Hz, -CH2-); 13C NMR (100 MHz, DMSO- d6), δ (ppm): 158.8 (C = N), 131.9 (arom. C), 129.3 (arom. C), 129.0 (arom. C), 128.5 (arom. C), 48.6 (-CH2), 45.5 (-CH2); HRMS (ESI) (m/z) Calcd for C11H15N3O 206.12878 found: 206.12971 (M + H)+.
(Z)-(4-Methylpiperazin-1-yl)(phenyl)methanone oxime (5c) [17]
Yield 55% (3 g) as colorless solid; m.p: 140–142 °C; IR (ATR) ʋmax 3145 (OH), 3057 (Ar. C-H), 2840 (Aliphatic C-H), 1624 (C = N), 1139 (C-N), 770, 693 (Ar. C-H) cm−1; 1H NMR (400 MHz, CDCl3), δ (ppm): 7.46–7.39 (5H, m, Ar–H), 3.06 (4H, t, J = 5.2 Hz, -CH2-), 2.40 (4H, t, J = 5.2 Hz, -CH2-), 2.28 (3H, s, -CH3); 13C NMR (100 MHz, CDCl3), δ (ppm): 160.3 (C = N), 130.8 (arom. C), 129.3 (arom. C), 128.9 (arom. C), 128.3 (arom. C), 54.67 (-CH2), 46.86 (-CH2), 46.15 (-CH3); HRMS (ESI) (m/z) Calcd for C12H17N3O 220.14443 found: 220.14419 (M + H)+.
(Z)-Furan-2-yl(4-((hydroxyimino)(phenyl)methyl)piperazin-1-yl)methanone (5d)
Yield 69% (5.78 g) as colorless solid; m.p: 125–127 °C; IR (ATR) ʋmax 3284 (OH), 3110 (arom. C-H), 2834 (Aliphatic C-H), 1660 (C = O) 1618 (C = N), 1147 (C-N), 765, 700 (arom. C-H) cm−1; 1H NMR (400 MHz, CDCl3), δ (ppm): 7.48–7.34 (5H, m, Ar–H), 7.38 (1H, d, J = 2.0 Hz, Ar–H), 7.01 (1H, d, J = 3.6 Hz, Ar–H), 6.47 (1H, dd, J = 3.2, 2.0 Hz, Ar–H), 3.82 (4H, s, -CH2-), 3.42 (4H, t, J = 5.2 Hz, -CH2-), 3.11 (1H, t, J = 5.2 Hz, OH); 13C NMR (100 MHz, CDCl3), δ (ppm): 159.3 (C = N), 153.9 (C = O), 147.8 (arom. C), 143.7 (arom. C), 129.8 (arom. C), 129.6 (arom. C), 128.8 (arom. C), 128.5 (arom. C), 116.6 (arom. C), 111.3 (arom. C), 49.0 (-CH2), 47.3 (-CH2); HRMS (ESI) (m/z) Calcd for C16H17N3O3 300.13426 found: 300.13566 (M + H)+.
Ethyl (Z)-4-((hydroxyimino)(phenyl)methyl)piperazine-1-carboxylate (5e)
Yield 54% (4.74 g) as colorless crystal; m.p: 131–133 °C; IR (ATR) ʋmax 3284 (OH), 3067 (arom. C-H), 2834 (Aliphatic C-H), 1665 (C = O), 1620 (C = N), 1136 (C-N), 1016 (C-O), 768, 694 (arom. C-H) cm−1; 1H NMR (400 MHz, CDCl3), δ (ppm): 7.46–7.43 (5H, m, Ar–H), 4.13 (2H, q, J = 6.8 Hz, O-CH2CH3), 3.46 (4H, t, J = 5.2 Hz, -CH2-), 3.00 (4H, t, J = 5.2 Hz, -CH2-), 1.24 (3H, t, J = 7.2 Hz, O-CH2CH3); 13C NMR (100 MHz, CDCl3), δ (ppm): 160.1 (C = N), 155.4 (C = O), 130.3 (arom. C), 129.6 (arom. C), 128.9 (arom. C), 128.4 (arom. C), 61.5 (-CH2), 46.9 (-CH2), 43.2 (-CH2), 14.6 (-CH3); HRMS (ESI) (m/z) Calcd for C14H19N3O3 278.14991 found: 278.14952 (M + H)+.
(Z)-Phenyl(4-phenylpiperazin-1-yl)methanone oxime (5f)
Yield 68% (5.76 g) as colorless solid; m.p: 114–116 °C; IR (ATR) ʋmax 3296 (OH), 3051 (arom. C-H), 2827 (Aliphatic C-H), 1629 (C = N), 1230 (C-N), 754, 688 (arom. C-H) cm−1; 1H NMR (400 MHz, CDCl3), δ (ppm): 7.52 (1H, dd, J = 8.0, 1.6 Hz, Ar–H), 7.46 (1H, dd, J = 2.4, 1.2 Hz, Ar–H), 7.39 (2H, d, J = 8.0 Hz, Ar–H), 7.29 (2H, d, J = 8.0 Hz, Ar–H), 6.95–6.86 (4H, m, Ar–H), 3.56 (2H, t, J = 4.8 Hz, -CH2-), 3.27–3.14 (6H, m, -CH2-); 13C NMR (100 MHz, CDCl3), δ (ppm): 154.3 (C = N), 151.5 (C = O), 133.9 (arom. C), 129.6 (arom. C), 129.1 (arom. C), 128.9 (arom. C), 128.4 (arom. C), 120.1 (arom. C), 116.4 (arom. C), 50.2 (-CH2), 48.8 (-CH2); HRMS (ESI) (m/z) Calcd for C17H19N3O 282.16008 found: 282.15959 (M + H)+.
(Z)-1-(4-Phenylpiperazin-1-yl)butan-1-one oxime (5g)
Yield 43% (0.436 g) as colorless crystal; m.p: 124–126 °C; IR (ATR) ʋmax 3279 (OH), 3061 (arom. C-H), 2827 (Aliphatic C-H), 1629 (C = N), 1234 (C-N), 755, 684 (arom. C-H) cm−1; 1H NMR (400 MHz, CDCl3), δ (ppm): 8.10 (1H, s, OH), 7.28 (2H, td, J = 7.2, 1.2 Hz, Ar–H), 6.94 (2H, d, J = 7.6 Hz, Ar–H), 6.88 (1H, t, J = 7.6 Hz), 3.27 (4H, dt, J = 5.6, 2.4 Hz, -CH2-), 3.20 (4H, dt, J = 5.6, 2.4 Hz, -CH2-), 2.49 (2H, dt, J = 8.0, 4.0 Hz, -CH2-), 1.65–1.55 (2H, m, -CH2-), 1.01 (3H, t, J = 7.6 Hz, -CH3); 13C NMR (100 MHz, CDCl3), δ (ppm): 161.6 (C = N), 151.2 (arom. C), 129.1 (arom. C), 120.1 (arom. C), 116.4 (arom. C), 49.1 (-CH2), 46.0 (-CH2), 26.4 (-CH2), 19.6 (-CH2), 14.2 (-CH3); HRMS (ESI) (m/z) Calcd for C14H21N3O 248.17573 found: 248.17662 (M + H)+.
(Z)-Phenyl(piperidin-1-yl)methanone oxime (5h) [18]
Yield 67% (6.68 g) as colorless crystal; m.p: 140–142 °C (lit m.p: 145 °C); 1H NMR (400 MHz, CDCl3), (Z/E): (98.5: 1.5); Z isomer δ (ppm): 7.47–7.39 (5H, m, Ar–H), 2.98 (4H, t, J = 5.6 Hz, -CH2-), 1.53 (6H, s, -CH2-).
(Z)-(2-Hydroxyphenyl)(piperidin-1-yl)methanone oxime (5i)
Yield 83% (0.182 g) as colorless solid; m.p: 156–158 °C; IR (ATR) ʋmax 3223 (OH), 3080 (arom. C-H), 2928 (Aliphatic C-H), 1638 (C = N), 1224 (C-N), 722 (Ar. C-H) cm−1; 1H NMR (400 MHz, DMSO- d6), δ (ppm): 9.32 (1H, s, -OH), 7.44 (2H, dt, J = 6.8, 2.0 Hz, Ar–H), 7.33 (2H, dt, J = 6.8, 2.0 Hz, Ar–H), 2.85 (4H, t, J = 5.2 Hz, -CH2-), 1.47 (6H, s, -CH2-); 13C NMR (100 MHz, DMSO- d6), δ (ppm): 157.8 (C = N), 133.6 (arom. C), 131.2 (arom. C), 131.1 (arom. C), 128.5 (arom. C), 48.4 (-CH2), 25.3 (-CH2), 24.6 (-CH2); HRMS (ESI) (m/z) Calcd for C12H16N2O2 239.14011 found: 239.09510 (M + H)+ + H2O.
(Z)-(4-Hydroxyphenyl)(piperidin-1-yl)methanone oxime (5j)
Yield 78% (5.0 g) as brown oil; IR (ATR) ʋmax 3205 (OH), 2932 (arom. C-H), 2853 (Aliphatic C-H), 1620 (C = N), 1223 (C-N), 749 (arom. C-H) cm−1; 1H NMR (400 MHz, DMSO- d6), δ (ppm): 9.59 (1H, s, -OH), 9.06 (1H, s, -OH), 7.16 (2H, dt, J = 6.8, 1.6 Hz, Ar–H), 6.74 (2H, dt, J = 6.8, 1.6 Hz, Ar–H), 2.84 (4H, t, J = 5.2 Hz, -CH2-), 1.46 (6H, s, -CH2-); 13C NMR (100 MHz, DMSO- d6), δ (ppm): 158.8 (C = N), 158.1 (arom. C), 130.8 (arom. C), 122.5 (arom. C), 115.1 (arom. C), 48.6 (-CH2), 25.4 (-CH2), 24.7 (-CH2); HRMS (ESI) (m/z) Calcd for C12H16N2O2 239.14011 found: 239.09617 (M + H)+ + H2O.
(Z)-(4-Methoxyphenyl)(piperidin-1-yl)methanone oxime (5k)
Yield 65% (1.8 g) as colorless solid; m.p: 174–176 °C; IR (ATR) ʋmax 3186 (OH), 2967 (arom. C-H), 2852 (Aliphatic C-H), 1624 (C = N), 1247 (C-N), 1121 (C-O), 732 (arom. C-H) cm−1; 1H NMR (400 MHz, DMSO- d6), (Z/E): (62:38); Z isomer δ (ppm): 9.33 (1H, s, -OH), 7.29 (2H, d, J = 2.4 Hz, Ar–H), 6.92 (2H, dd, J = 6.8, 2.4 Hz, Ar–H), 3.85 (3H, s, -OCH3), 1.47 (10H, s, -CH2-); 13C NMR (100 MHz, CDCl3), δ (ppm): 160.1 (C = N), 159.1 (arom. C), 130.5 (arom. C), 113.6 (arom. C), 111.5 (arom. C), 48.3 (-CH3), 48.2 (-CH2-), 25.43 (-CH2), 24.50 (-CH2); HRMS (ESI) (m/z) Calcd for C13H18N2O2 235.14410 found: 235.14319 (M + H)+.
(Z)-1-(Piperidin-1-yl)butan-1-one oxime (5l) [19]
Yield 82.5% (0.30 g) as yellow oil; IR (ATR) ʋmax 3273 (OH), 2871 (Aliphatic C-H), 1638 (C = N), 1255 (C-N) cm−1; 1H NMR (400 MHz, CDCl3), δ (ppm): 3.07 (4H, s, -CH2-), 2.43 (2H, t, J = 8.0 Hz, -CH2-), 1.54 (8H, s, -CH2-), 0.96 (3H, t, 7.2 Hz); 13C NMR (100 MHz, CDCl3), δ (ppm): 162.5 (C = N), 46.9 (-CH2), 46.9 (-CH2), 26.5 (-CH2), 25.5 (-CH2), 19.75 (-CH2), 14.2 (-CH3); HRMS (ESI) (m/z) Calcd for C9H18N2O 171.14919 found: 171.14860 (M + H)+.
(Z)-N,N-Diethyl-N'-hydroxybutyrimidamide (5m)
Yield 80% (2 g) as yellow oil; IR (ATR) ʋmax 3273 (OH), 2871 (Aliphatic C-H), 1638 (C = N), 1255 (C-N) cm−1; 1H NMR (400 MHz, CDCl3), δ (ppm): 3.14 (4H, q, J = 6.8 Hz, -CH2-), 2.42 (2H, t, J = 8.4 Hz, -CH2-), 1.61–1.51 (2H, m, -CH2-), 1.07 (6H, t, J = 6.8 Hz, -CH3), 0.97 (3H, t, J = 7.6 Hz, -CH3); 13C NMR (100 MHz, CDCl3), δ (ppm): 165.1 (C = N), 42.1 (-CH2), 35.3 (-CH2), 20.5 (-CH2), 14.1 (-CH3), 13.3 (-CH3); HRMS (ESI) (m/z) Calcd for C8H18N2O 159.14919 found: 159.14986 (M + H)+.
(Z)-N,N-Diethyl-N'-hydroxybenzimidamide (5n) [24]
Yield 74% (0.29 g) as yellow crystal; m.p: 68–70 °C (lit m.p: 79 °C 50); 1H NMR (400 MHz, CDCl3), δ (ppm): 7.45–7.35 (5H, m, Ar–H), 3.06 (4H, q, J = 6.8 Hz, -CH2-), 1.03 (6H, t, J = 6.8 Hz, -CH3).
(Z)-Morpholino(phenyl)methanone oxime (5o) [25]
Yield 50% (6.0 g) as colorless crystal; m.p: 118–120 °C (lit m.p: 121–122°C51); 1H NMR (400 MHz, CDCl3), δ (ppm): 7.47–7.41 (5H, m, Ar–H), 3.68 (4H, t, J = 5.2 Hz, -CH2-), 3. 00 (4H, t, J = 4.8 Hz, -CH2-).
(Z)-N'-Hydroxybenzimidamide (5p) [16]
Yield 45% (0.195 g) as yellow oil; 1H NMR (400 MHz, DMSO- d6), δ (ppm): 9.61 (1H, s, -OH), 7.66–7.63 (2H, m, Ar–H), 7.36–7.34 (3H, m, Ar–H), 5.81 (2H, s, NH2).
(Z)-N'-Hydroxy-N-propylbenzimidamide (5q) [26]
Yield 65% (0.75 g) as orange oil; 1H NMR (400 MHz, CDCl3), δ (ppm): 7.47–7.35 (5H, m, Ar–H), 5.33 (1H, d, J = 2.8 Hz, NH), 2.96 (2H, t, J = 6.0 Hz, -CH2-), 1.43 (2H, q, J = 7.2 Hz, -CH2-), 0.8 (3H, t, J = 7.2 Hz, -CH3).
DPPH radical scavenging method
The DPPH radical scavenging method was carried out according to slightly modified Brand-Williams method [27]. Different concentrations of antiradical compounds and Trolox as standart and 6 × 10−5 M DPPH solution were prepared in methanol.
For the method, 0.1 mL of antiradical compound at desired concentration was added to 3.9 mL of 6.5 × 10−5 M DPPH. The decrease in absorbance was monitored by UV–VIS spectrophotometer at 515 nm at 0 min, 1 min, and every 15 min until reaction reached a plateu. Exact concentration of DPPH ([CDPPH]) in reaction medium was calculated from the calibration curve with the equation:
Remaining percentage of DPPH in reaction medium was calculated as follows:
where [CDPPH]T=0 is the starting concentration of DPPH. Percentage of remaining DPPH as a function of time for each antiradical concentration was plotted graphically to calculate the time needed for the reaction to reach the steady state. Then, the percentage of remaining DPPH at the steady state was plotted as a function of antiradical concentration to calculate EC50 values. The antiradical activity (EC50) was expressed as conc. of antiradical sample/conc. of DPPH needed to reduce DPPH concentration by 50%. EC50 is inversely related to antiradical power, thus low EC50 means better antiradical efficiency. Also, the time needed to reach the steady state was calculated and reaction time was presented for each antiradical compound as fast (< 30 min), medium (30–60 min), and slow (> 60 min).
H2O2 scavenging method
Peroxide scavenging activity of N-substituted oxime derivatives was carried out according to modified literature method [28]. A total of 40 mM H2O2 solution was prepared in phosphate buffer (100 mM, pH: 7.4). Test compounds and Trolox standart were prepared in 10 and 20 µM concentrations in DMSO.
Firstly, absorbance of 40 mM H2O2 solution without test compound was monitored at 230 nm with UV–VIS spectrophotometer as control absorbance. A total of 3.4 mL of test compound at desired concentration was added to 0.6 mL of H2O2 solution. Absorbance of reaction was monitored after 10 min at 230 nm. Percentage of scavenged H2O2 was calculated with the equation:
where At is the absorbance of H2O2 solution with test compound and A0 is the absorbance of control. Peroxide scavenging ability was expressed as percent scavenged H2O2 for 10 and 20 µM concentrations of test compounds.
Molecular docking method
Three dimensional structures of CYP1A2, NADPH oxidase, and Xanthine oxidase were obtained from Protein data bank. All co-crystallized ligands, metal atoms, and water molecules were removed; polar hydrogens and Kollman charges were added using Autodock Tools 1.5.7. Active site coordinates were determined with Discovery Studio Visualizer 2020. All docking calculations were performed with AutoDock Vina 1.1.2 [29]. Docking results were analyzed with Discovery Studio Visualizer 2020. PDB codes of receptors, gridcenter coordinates, and grid size used for docking protocol are presented in Table 6.
In order to prepare ligand molecules (5a-q), conformational analysis and geometry optimizations were performed with Avogadro 1.1 software [30]. Optimized ligands were converted to pdbqt format with Autodock Tools 1.5.7.
Data availability
All experimental data were included in the manuscript. All spectral data can be found in Supplementary Material.
Code availability
Not applicable.
References
Ray PD, Huang BW, Tsuji Y (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24:981–990
Snezhkina AV, Kudryavtseva AV, Kardymon OL, Savvateeva MV, Melnikova NV et al (2019) ROS generation and antioxidant defense systems in normal and malignant cells. Oxid Med Cell Longev 1–17
Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F et al (2013) Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev 1–13
Costa JDS, Ramos RDS, Costa KDSL, Brasil DDSB, Silva CHTD (2018) An in silico study of the antioxidant ability for two caffeine analogs using molecular docking and quantum chemical methods. Molecules 23(11):2801
Liu XH, Cui P, Song BA, Bhadury PS, Zhu HL et al (2008) Synthesis, structure and antibacterial activity of novel 1-(5-substituted-3-substituted-4,5-dihydropyrazol-1-yl)ethanone oxime ester derivatives. Bioorganic Med Chem 16:4075–4082
Zhao H, Zhou M, Duan L, Wang W, Zhang J, Liang X et al (2013) Efficient synthesis and anti-fungal activity of oleanolic acid oxime esters. Molecules 18:3615–3629
Kala TS, Latha DS, Sowjanya G, Swathi P, Bharathi V et al (2016) Synthesis, characterization and pharmacological evaluation of substituted N-benzyl isatin 3-oximes. J Glob Trends Pharm Sci 7:3057–3064
Harini ST, Kumar HV, Peethambar SK, Peethambar SK, Rangaswamy J et al (2013) Novel 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters: synthesis and a new insight into their antioxidant and antimicrobial potential. Med Chem Res 23:1887–1898
Özen T, Taş M (2009) Screening and evaluation of antioxidant activity of some amido-carbonyl oxime derivatives and their radical scavenging activities. J Enzyme Inhib Med Chem 24(5):1141–1147
Bolotin DS, Demakova MY, Legin AA, Suslonov VV, Nazarov AA et al (2017) Amidoxime platinum(II) complexes: pH-dependent highly selective generation and cytotoxic activity. New J Chem 41:6840–6848
Worek F, Thiermann H, Wille T (2016) Oximes in organophosphate poisoning: 60 years of hope and despair. Chem Biol Interact 259:93–98
Markandewar RA, Baseer MA (2016) Exploring pharmacological signifıcance of piperazine scaffold. World J Pharm Res 5(7):1409–1420
Salat K, Moniczewski A, Salat R, Janaszek M, Filipek B et al (2012) Analgesic, anticonvulsant and antioxidant activities of 3-[4-(3-trifluoromethyl-phenyl)-piperazin-1-yl]-dihydrofuran-2-one dihydrochloride in mice. Pharmacol Biochem Behav 101:138–147
Prashanth MK, Revanasiddappa HD, Lokanatha Rai KM, Veeresh B (2012) Synthesis, characterization, antidepressant and antioxidant activity of novel piperamides bearing piperidine and piperazine analogues. Bioorganic Med Chem Lett 22:7065–7070
Paulrasu K, Duraikannu A, Palrasu M, Shanmugasundaram A, Kuppusamy M (2014) Synthesis of 4-methyl-N′-(3-alkyl-2r,6c-diarylpiperidin-4-ylidene)-1,2,3-thiadiazole-5-carbohydrazides with antioxidant, antitumor and antimicrobial activities. Org Biomol Chem 12:5911–5921
Lin CC, Hsieh TH, Liao PY, Liao ZY, Chang CW (2014) Practical synthesis of N-substituted cyanamides via tiemann rearrangement of amidoximes. Org Lett 16:892–895
Li Y, Jian L, Dedou Z, Hui L, Hu Z; Preparation of novel oxime derivative and its application as agricultural fungicide. CN112624965A. 2012 April 9
Kallury RKMR, Rao PLKM (1977) Electron impact studies of some aryl heteryl ketoximes. Org Mass Spectrom 12(6):411–415
Dornow A, Jordan HD, Muller A (1961) Aliphatic nitro compounds. XXI. The preparation of α-chloroöximes from nitroölefins. Chem Ber 94:67–76
Dutkiewicz Z, Mikstacka R (2018) Structure-based drug design for cytochrome P450 family 1 ınhibitors. Bioinorg Chem Appl 1–21
Yim S-K, Kim K, Chun S, Oh T, Jung W et al (2013) Screening of human CYP1A2 and CYP3A4 inhibitors from seaweed in silico and in vitro. Mar Drugs 18:603
Schäfer RJB, Monaco MR, Tirla LM, A, Rivera-Fuentes P, Wennemers H, (2019) The bioorthogonal isonitrile–chlorooxime ligation. J Am Chem Soc 141:18644–18648
Tran NC, Dhondt H, Flipo M, Deprez B, Willand N (2015) Synthesis of functionalized 2-isoxazolines as three-dimensional fragments for fragment-based drug discovery. Tetrahedron Lett 56:4119–4123
Exner O, Motekov N (1982) Configuration and conformation of amidoximes. N. N-Dialkyl derivatives Collect Czechoslov Chem Commun 47:814–827
Okecha SA (1979) Reactions of N, N, S, S’-tetramethyldithiocarbamidium iodide. Chem Ind-London 15:526
Thipyapong K, Uehara T, Tooyama Y, Braband H, Alberto R, Arano Y (2011) Insight into technetium amidoxime complex: oxo technetium(V) complex of N-substituted benzamidoxime as new basic structure for molecular ımaging. Inorg Chem 50:992–998
Brand-Williams W, Cuvelier ME, Berset C (1995) Use of a free radical method to evaluate antioxidant activity. LWT Food Sci Technol 28:25–30
Ruch RJ, Cheng SJ, Klaunig JE (1989) Prevention of cytotoxicity and inhibition of intercellular communication by antioxidant catechins isolated from Chinese green tea. Carcinogenesis 10:1003–1008
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison J (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 4:17
Funding
This study was financially supported by Kocaeli University BAP (FBA-2020–2072 and FYL-2020–2208).
Author information
Authors and Affiliations
Contributions
Sait Sari: methodology, investigation, experimental studies, data analysis, writing original draft, and in silico studies. Nazlıcan Kılıç: investigation and experimental studies. Mehmet Yilmaz: methodology, data analysis, supervision, writing original draft, and review. Final version of the manuscript submitted was approved by all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sarı, S., Kılıç, N. & Yılmaz, M. In vitro antioxidant activities and in silico molecular docking studies of N-substituted oxime derivatives. Struct Chem 34, 605–616 (2023). https://doi.org/10.1007/s11224-022-01978-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-022-01978-0