
Abstract
Objective: This research aims to synthesize and characterize a new series of bioactive quinazoline derivatives, including Schiff bases, biquinazolinone, and benzothiazine, and to evaluate their bioactivity as antioxidants through both in silico and in vitro investigations. Methods: The new derivatives were synthesized with high yields by reacting Schiff base derivatives of quinazoline with 2-aminobenzoic acid and 2-mercaptobenzoic acid. The characterization of the prepared derivatives was carried out using FTIR, 1H NMR, and 13C NMR techniques. In silico investigations included assessing pharmacokinetic and pharmacodynamic properties through the SwissADME online server and conducting molecular docking studies with the tyrosinase enzyme. In vitro assessments involved evaluating antioxidant activity using the total antioxidant capacity method and the DPPH scavenging activity method. Results and Discussion: In silico analysis revealed favorable pharmacokinetic and pharmacodynamic properties for all compounds. Compounds (III) and (IV) exhibited acceptable pharmacological characteristics. Molecular docking studies showed good docking scores for all compounds with the tyrosinase enzyme. In vitro assessments demonstrated noteworthy to excellent antioxidant activity for all compounds, with compounds (III) and (IV) exhibiting very strong activity compared to the standard reference, ascorbic acid. Conclusions: The synthesized quinazoline derivatives exhibit promising bioactivity as antioxidants, supported by both computational and experimental assessments. These findings suggest their potential for further exploration in the development of antioxidant agents, with particular emphasis on the notable performance of compounds (III) and (IV).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Oxidative stress arises from an imbalance between the generation of reactive oxygen species (ROS) and the body’s antioxidant defense mechanisms [1]. It has been linked to the pathogenesis of several chronic diseases, including neurodegenerative disorders [2], cardiovascular diseases [3], cancer [4], and the aging process [5]. As a consequence, the pursuit of efficacious antioxidants has become a pivotal focus of research to ameliorate the deleterious effects of oxidative stress and enhance overall human health. Therefore, the field of medicinal chemistry has witnessed a surge of interest in the exploration and design of novel antioxidant agents based on bioactive organic molecules to combat oxidative stress-related disorders [6]. Among the diverse classes of organic molecules that have been scrutinized for their antioxidant potential, quinazolinone molecules, characterized by a heterocyclic core, have emerged as promising candidates due to their structural versatility and documented biological activities. Importantly, the integration of specific functional groups onto the quinazolinone scaffold has yielded derivatives with enhanced antioxidant capabilities, opening up avenues for potential therapeutic interventions [7]. Additionally, quinazolinone has served as a foundation for various bioactive compounds with a wide array of pharmacological activities, including antioxidative [8], antimicrobial [9–11], anticonvulsant [12], anticancer [13], anti-inflammatory [14], antidiabetic [15], dihydrofolate reductase inhibition [16], and kinase inhibitory activities [17]. Furthermore, recent studies have demonstrated that specific quinazoline derivatives can function as tyrosinase enzyme inhibitors. The tyrosinase enzyme stimulates the melanogenesis process, which involves the conversion of the amino acid tyrosine into melanin. This process leads to the production of free radicals as secondary products, which, in turn, contribute to the balance of the body’s oxidative stress. Therefore, inhibiting the tyrosinase enzyme can reduce the production of free radicals, lower oxidative stress, and indirectly mitigate its harmful effects [18–19].
Drawing from the known properties of the quinazolinone moiety, the research aimed to synthesize novel quinazolinone derivatives with the potential for potent antioxidant activity. Subsequently, both in silico and in vitro evaluations were conducted to investigate the activity of these quinazolinone derivatives and elucidate their antioxidant efficacy, unveiling their potential therapeutic benefits in the management of oxidative stress-related disorders.
RESULTS AND DISCUSSION
The aimed quinazolinone derivatives were prepared by reaction of compound (A) with different aromatic aldehydes to produce Schiff bases derivatives (I–IV). Then, Schiff bases derivatives undergo the cyclization reactions with anthranilic acid and 2-mercptobenezoic acid to produce biquinazolinone (V–VIII) and thiazine (IX–XII) derivatives respectively. Synthetic strategy of compounds (I–XII) is represented in Scheme 1.

Synthetic strategy of compounds (I–XII).
Synthesis of compounds (I–XII) were characterized by using FT-IR, 1H NMR, and 13C NMR spectra. FT-IR spectra confirm the synthesis of compounds (I–XII) by disappearance of symmetric and asymmetric stretching bands of –NH2 group and appearance of stretching bands at 3036–2902 and 1602–1600 cm–1 due to formation of –C–H and C=N bonds of imine groups. In addition, FT-IR spectra showed appearance of some characteristic bands for the formation of derivatives (I–XII), such as the nitro group bands of compound (II) at asy. 1446, sym. 1342 cm–1, the hydroxyl stretching band of compound (III) at 3259 cm–1, and the C–Haliphatic band of compound (IV) at 2902, 2858 cm–1. The other bands of compounds (I–IV) appeared at 3064–3041, 1650–1645, and 1590–1512 cm–1 due to presence of C–H aromatic, carbonyl groups, and C=C groups respectively.
1H NMR spectra confirm the synthesis of compounds (I–IV) by appearance multiple signals in the range 6.68–8.92 ppm due to C–H aromatic proton and singlet signals at 9.05–9.67 ppm due to C–H proton of imine groups. In addition, 1H NMR spectra showed characteristic signals of compounds (I–IV) such as singlet signals at 9.34 and 9.34 ppm due to OH protons of compound (III) and singlet signals at 2.99 ppm due to N–CH3 protons of compound (IV).
13C NMR spectra confirm the synthesis of compounds (I–IV) by appearance signals at 164.93–165.72 ppm due to carbon of carbonyl groups (C=O), signals at 165.06–164.91 ppm due to C=N of imine groups, signals at 148.43–152.17 ppm due to C=N of quinazolinone ring, signals in the range 112.23–150.53 ppm due to aromatic carbons (C=C) [20].
FT-IR spectra confirm the synthesis of compounds (V–VIII) appearance of stretching bands at 3221–3197 and 1678–1662 cm–1 due to the new amine and carbonyl groups of the formed quinazolinone rings. In addition, FT-IR spectra showed appearance of some characteristic bands for the formation of derivatives (V–VIII), such as the nitro group bands of compound (VI) at asy. 1448, sym. 1342 cm–1, the hydroxyl stretching band of compound (VII) at 3251 cm–1, and the CHaliphatic band of compound (VIII) at 3004, 2893 cm–1. The other bands of compounds (V–VIII) appeared at 3064–3053, 1654–1647, and 1589–1518 cm–1 due to presence of C–H aromatic, carbonyl groups, and C=C groups respectively.
1H NMR spectra confirm the synthesis of compounds (V–VIII) by appearance of singlet signals at 5.68 ppm due to N–H protons, multiple signals in the range 8.67– 6.32 ppm due to C–H aromatic proton and singlet signals at 8.61–8.75 ppm due to C–H proton of quinazoline ring. In addition, 1H NMR spectra showed characteristic signals of compounds (V–VIII) such as singlet signals at 9.36 and 9.82 ppm due to OH protons of compound (VII) and singlet signals at 2.99 ppm due to N–C–H3 protons of compound (VIII).
13C NMR spectra confirm the synthesis of compounds (V–VIII) by appearance signals at 170.48–170.04 ppm due to new carbonyl groups (C=O) of quinazolinone ring, signals at 165.12–164.93 ppm due to carbon of carbonyl groups (C=O), signals at 158.54–158.37 ppm due to C=N of imine groups and signals in the range 153.74–111.94 ppm due to aromatic carbons (C=C).
FT-IR spectra confirm the synthesis of compounds (IX–XII) appearance of stretching bands at 1681– 1670 cm–1 due to the new carbonyl groups of the formed thiazine rings. In addition, FT-IR spectra showed appearance of some characteristic bands for the formation of derivatives (IX–XII), such as the nitro group bands of compound (X) at asy. 1447, sym. 1344 cm–1, the hydroxyl stretching band of compound (XI) at 3280 cm–1, and the C–Haliphatic band of compound (XII) at 2904, 2882 cm–1. The other bands of compounds (IX–XII) appeared at 3062–3037, 1650–1646, and 1593–1519 cm–1 due to presence of C–H aromatic, carbonyl groups, and C=C groups respectively.
1H NMR spectra confirm the synthesis of compounds (IX–XII) by appearance multiple signals in the range 8.61–6.59 ppm due to C–H aromatic proton and singlet signals at 8.67–8.71 ppm due to C–H proton of thiazine ring. In addition, 1H NMR spectra showed characteristic signals of compounds (IX–XII) such as singlet signals at 9.42 and 9.60 ppm due to OH protons of compound (XI) and singlet signals at 3.00 ppm due to N–C–H3 protons of compound (XII).
13C NMR spectra confirm the synthesis of compounds (IX–XII) by appearing signal due to amid carbon of the new thiazine ring at 170.48–170.03 ppm while the signals at 165.06–164.91 ppm due to imine groups have disappeared. In addition, 13C NMR spectra showed the other characteristic signals of compounds (XI–XII) at 162.72–161.70 ppm due to carbon of carbonyl groups signals at 158.54–158.37 ppm due to C=N of imine groups, and multi signals in the rang 156.30–111.94 ppm due to aromatic carbons.
In silico ADME Study
The online tool SwissADME was used to calculate the ADME profiles and drug-like nature of all prepared compounds (I–XII). This tool proved many useful parameters such as physicochemical properties, lipophilicity, water solubility, pharmacokinetics, drug likeness and medicinal chemistry [21]. If the compounds have an acceptable pharmacokinetic profile in addition to the desired biological activity, they are considered potential candidates for drug discovery. The results of the study on the physicochemical properties of the prepared compounds (I–XII), including size, flexibility, unsaturation, and solubility, are represented in Table 1. The study revealed that some compounds exhibit acceptable properties. Regarding size, all compounds meet the acceptable size criteria, except for compounds (X) and (XII), whose molecular weights exceed 500 Daltons. In terms of flexibility, all molecules exhibit acceptability, with the number of rotatable bonds ranging from 1 to 5, falling within the acceptable range of 0 to 9 rotatable bonds. On the other hand, concerning lipophilicity and water solubility properties, only compounds (I–IV) demonstrated acceptable characteristics. However, all prepared compounds exhibited unacceptable levels of unsaturation, which is consistent with the profile of known pharmaceutical compounds [21].
In dissuasion of Pharmacokinetics properties, all compounds showed high gastrointestinal absorption (GI) because they have acceptable topological polar surface areas (TPSA) that ranged between 47.25– 126.32 Å2. All compounds except (I), (IV), (V), and (VIII) may not penetrate the BBB and therefore can be used safely without CNS complications, whereas the other compounds may penetrate the BBB as shown in Table 2. In a similar case to paracetamol and metformin, none of the prepared compounds (I–XII) act as substrates for P-glycoprotein (P-gp). P-gp is responsible for pumping drugs and toxins out of cells into the lumen, which suggests that the bioavailability of these compounds within the cells might increase. This would make them less susceptible to drug-drug interactions involving P-gp inhibition or induction. However, it could also impact their overall pharmacokinetics, potentially affecting dosing regimens and therapeutic outcomes. Additionally, their clearance may differ. On the other hand, the metabolism of all prepared compounds (I–XII), as with most drugs, is determined in the liver by CYTP450 enzymes. Therefore, it is recommended to administer them alone to minimize possible drug-drug interactions. Table 2. Represents the pharmacokinetics properties of the prepared compounds (I–XII) [22].
As shown in Table 3, compounds (I–IV) exhibited good compliance with the drug-likeness rules (Lipinski, Ghose, Veber, Egan, and Muegge). Conversely, the remaining compounds exhibited some rule violations, ranging from high partial weight to high Lipophilicity.
Furthermore, all compounds triggered several medicinal chemistry alerts in accordance with the rules of PAINS, Brenk, and lead likeness. These alerts encompassed high molecular weight, elevated lipid content, and the presence of certain functional groups, including nitro, dialkyl aniline, N–O, and Mannich base. While all of the prepared compounds displayed high bioavailability and good synthetic accessibility (Table 3). Represents the drug-likeness rules and medicinal chemistry properties of the prepared compounds (I–XII) [23].
Molecular Docking
The structure of the Tyrosinase enzyme consists of two subunits, H and L, with 392 and 150 amino acid residues, respectively. The H structure of tyrosinase contains a binuclear copper site where three histidine residues, namely His61, His85, and His94, interact with the first copper ion, while His259, His263, and His296 coordinate the second copper ion. The critical factor in the tyrosinase catalytic activity is the oxygen-binding pocket and the coordinated histidine residue interacting with the copper ion, which plays a vital role in the enzymatic activity [24].
To gain insights into the interactions and binding mode of synthesized compounds (I–XII) at the tyrosinase site for enzymatic inhibition, a molecular docking analysis was performed. The three-dimensional coordinates of tyrosinase (PDB ID: 5OAE) were retrieved from the Protein Data Bank (PDB) website. Prior to docking with synthetic ligands, the best binding pose with the lowest binding energy was selected to enhance the analysis. The test results indicated that all derivatives exhibited high binding energies ranging from –4.74 to –3.699 kcal/mol, approximately equivalent or higher to the binding energy of the co-crystallized ligand –3.355 kcal/mol (Fig. 1). The docking scores displayed minimal fluctuations, and the comparison revealed no significant differences in energy among all the ligands due to the similar basic structure of the binding sites. Additionally, compound (III) exhibited a higher binding energy compared to the co-crystalized ligand due to forming numerous hydrogen bonds with VAL218, MET215, VAL217, GLY216, ARG209, as well as other types of hydrophobic and electrostatic interactions with copper atoms (Fig. 2). While the co-crystalized form only hydrophobic and electrostatic interactions as represented in Table 4.

3D interaction of tyrosinase with co-crystalized ligand.

3D interaction of tyrosinase with compound (III).
The docked orientations revealed that all ligands were situated within the hydrophobic binding pocket surrounding the binuclear copper site. All compounds occupied the same binding site as the crystallized ligand and form many hydrophobic interactions with histidine residues, which may contribute to their tyrosinase inhibitory potency.
As a test of binding accuracy, we re-docked the crystallized tropolone ligand with the enzyme (PDB ID: 5OAE). The binding position with the lowest energy for the compound occupied the same site as the active site, but the orientation was reversed as shown in Fig. 3. The rootmean-square deviation (RMSD) of the upright compound compared to the crystallized compound was 3.138 Å.

(a) Scavenging activity standard carve of ascorbic acid; (b) scavenging activity carve of compounds (III) and (VII).
In Vitro Antioxidant Study
The inhibition activities of synthesized compounds as potential antioxidants were evaluated through two different in vitro methods: the total antioxidant capacity method using molybdenum complex and the free radical scavenging method using DPPH (2,2-diphenyl-1-picrylhydrazyl) with varying concentrations 100, 150, and 200 μg of the compounds (I–XII). The results were listed in Table 5.
Total Antioxidant Capacity
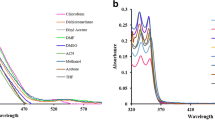
The results presented in Table 5 and Fig. 4 demonstrate that all synthesized compounds possess potential antioxidant activity. The activities ranged from moderate to strong compared to the standard antioxidant ascorbic acid. Compounds containing amine and hydroxyl groups (III), (IV), (VII), (VIII), (XI), (XII) exhibited higher antioxidant activity than the other derivatives, with compounds (III) and (IV) showing particularly promising results, warranting further detailed investigation.

(a) Total antioxidant capacity of compounds (I–XII); (b) standard carve of compounds (III), (IV), and ascorbic acid.
DPPH Free Radical Scavenging
As shown in Table 5 and Fig. 5, all synthesized compounds exhibited moderate to excellent inhibition activity compared to the standard ascorbic acid. The results indicated that substituted groups on the benzene rings, such as hydroxyl, amine, and nitro groups, played a significant role in the inhibition and scavenging of free radicals. These groups efficiently captured free radicals, forming stable intermediate compounds. Compounds containing hydroxyl groups (III), (VII), and (XI) demonstrated the highest inhibition activity, followed by derivatives containing amine groups (IV), (VIII), and (XII), and then those with nitro groups (II), (VI), and (X). Unsubstituted derivatives (I), (V), and (IX) exhibited weaker activity. It was also observed that compounds containing thiazine rings (IX–XII) exhibited greater inhibition activity than their counterparts with quinazoline rings (V–VIII), attributed to the presence of sulfur atom, which facilitated the scavenging of free radicals and enhanced compound stability.

Free radical scavenging activity by DPPH of compounds (I–XII).
Ascorbic acid was used as standard for DPPH scavenging activity, and the standard curve is presented in Fig. 3a. The DPPH scavenging activity of compounds (III) and (VII) revealed an unexpected peculiar pattern. They higher activities but tends to decrease beyond the concentration 100 μg as in Fig. 3b. The usual trend to show scavenging activity is by indicating IC50 (halfmaximal inhibitory concentration) which is unable to show for these compounds. The reasons may be of interference by other chemical molecules competing for reduction by DPPH or having higher internal activity scavenging chain reaction, not permitting DPPH be donated with an electron [25]. IC50 for ascorbic acid was 29.24 μg.
EXPERIMENTAL
Material. The chemicals were sourced from BDH, Merck, Fluka, and Sigma Aldrich. Melting points were measured using an SMP3 melting point apparatus without correction. FT-IR spectra, spanning the range of 4000 to 600 cm–1, were analyzed employing a Shimadzu FT-IR 8400 spectrophotometer with a KBr disc. For 1H NMR and 13C NMR spectra recorded at 400 MHz, TMS served as a reference and DMSO-d6 as the solvent, using an ECA. UV-Vis spectra were obtained utilizing a Shimadzu UV-1900i spectrophotometer from Japan and Nibgen micro reader from Canada. CNHSO analysis done by EMA 502 Elemental Analyzer/Italy.
Synthesis of 3-( substituted-benzylideneamino)-2phenylquinazolin-4(3H)-one (I–IV) [26]. 1 g (0.0041 mol) of quinazoline derivative (A) was dissolved in 5 mL of dimethylformamide (DMF). To the solution was added 0.0041 mol of a variously substituted aromatic aldehyde and three drops of glacial acetic acid. The mixture was refluxed for 8–10 h. After cooling, the reaction mixture was poured into ice water, and the precipitated solid was filtered, washed with water, and recrystallized from ethanol/water to obtain compounds (I–IV).
3-(Benzylideneamino)-2-phenylquinazolin-4(3H)one (I). White solid; Yield: 90%, mp: 240–242°C; FT-IR νmax, cm−1: 3060 (Ar–H), 3036 (N=C–H), 1649 (C=O), 1602 (C=N), 1590–1520 (C=C); 1H NMR (400 MHz, DMSO-d6), ppm: 9.05 (s, 1H, N=C–H), 8.59–7.29 (m, Ar–H); 13C NMR (101 MHz, DMSO-d6), ppm: 165.48, 165.01, 149.55, 139.81, 134.86, 134.50, 133.11, 132.61, 130.87, 130.22, 129.57, 129.41, 129.37, 129.10, 128.15, 127.77, 127.54, 123.62, 121.52, 120.94; C21H15N3O (325.36); Calculated, %: C, 77.52; H, 4.65; N, 12.91; O, 4.92; Found, %: C, 77.31; H, 4.83; N, 12.80; O, 4.79.
3-((4-Nitrobenzylidene)amino)-2-phenylquinazolin-4(3H)-one (II). Yellow solid; Yield: 95%; mp: 206– 208°C; FT-IR νmax, cm−1: 3062 (Ar–H), 2935 (N=C–H), 1650 (C=O), 1602 (C=N), 1585–1517 (C=C), asy. 1446, sym. 1342 (NO2); 1H NMR (400 MHz, DMSOd6), ppm: 9.32 (s, 1H, N=C–H), 8.54–7.30 (m, Ar–H); 13C NMR (101 MHz, DMSOd6), ppm: 165.72, 165.06, 148.43, 146.78, 140.77, 139.80, 134.82, 133.30, 132.59, 130.23, 130.00, 129.38, 129.19, 128.65, 128.19, 127.56, 124.66, 124.53, 123.67, 121.72, 121.40, 120.97; C21H14N4O3 (370.36); Calculated, %: C, 68.10; H, 3.81; N, 15.13; O, 12.96; Found, %: C, 67.75; H, 3.73; N, 15.25; O, 13.09.
3-((2,4-Dihydroxybenzylidene)amino)-2-phenylquinazolin-4(3H)-one (III). Off-white solid; Yield: 85%; mp: 249–250°C; FT-IR νmax, cm−1: 3259 (O–H), 3064 (Ar–H), 2974 (N=C–H), 1645 (C=O), 1602 (C=N), 1583–1512 (C=C); 1H NMR (400 MHz, DMSOd6), ppm: 9.51 (s, 1H, OH), 9.34 (s, 1H, OH), 9.20 (s, 1H, N=C–H), 8.61–6.81 (m, Ar–H); 13C NMR (101 MHz, DMSOd6), ppm: 165.14, 164.91, 150.17, 148.78, 146.25, 139.83, 134.86, 132.97, 132.62, 129.44, 128.98, 127.49, 125.89, 123.54, 121.43, 121.28, 120.79, 116.07, 113.26; C21H15N3O3 (357.36); Calculated, %: C, 70.58; H, 4.23; N, 11.76; O, 13.43; Found, %: C, 70.71; H, 4.11; N, 11.92; O, 13.35.
3-((4-(Dimethylamino)benzylidene)amino)-2phenylquinazolin-4(3H)-one (IV). Orange solid; Yield: 87%; mp: 254–256°C; FT-IR νmax, cm−1: 3041 (Ar–H), 2902, 2858 (C–Haliphatic), 2902 overlapped (N=C–H), 1647 (C=O), 1600 (C=N), 1585–1521 (C=C); 1H NMR (400 MHz, DMSOd6), ppm: 9.67 (s, 1H, N=C–H), 8.92–6.68 (m, Ar–H), 2.99 (s, 6H, N–C–H3); 13C NMR (101 MHz, DMSOd6), ppm: 164.93, 164.91, 152.17, 150.53, 139.81, 134.87, 132.87, 132.61, 130.24, 129.44, 129.20, 128.95, 128.06, 127.49, 123.52, 121.59, 121.24, 120.87, 112.23, 40.59; C23H20N4O (368.43); Calculated, %: C, 74.98; H, 5.47; N, 15.21; O, 4.34; Found, %: C, 75.15; H, 5.62; N, 15.05; O, 4.22.
2-(Substituted-phenyl)-2′-phenyl-1H,4′H-(3,3′-biquinazoline)-4,4′(2H)-dione (V–VIII) [26]. 0.00025 mol of Schiff base derivatives was dissolved in 10 mL of dimethylformamide (DMF). Then, 0.00025 mol of 2-aminobenzoic acid was added to the mixture, and the mixture was refluxed for 18–20 h. After cooling, the reaction mixture was poured into ice water, and the precipitated solid was filtered, washed with 5% sodium bicarbonate solution and water, and recrystallized from ethanol to obtain compounds (V–VIII).
2,2′-Diphenyl-1H,4′H-(3,3′-biquinazoline)-4,4′(2H)dione (V). Light brown solid; Yield: 81%; mp: 168–170°C; FT-IR νmax, cm−1: 3220 (N–H), 3058 (Ar–H), 3035 (N–C–H), 1675 (C=O), 1647 (N–C=O), 1602 (C=N), 1525–1518 (C=C); C28H20N4O2 (444.48); Calculated, %: C, 75.66; H, 4.54; N, 12.60; O, 7.20; Found, %: C, 75.55; H, 4.68; N, 12.68; O, 7.01.
2-(4-Nitrophenyl)-2′-phenyl-1H,4′H-(3,3′-biquinazoline)-4,4′(2H)-dione (VI). Light yellow solid; Yield: 85%; mp: 195–197°C; FT-IR νmax, cm−1: 3221 (N–H), 3064 (Ar–H), 3937 (N–C–H), 1677 (C=O), 1650 (N–C=O), 1602 (C=N), 1585–1519 (C=C), 1448 (asy. NO2), 1342 (sym. NO2); C28H19N5O (489.48); Calculated,%: C, 68.71; H, 3.91; N, 14.31; O, 13.07; Found, %: C, 68.84; H, 3.78; N, 14.41; O, 12.88.
2-(2,4-Dihydroxyphenyl)-2-methyl-2′-phenyl-1H,4′H(3,3′-biquinazoline)-4,4′(2H)-dione (VII). Brown solid; Yield: 80%; mp: 172–174°C; FT-IR νmax, cm−1: 3251 (O–H), 3213 (N–H), 3062 (Ar–H), 2962 (N–C–H), 1678 (C=O), 1654 (N–C=O), 1602 (C=N), 1589–1519 (C=C); 1H NMR (400 MHz, DMSOd6), ppm: 9.82 (s, 2H, OH), 9.36 (s, 1H, OH), 8.61 (s, 1H, N–C–H), 8.28–6.73 (m, Ar–H), 5.68 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6), ppm: 170.48, 165.12, 158.37, 153.60, 150.92, 150.16, 148.78, 147.18, 146.86, 146.25, 139.83, 135.36, 135.15, 134.86, 134.79, 132.97, 132.63, 130.22, 130.08, 129.96, 129.44, 128.98, 128.13, 128.01, 127.90, 127.87, 127.49, 127.24, 126.51, 125.88, 124.01, 123.70, 123.54, 121.54, 121.42, 121.27, 120.78, 116.07, 113.25; C29H22N4O4 (490.51); Calculated, %: C, 71.01; H, 4.52; N, 11.42; O, 13.05; Found, %: C, 70.90; H, 4.64; N, 11.60; O, 12.93.
2-(4-(Dimethylamino)phenyl)-2′-phenyl-1H,4′H(3,3′-biquinazoline)-4,4′(2H)-dione (VIII). Yellow solid; Yield: 81%; mp: 183–184°C; FT-IR νmax, cm−1: 3197 (N–H), 3053 (Ar–H), 3004, 2893 (C–Haliphatic), 3004 (N–C–H, overlapped), 1662 (C=O), 1647 (N–C=O), 1602 (C=N), 1589–1523 (C=C); 1H NMR (400 MHz, DMSOd6), ppm: 8.75 (s, 1H, N–C–H), 8.67–6.32 (m, Ar–H), 5.68 (s, 1H, NH), 2.99 (s, 6H, N–C–H3); 13C NMR (101 MHz, DMSOd6), ppm: 170.04, 164.93, 158.54, 153.74, 153.52, 152.17, 150.53, 146.90, 139.82, 135.24, 134.87, 132.86, 132.60, 130.79, 130.25, 130.09, 129.96, 129.44, 129.20, 128.95, 128.05, 127.99, 127.49, 127.35, 127.09, 123.51, 121.60, 121.23, 120.85, 119.47, 112.23, 111.94, 40.60, 40.21; C30H25N5O2 (487.55); Calculated, %: C, 73.90; H, 5.17; N, 14.36; O, 6.56; Found, %: C, 74.04; H, 5.28; N, 14.08; O, 6.53.
3-(2-(Substituted-phenyl)-2H-benzo[e]-[1,3]thiazin3(4H)-yl)-2-phenylquinazolin-4(3H)-one (IX–XII) [26]. 0.00025 mol of a Schiff base derivative was dissolved in 10 mL of dimethylformamide (DMF). Then, 0.00025 mol of 2-mercaptobenzoic acid was added, and the mixture was refluxed for 18–20 h. After cooling, the reaction mixture was poured into ice water, and the precipitated solid was filtered, washed with a 5% sodium bicarbonate solution and water, and recrystallized from ethanol to obtain compounds (IX–XII).
3-(2-Phenyl-4-oxo-2H-benzo[e][1,3]thiazin-3(4H)-yl)2-phenylquinazolin-4(3H)-one (IX). White solid; Yield: 83%; mp: 153–155°C; FT-IR νmax, cm−1: 3060 (Ar–H), 3037 (N–C–H), 1681 (C=O), 1649 (N–C=O), 1606 (C=N), 1582–1556 (C=C); C28H19N3O2S (461.53); Calculated, %: C, 72.87; H, 4.15; N, 9.10; O, 6.93; S, 6.95; Found, %: C, 72.75; H, 4.24; N, 9.21; O, 7.11; S, 6.88.
3-(2-(4-Nitrophenyl)-4-oxo-2H-benzo[e][1,3]thiazin3(4H)-yl)-2-phenylquinazolin-4(3H)-one (X). Orange solid; Yield: 90%; mp: 174–176°C; FT-IR νmax, cm−1: 3060 (Ar–H), 2925 (N–C–H), 1679 (C=O), 1649 (N–C=O), 1600 (C=N), 1560–1519 (C=C), 1447 (asy. NO2), 1344 (sym. NO2); C28H18N4O4S (506.53); Calculated, %: C, 66.39; H, 3.58; N, 11.06; O, 12.63; S, 6.33; Found, %: C, 66.46; H, 3.49; N, 10.95; O, 12.78; S, 6.40.
3-(2-(2,4-Dihydroxyphenyl)-4-oxo-2H-benzo[e][1,3]thiazin-3(4H)-yl)-2-phenylquinazoli n-4(3H)-one (XI). Brown solid; Yield: 82%; mp: 148–150°C; FT-IR νmax, cm−1: 3280 (O–H), 3062 (Ar–H), 2935 (N–C–H), 1670 (C=O), 1650 (N–C=O), 1602 (C=N), 1587–1519 (C=C); 1H NMR (400 MHz, DMSOd6), ppm: 9.60 (s, 2H, OH), 9.42 (s, 1H, OH), 8.71 (s, 1H, N–C–H–S), 8.61–6.59 (m, Ar–H); 13C NMR (101 MHz, DMSOd6), ppm: 170.48, 161.70, 158.37, 156.30, 153.60, 150.94, 147.18, 146.86, 146.27, 135.36, 135.15, 134.92, 134.79, 130.22, 130.08, 129.96, 129.43, 129.10, 128.24, 128.13, 128.01, 127.90, 127.87, 127.45, 127.24, 127.16, 126.51, 124.00, 123.70, 121.54, 120.54, 116.09, 114.13. C28H19N3O4S (493.53); Calculated, %: C, 68.14; H, 3.88; N, 8.51; O, 12.97; S, 6.50; Found, %: C, 68.33; H, 3.74; N, 8.59; O, 13.06; S, 6.49.
3-(2-(4-(Dimethylamino)phenyl)-4-oxo-2H-benzo[e][1,3]thiazin-3(4H)-yl)-2-phenylquin azolin-4(3H)one (XII). Light brown solid; Yield: 83%; mp: 158–160°C; FT-IR νmax, cm−1: 3062 (Ar–H), 2904, 2882 (C–Haliphatic), 2904 (N–C–H, overlapped), 1676 (C=O), 1646 (N–C=O), 1600 (C=N), 1593–1533 (C=C); 1H NMR (400 MHz, DMSOd6), ppm: 8.67 (s, 1H, N–C–H–S), 8.20–6.74 (m, Ar–H), 5.69 (s, 1H, NH), 3.00 (s, 6H, N–C–H3); 13C NMR (101 MHz, DMSOd6), ppm: 170.03, 162.72, 158.54, 153.74, 153.52, 152.77, 149.21, 146.90, 135.24, 135.09, 134.79, 133.18, 131.88, 130.80, 130.25, 130.10, 129.09, 128.24, 128.05, 127.99, 127.35, 127.08, 126.33, 121.59, 121.46, 119.48, 111.94, 40.09; C30H24N4O2S (504.60); Calculated, %: C, 71.41; H, 4.79; N, 11.10; O, 6.34; S, 6.35; Found, %: C, 71.52; H, 4.71; N, 10.97; O, 6.42; S, 6.23.
In silico studies. ADME study. The Swiss ADME online server (http://www.swissadme.ch/) was used to determine the physicochemical parameters of each compound. The server uses an accurate and efficient iLOG algorithm to calculate the physicochemical parameters of small molecules. It provides a wealth of important information about pharmacokinetic profiles and drug-like nature [21].
Preparation of ligands. The two-dimensional structures of the compounds were drawn in ChemBioDraw 12.0, converted to three-dimensional structures, and saved in MOL file format. All ligands (I–XII) were energyminimized using Avogadro 1.2 with the MMFF94S force field.
Preparation of proteins for docking. The protein 3D structure (Tyrosinase PDB ID: 5OAE) [27] used for docking were retrieved from the Protein Data Bank (PDB) and prepared using the Protein Preparation Wizard of Maestro v12.5. Preparation included hydrogen bond assignment, bond ordering, hydrogen addition, optimization, protein minimization, and deletion of water molecules within 5 Å units of the het group. The Glidegrid wizard was used to create a protein-receptor grid. The high-affinity binding site of the ligands was determined by selecting an atom of the protein co-crystallized ligand. The Van der Waals scaling factor was set to 1.0, and the partial charge cutoff was set to 0.25. In addition, docking of all ligands was performed using the ligand docking module of Glide in standard-precision (SP) mode, with all other settings used as default. Visualization and analysis of the docking results were performed using BIOVIA Discovery Studio Visualizer 2021.
In vitro study. Total antioxidant capacity [28]. The total antioxidant activity of the sample was calculated using a modified phosphomolybdenum complex formation method. Briefly, 100 mL aliquots of the sample at various concentrations (200, 150, 100, and 50 mg/mL) were mixed with 500 mL of a reagent solution (0.6 M sulfuric acid, 28 mM sodium phosphate, and 4 mM ammonium molybdate) and incubated at 95°C for 90 min. The mixture was then cooled to 25°C, and the absorbance of 300 mL of the sample solution was measured at 695 nm using a microplate reader. The antioxidant activity was calculated based on the optical density of the sample.
Determination of the free radical scavenging activity by DPPH [27]. In a microplate, 100 μL of ethanolic solutions of each compound at different concentrations (200, 150, 100, and 50 mg/mL) were mixed with 200 μL of an ethanolic solution of DPPH (0.2 M). The reaction mixture was shaken for 1 min and incubated at room temperature for 30 min to measure the optical density (OD). The OD was measured at a wavelength of 517 nm using a microplate reader. Ascorbic acid was used as a positive control. The percent inhibition of the DPPH radical was calculated using the following formula:
CONCLUSIONS
In this study, a series of quinazolinone derivatives (I–XII) were successfully synthesized through a multi-step synthetic strategy involving Schiff base formation and subsequent cyclization reactions with anthranilic acid and 2-mercaptobenzoic acid. The chemical structures of these compounds were characterized and confirmed through FT-IR, 1H NMR, and 13C NMR spectra. Furthermore, in silico ADME (Absorption, Distribution, Metabolism, Excretion) studies were conducted to assess the drug-like properties of the synthesized compounds. The results find that certain compounds, particularly (I–IV), exhibit promising ADME properties, including favorable absorption, flexibility and, making them suitable for subsequent drug development. Molecular docking analyzes were performed to investigate the binding poses and interaction energies of the synthesized compounds (I–XII) with the tyrosinase enzyme and showed robust binding energies for all derivatives (I–XII). Compound (III) exhibited a particularly high binding energy due to multiple hydrogen bonding with specific residues, suggesting its potential as an inhibitor of tyrosinase enzyme. In vitro antioxidant activity assessments using total antioxidant capacity and the DPPH free radical scavenging methods demonstrated varied antioxidant activity, with compounds containing amine and hydroxyl groups (III), (IV), (VII), (VIII), (XI), and (XII) exhibiting heightened potential. In summary, compounds (III) and (IV) exhibited promising ADME properties, high docking scores with tyrosinase enzyme and remarkable antioxidant activities, positioning them as promising subjects for further research in developing therapeutic agents for oxidative stress-related diseases.
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
Adwas, A.A., Elsayed, A., Azab, A., and Quwaydir, F.J.J.A.B.B., J. Appl. Biotechnol. Bioeng., 2019, vol. 6, pp. 43–47. https://doi.org/10.15406/jabb.2019.06.00173
Hassan, W., Noreen, H., Rehman, S., Kamal, M.A., and Rocha, J.B.T.D., Curr. Neuropharmacol., 2022, vol. 20, pp. 1046–1072. https://doi.org/10.2174/1570159x19666211111141246
Dubois-Deruy, E., Peugnet, V., Turkieh, A., and Pinet, F., Antioxidants, 2020, vol. 9, pp. 864–879. https://doi.org/10.3390/antiox9090864
Hayes, J.D., Dinkova-Kostova, A.T., and Tew, K.D., Cancer Cell, 2020, vol. 38, pp. 167–197. https://doi.org/10.1016/j.ccell.2020.06.001
Hano, C. and Tungmunnithum, D., Medicines, 2020, vol. 7, pp. 26–35. https://doi.org/10.3390/medicines7050026
Yernale, N.G., Mathada, B.S., Baburao, G., Sahane, S., Patil, P., Karunakar, P., and Venkatesulu, A., J. Mol. Struct., 2023, vol. 1294, Article ID: 136512. https://doi.org/10.1016/j.molstruc.2023.136512
Dube, P.S., Legoabe, L.J., and Beteck, R.M., Mol. Divers., 2023, vol. 27, pp. 1501–1526. https://doi.org/10.1007/s11030-022-10581-8
Perveen, S., Saad, S.M., Khan, K.M., and Choudhary, M.I., Lett. Drug Des. Discov., 2021, vol. 18, pp. 806–815. https://doi.org/10.2174/1570180818666210427092319
Ghorab, M.M., Alqahtani, A.S., Soliman, A.M., and Askar, A.A., Int. J. Nanomed., 2020, vol. 15, pp. 3161–3180. https://doi.org/10.2147/IJN.S241433
Gandomi, F., Dilmaghani, K.A., Massoumi, B., and Ahrabi, Y.S., Russ. J. Bioorg. Chem., 2023, vol. 49, pp. 1089–1099. https://doi.org/10.1134/S1068162023050163
Ibrahim, O.F., Bakhite, E.A., Metwally, S.A.M., El-Ossaily, Y.A., Abdu-Allah, H.H.M., Al-Taifi, E.A., and Kandel, M., Russ. J. Bioorg. Chem., 2021, vol. 47, pp. 918–928. https://doi.org/10.1134/S1068162021040117
Mohammadi, A.A., Taheri, S., Shisheboran, S., Ahdenov, R., Mohammadi-Khanaposhtani, M., Darjani, P.S., Masihi, P.H., Shakiba, A., Larijani, B., Mahdavi, M., and Ahangar, N., J. Biochem. Mol. Toxicol., 2023, vol. 37, Article ID: e23234. https://doi.org/10.1002/jbt.23234
Gatadi, S., Pulivendala, G., Gour, J., Malasala, S., Bujji, S., Parupalli, R., Shaikh, M., Godugu, C., and Nanduri, S., J. Mol. Struct., 2020, vol. 1200, Article ID: 127097. https://doi.org/10.1016/j.molstruc.2019.127097
Verma, S.K., Verma, R., Rakesh, K.P., and Gowda, D.C., Eur. J. Med. Chem. Rep., 2022, vol. 6, Article ID: 100087. https://doi.org/10.1016/j.ejmcr.2022.100087
Khalifa, M.M., Sakr, H.M., Ibrahim, A., Mansour, A.M., and Ayyad, R.R., J. Mol. Struct., 2022, vol. 1250, Article ID: 131768. https://doi.org/10.1016/j.molstruc.2021.131768
Bhagat, K., Kumar, N., Kaur Gulati, H., Sharma, A., Kaur, A., Singh, J. V., Singh, H., and Bedi, P.M.S., Exp. Opin. Ther. Pat., 2022, vol. 32, pp. 1079–1095. https://doi.org/10.1080/13543776.2022.2130752
Mortazavi, M., Divar, M., Damghani, T., Moosavi, F., Saso, L., Pirhadi, S., Khoshneviszadeh, M., Edraki, N., and Firuzi, O.J.F.I.C., Front. Chem., 2022, vol. 10, Article ID: 969559. https://doi.org/10.3389/fchem.2022.969559
Huang, Y., Yang, J., Chi, Y., Gong, C., Yang, H., Zeng, F., Gao, F., Hua, X., and Wang, Z., Molecules, 2022, vol. 27, Article ID: 5558. https://doi.org/10.3390/molecules27175558
Fedarkevich, A.N., Sharko, O.L., and Shmanai, V.V., Russ. J. Bioorg. Chem., 2020, vol. 46, pp. 187–198. https://doi.org/10.1134/S1068162020020089
Silverstein, R.M. and Bassler, G.C., J. Chem. Ed., 1962, vol. 39, Article ID: 546. https://doi.org/10.1021/ed039p546
Daina, A., Michielin, O., and Zoete, V., Sci. Rep., 2017, vol. 7, Article ID: 42717. https://doi.org/10.1038/srep42717
Daina, A. and Zoete, V., ChemMedChem, 2016, vol. 11, pp. 1117–1121. https://doi.org/10.1002/cmdc.201600182
Lipinski, C.A., Lombardo, F., Dominy, B.W., and Feeney, P.J., Adv. Drug Del. Rev., 2001, vol. 46, pp. 3–26. https://doi.org/10.1016/S0169-409X(00)00129-0
Ismaya, W.T., Rozeboom, H.J., Weijn, A., Mes, J.J., Fusetti, F., Wichers, H.J., and Dijkstra, B.W., Biochemistry, 2011, vol. 50, pp. 5477–5486. https://doi.org/10.1021/bi200395t
Athavale, A., Jirankalgikar, N., Nariya, P., and De, S., Int. J. Pharm. Sci. Res., 2012, vol. 3, pp. 2543–2549. https://doi.org/10.13040/IJPSR.0975-8232.3(8).2543-49
Al-Khuzaie, M.G. and Al-Majidi, S.M., J. Global Pharma Technol., 2018, vol. 10, pp. 415–423.
Ferro, S., Deri, B., Germanò, M.P., Gitto, R., Ielo, L., Buemi, M.R., Certo, G., Vittorio, S., Rapisarda, A., Pazy, Y., Fishman, A., and De Luca, L., J. Med. Chem., 2018, vol. 61, pp. 3908–3917. https://doi.org/10.1021/acs.jmedchem.7b01745
Elgubbi, A.S., Alzahrani, A.Y.A., El-Helw, E.A.E., and Shaban, S.S., Polycyclic Aromat. Comp., 2023, vol. 44, pp. 1–14. https://doi.org/10.1080/10406638.2023.2210729
ACKNOWLEDGMENTS
We extend our sincere appreciation to the Chemistry Department’s laboratory at the College of Science, University of Baghdad, and the Advanced Research Laboratory at the Technical Institute – Diwaniya, University of Middle Euphrates. Their invaluable assistance in conducting laboratory experiments greatly contributed to the success of this research. We are grateful for their expertise and support, which enhanced the quality of our work.
Funding
This work was supported by regular institutional funding, and no additional grants were obtained.
Author information
Authors and Affiliations
Contributions
The authors MAl-K and LH carried out the experimental work. The author SMH contributed to manuscript preparation.
All authors participated in the discussion of the results.
Corresponding author
Ethics declarations
This article does not contain any studies involving patients or animals as test objects.
Informed consent was not required for this article. No conflicts of interest was declared by the authors.
Additional information
Publisher's Note. Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Al-Khuzaie, M.G.A., Hama, L.H.k.k. & Al-Majidi, S.M.H. Synthesis, In Silico, and In Vitro Evaluation of the Potential Antioxidant Activity of New Quinazoline Derivatives. Russ J Bioorg Chem 50, 1476–1490 (2024). https://doi.org/10.1134/S1068162024040228
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162024040228