
Abstract
The cycloaddition reactions of 2-sulfonyl dienes with some alkenes have been investigated using density functional theory (DFT)-based reactivity indices and activation energy calculations at the MPWB1K/cc-pVDZ level of theory. Two modes of [4+2] and [2+4] cycloaddition reactions can occur from the results of cross-Diels-Alder reactions of 2-sulfonyl dienes, with 2,3-dimethyl butadiene and or cyclopentadiene. The energy results indicated that the [2+4] cycloaddition reactions are the most favorable pathways. The considerable difference in the electron deficiency can lead to the different reactivity of the two C-C double bonds of the 2-sulfonyl diene. Moreover, the phenylsulfanyl group is a much more powerful directing element than the phenylsulfonyl group (SO2Ph) for the control of the regioselectivity of cycloaddition reactions. The reactions take place via an asynchronous one-step mechanism with a polar character, and an analysis of the conceptual DFT indices explains the polar character of these reactions. The electron reorganization along the most preferred pathway of the [2+4] cycloaddition reaction between 2-phenylsulfonyl-1,3-butadienes and cyclopentadiene have been studied using the topological analysis of the electron localization function (ELF). The ELF analysis revealed that this reaction proceeds through a two-stage one-step mechanism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sulfonyl dienes are interesting as Diels-Alder (DA) dienes [1,2,3,4,5,6,7] and are useful building blocks in organic synthesis. These reagents may act as Michael acceptors with nucleophiles, [8,9,10] or be transformed into 1,4-difunctionalized olefins [8] and also undergo regioselective [4+2] cycloaddition reactions with both electron-rich and electron-deficient olefins to give functionalized cyclic systems [8,9,10]. Accordingly, the sulfonylated dienes belong to the category of conjugated dienes with both a normal and inverse electron demand. Alternatively, these sulfonylated dienes can be viewed as dienophiles because they are olefins with electron-withdrawing sulfonyl and vinyl groups. Therefore, these dienes are ideal candidates to react in the cross-DA (CDA) reactions as both the diene and the dienophile. 2-arylsulfonyl-1,3-dienes are an example of these dienes which have generated much attention in DA reactions [8,9,10] due to their reasonably good reactivity despite electron deficiency.
2-arylsulfonyl-1,3-dienes have two double bonds with different reactivity; one of the double bonds is almost electron-rich, while the other is electron-deficient [11]. Regioselectivity of 2-arylsulfonyl-1,3-diene at each double bond can be achieved by taking advantage of their electron density, where only the double bond directly attached to the sulfonyl group can react as the dienophile part. Also, in the cycloaddition reactions of 2-arylsulfonyl-1,3-dienes with unsymmetrical dienophiles, the sulfonyl group is an effective directing element favoring the formation of para adducts [8,9,10]. These reactions are highly stereoselective with the vinyl group or the sulfonyl group on the endo or exo face.
In this regard Chou et al. describe their findings on the CDA reactions of substituted 2-phenylsulfonyl-1,3-butadienes, 1, as both dienes and dienophiles with a variety of electron-rich alkenes [12] (Scheme 1). When the 2-phenylsulfonyl-1,3-butadienes, 1a, were reacted with 2,3-dimethyl-1,3-butadiene, 2, two types of cycloadducts, 3a-p and 4a-A, in a 1:3 ratio were produced. Moreover, reaction of 2-phenylsulfonyl-1,3-butadienes, 1a, with cyclopentadiene, 5 at r.t, led to the formation of the bicyclic compound 7a-A (55%) as the major product and 6a-P as the minor component (16%), while the CDA reaction of cyclopentadiene, 5, with 1a at 130°C produced only one product 6a-p, in good yield.

The cycloaddition reactions of 2-phenylsulfonyl-1,3-butadiene, 1a, with 2,3-dimethyl-1,3-butadiene, 2, and cyclopentadiene 5 in benzene solvent [12]
As pointed to the experimental results [12], 2-phenylsulfonyl-1,3-butadiene, 1a, can act not only as dienes [4+2] but also as dienophiles [2+4] in the cycloaddition reactions. Therefore, the purpose of the present study is to clarify whether 2-phenyl sulfonyl-1,3-butadiene, 1a, and 2-phenyl sulfonyl-3-phenylsulfanyl-1,3-butadiene 1b prefer to participate as diene or dienophile in the cycloaddition reactions and to a better understanding of the influence of sulfonyl and sulfanyl substituents on the stereoselectivity of these reactions. Herein, we first studied the cycloaddition reaction of 2-phenyl sulfonyl-1,3-butadiene 1a with 2,3-dimethyl-1,3-butadiene, 2, and cyclopentadiene, 5, which reported by Chou et al. [12], and then extended our experimental investigations by assessing the influence of SPh substituent on the selectivity of reactions. Therefore, the cycloaddition reaction of 2-phenyl sulfonyl-3-phenylsulfanyl-1,3-butadiene 1b, which are experimentally untried, are also studied as a suggestion reaction.
The mechanistic details were explored through computational analysis by the DFT method, as well as the origin of the selectivity in the experimental observations was investigated through the analysis of the global and local indices and Parr functions. Finally, the mechanism and selectivity of the [2+4] cycloaddition reaction between 2-phenylsulfonyl-1,3-butadiene and cyclopentadiene have been studied using the electron localization function (ELF) [13,14,15].
Computational methods
We optimized all species of the aforementioned cycloaddition reactions using MPWB1K exchange-correlation functionals [16] together with the standard cc-pVDZ basis set. The intrinsic reaction coordinate (IRC) paths were traced [17]. The solvent effects of benzene were calculated using full optimization of the gas phase structures using new solvation model density (SMD) solvent model, which is based on the polarized continuous quantum mechanical charge density of the solute [18, 19]. Values of thermodynamic parameters, enthalpies, entropies, and Gibbs free energies were calculated at 298 K and 1 atm [20]. The global electron density transfer (GEDT) [21, 22] at the TSs was computed through a natural population analysis (NPA) [23]. The global electrophilicity index, ω, is given in terms of the electronic chemical potential, μ, and the chemical hardness, η, ω=μ2/2η [24]. These two quantities are evaluated in terms of the energies of the frontier molecular orbitals HOMO and LUMO, ɛH and ɛL, as μ= (ɛH+ɛL)/2 and η=εL˗ɛH, respectively [25, 26]. The global nucleophilicity index, N [27, 28], based on the HOMO energies [29] is defined as N=ɛH(NU)-ɛH(TCE), with tetracyanoethylene (TCE) as a reference. In 2013, the nucleophilic, Pkˉ, and electrophilic, Pk+, Parr functions, [30] proposed by Domingo, are based on the atomic spin density (ASD) at the radical cation and radical anion. The local electrophilicity indices, ωk [31], and the local nucleophilicity indices, Nk [31], were calculated using ωk=ωPk+ and Nk=NPkˉ, respectively. All computations were carried out with the Gaussian 09 suite of programs [30].
The ELF topological analysis was carried out on the selected points of the IRC profile of TS4-Ae using the TopMod program [32].
Results and discussion
In the first part, energy aspects, transition states (TSs), and their electronic structures in terms of bond orders and global electron density transfer (GEDT) for the cycloaddition reactions of 2-phenylsulfonyl-1,3-butadiene, 1a, and 2-phenylsulfonyl-3-phenylsulfanyl-1,3-butadiene, 1b, with some alkenes, 2 and 5, are analyzed. Next the global and local DFT reactivity indices of the reactants are calculated in order to determine the electronic character and the regio- and chemoselectivity of the reactions. Finally, ELF topological analysis carried out for the cycloaddition reaction between 1a and 5.
Mechanistic study of the cycloaddition reactions of 2-phenylsulfonyl-1,3-butadiene, 1a, and 2-phenylsulfonyl-3-phenylsulfanyl-1,3-butadiene, 1b, with 2,3-dimethyl-1,3-butadiene, 2.
Due to the presence of two possible roles of 1, as diene or dienophile, and two reactive sites in 1, C=C-SO2Ph and C=C-R, two regioisomeric [4+2] reaction pathways and two chemoisomeric [2+4] reaction pathways may occur, respectively. When sulfonyl alkenes 1a and 1b act as diene, the regioselectivity of the cycloadducts are associated with the formation of the C1-C5 and C4-C6 single bond, para (denoted p), and the formation of the C1-C6 and C4-C5 single bond, meta (denoted m). On the other hand, a significant difference in electron deficiency between the two double bonds of 1 can lead to the two possible chemoselectivity pathways through two approach modes of the C=C-SO2Ph (A) and C=C-R (B) of 1 (as dienophile) with butadiene 2 (Scheme 2). For each pathway, the two stereoisomeric approach modes of the vinyl group of dienophiles relative to dienes produced two stereoisomeric, endo (e) and exo (x). Therefore, along the endo pathway, the vinyl group is placed over the double bond of diene framework. Also, the stereoselectivity in the [2+4] and [4+2] reaction pathways are considered with respect to the sulphonyl and vinyl groups, respectively.

The possible reaction pathways for the cycloaddition reactions of 2-phenylsulfonyl-1,3-butadiene, 1a, and 2-phenylsulfonyl-3-phenylsulfanyl-1,3-butadiene, 1b, with 2,3-dimethyl-1,3-butadiene 2
Moreover, in order to obtain the best results, a conformational analysis was performed on the reactants. The calculation results indicated that the sulfonyl alkenes 1a and 1b can approach to the dienes through two possible conformers; s-trans and s-cis, which the (E)-1a and (E)-1b are more stable than (Z)-1a and (Z)-1b. The energy barriers for these processes are 3 and 2 kcalmol−1, respectively. However, both isomers (E)- and (Z) are formed in the process, and the (E)-isomer is a much more reactive electrophile than the (Z)-isomer in the [2+4] cycloaddition reaction. Moreover, the conformational analysis of the 1,3-butadiene 2 indicated two conformations, s-cis and s-trans, of which due to the steric effect of the methyl substituents the s-trans conformer is preferred to the s-cis conformer in the [4+2] cycloaddition reaction.
Firstly, we investigated the selectivity of the [4+2] and [2+4] cycloaddition reactions of the sulfonyl alkenes 1a (R=H) and 1b (R=SPh) with butadiene, 2. When the sulfonyl alkene, 1, acts as diene, it can take four possible approaches, para, meta, endo, and exo to butadiene 2 and can proceed via four transition states, TS1-pe, TS1-px, TS1-me, and TS1-mx, respectively. Moreover, when the sulfonyl alkenes, 1, approach to the dienic system as dienophile, four cycloadducts of 4-Ae, 4-Ax, 4-Be, and 4-Bx can be produced through TS2-Ae, TS2-Ax, TS2-Be, and TS2-Bx, respectively. The stationary points corresponding to these cycloaddition reactions are presented in Scheme 2, together with the atom numbering, and the relative energies are summarized in Table 1 and Table S1. Thus, as can be seen in Table 1, the most stable regio- and chemoselectivity pathways can take place through exo approaches of dienophiles to dienes. It is clear that the formation of exo cycloadducts with a lower energy barrier than the endo one originates from the reduced steric effect of the SO2Ph with the methyl group at TSs.
The calculated energy results in Table 1 indicated that for the [4+2] cycloaddition reaction of 1a+2, the energy barrier of the para/exo approach mode (TS1a-px) are lower than for the meta/exo ones (TS1a-mx) by 3.47 kcal mol−1. Moreover, the presence of the phenyl sulfanyl group on 1b cannot change the regioselectivity, and so the para/exo approach mode (TS1b-px) is more stable than the meta ones by 2.7 kcal mol-1. It can be concluded that the phenyl sulfonyl group (SO2Ph) is a much more powerful directing element than the phenyl sulfanyl group (SPh) for the control of the regioselectivity of these reactions. Moreover, in the [2+4] cycloaddition reactions of 1b+2, the relative energy of the TS2b-Ax at 6.7 kcal mol−1 is lower than that of TS2b-Bx at 9.7 kcal mol−1, and TS2a-Ax at 7.8 kcal mol−1 is more stable than TS2a-Bx at 12.1 kcal mol−1 in the reaction of 1a+2. Based on the relative energies, the electron-withdrawing nature of the sulfonyl group results in a considerable difference in the dienophilicity of the two double bonds of 1 so that the double bond directly attached to the sulfonyl group is more reactive as a dienophile.
Comparing the two regioselective and two chemoselective pathways of these reactions, it can be concluded that the formation of 4a-Ax and 4b-Ax via the TS2a-Ax and TS2b-Ax, respectively, are the most favorable reaction pathways. These observations are in good agreement with the experimental results of 1a+2, in which the stereoisomer of 4a-Ax has the highest yield [12], and also it can be concluded that the 4b-Ax can produced through the suggestion cycloaddition reaction with acceptable yield. All of the processes are strongly exothermic with ∆Er between −28.1 and −38.3 kcal mol−1, so they can be considered irreversible. Therefore, a comparison of the relative energies shows that the [2+4] pathways are more favorable than the [4+2] pathways, both thermodynamically and kinetically.
Moreover, the values of thermodynamic parameters associated with the four reactive pathways are given in Table 1 and Table S1 in the supporting information. Analysis of the activation Gibbs free energies and the activation enthalpies shows a preference of the [2+4] pathway, in complete agreement with the calculated activation energy barriers.
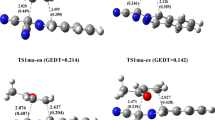
The comparison between the lengths of the two forming bonds at the TSs reveals that the para TSs are more asynchronous than meta ones. Accordingly, the most asynchronous transition state for the formation of the [2+4] cycloadducts is TS2b-Bx, with the lengths of the two forming bonds of C3-C5 (2.598 Å) and C4-C8 (1.994 Å) (Fig. 1). The extent of bond formation along reaction pathways is also provided by the concept of bond order (BO) [23]. Analysis of the C–C BO values indicates that the steric effect of the substituents in TSs delayed the formation of the C1-C6, C4-C6, C2-C8, and C3-C5 bonds relative to C4-C5, C1-C5, C1-C5, and C4-C8 bonds, respectively. The BO values validate the main conclusions, which are obtained from the analysis performed on the geometrical parameters.

The geometry optimized transition states for the [4+2] and [2+4] cycloaddition reactions of 2-phenylsulfonyl-1,3-butadiene, 1a, and 2-phenylsulfonyl-3-phenylsulfanyl-1,3-butadiene, 1b, with 2,3-dimethyl-1,3-butadiene, 2, along the most stable pathways at the MPWB1K/cc-pVDZ level of theory. Bond distances are given in Å, Wiberg bond indices are given in parentheses, and the GEDT of TSs are also given (for a full comparison of geometries, see supporting information)
Moreover, the electronic nature of these reactions is evaluated by computing the global electron density transfer (GEDT) [21, 22] at the TSs associated with the four reactive pathways. Reactions with GEDT values of 0.0e correspond to nonpolar processes, while values higher than 0.2e correspond to polar processes [21, 22]. As can be seen in Fig. 1, the GEDT values computed at the most stable pathways varied from 0.035 to 0.227 e. These values indicate that the processes have slightly polar character and the polarity of the preferred exo pathways is higher than the endo ones.
In addition, the energy calculations at MPWB1K/cc-pVDZ level of theory were utilized to study the solvent effect on the regio-, chemo-, and stereoselectivity of these reactions. As shown in Tables S3 and S4, inclusion of solvent effects of benzene in the geometry optimization does not produce appreciable changes in the gas-phase energies and does not change the low selectivity obtained in the gas phase. This convinces us that the accuracy of the calculations in the gas phase is enough to address the selectivity and mechanistic details.
Mechanistic study of the competitive reaction paths associated with cycloaddition reactions of 2-phenylsulfonyl-1,3-butadiene, 1a, and, 2-phenylsulfonyl-3-phenylsulfanyl-1,3-butadiene, 1b, with cyclopentadiene 5
In this section, an exhaustive exploration of the cycloaddition reactions of sulfonyl alkenes 1a and 1b with cyclic conjugated diene 5 allowed us to find several reactive paths associated with the formation of formal [4+2] and [2+4] CAs (Scheme 3). The approach of the sulfonyl dienes 1 to 5 could lead to eight reactive pathways; two regioisomeric (p/m) and two chemoisomeric (A/B) possibilities, with endo/exo approaches for each pathway named in the standard way based on the orientation of the dienophile with respect to the diene system.

The possible reaction pathways for the cycloaddition reactions of 2-phenylsulfonyl-1,3-butadiene, 1a, and 2-phenylsulfonyl-3-phenylsulfanyl-1,3-butadiene, 1b, with cyclopentadiene 5
Therefore, eight cycloadducts, 6-me, 6-mx, 6-pe, 6-px, 7-Ae, 7-Ax, 7-Be, and 7-Bx, and their corresponding TSs, TS3-me, TS3-mx, TS3-pe, TS3-px, TS4-Ae, TS4-Ax, TS4-Be, and TS4-Bx, were considered for each reaction of 1a+5 and 1b+5. The activation and relative energies are given in Table 2 and Table S2 in the supporting information, and the geometries of TSs are shown in Fig. 2 and Fig. S1 in the supporting information.

The optimized geometry of the transition states of [4+2] and [2+4] cycloaddition reactions of 2-phenylsulfonyl-1,3-butadiene, 1a, and 2-phenylsulfonyl-3-phenylsulfanyl-1,3-butadiene, 1b, with cyclopentadiene, 5, along the most stable pathways at the MPWB1K/cc-pVDZ level of theory. Bond distances are given in Å, Wiberg bond indices are given in parentheses, and the GEDT of TSs are also given (for a full comparison of geometries, see supporting information)
Firstly, the [4+2] cycloaddition reaction of sulfonyl alkenes 1 with cyclopentadiene 5 is investigated. As shown in Scheme 2, sulfonyl alkene 1 can act as a diene via two regioselective, para and meta, and two stereoselective, endo and exo, pathways. The relative energy results in Table 2 indicate that the reaction pathways are completely para regioselective, as para transition states lie lower in energy than the corresponding meta ones. Moreover, the cycloadducts formed from the exo approach in para pathways are more stable than that of formed from the endo ones. It can be concluded that the sulfonyl group is an effective directing element favoring the formation of “para” adducts. Additionally, the presence of the –SPh group in 1b causes the stereoselectivity of the para pathways to be increased for the cycloaddition reaction of 1b with 5 relatives to 1a with 5 (ΔΔE#1a/5=2.3 kcal mol−1, ΔΔE#1b/5=3.8 kcal mol−1). Therefore, the para/exo (px) cycloadducts of 6a-px and 6b-px, with relative energies of −30.88 and −32.06 kcal mol−1, are more favorable than meta ones.
Secondly, the [2+4] cycloaddition reaction of 1 with cyclopentadiene 5 is studied through two chemoselective and two stereoselective pathways (Scheme 3). Therefore, for these cycloaddition reaction pathways, four TSs, TS4-Ae, TS4-Ax, TS4-Be, and TS4-Bx, and four cycloadducts, 7-Ae, 7-Ax, 7-Be, and 7-Bx, are considered. The relative energies in Tables 1 and 2 show that these cycloaddition reactions present a total chemoselectivity, as TS4a-A and TS4b-A are lower in energy than TS4a-B and TS4b-B. Therefore, we can conclude that the C-C double bond directly attached to the sulfonyl group of 1 (C-C-SO2Ph) is more reactive than C-C-R double bond as the dienophilic part. The significant difference in electron deficiency between the two C-C double bonds of 1 can affect the reactivity of these C-C double bonds. Moreover, two stereoselective pathways, namely, endo and exo, can be observed for each chemoselective pathway. Therefore, comparing the most stable pathways, while TS4b-Ae has a higher activation energy than TS4b-Ax by 2.0 kcal mol−1, TS4a-Ae is more stable than TS4a-Ax by 1.9 kcalmol−1. Indeed, the presence of SPh group on 1 can change the stereoselectivity from endo to exo, which is probably due to the steric effect and secondary orbital overlap.
An analysis of the energetic results of the [2+4] and [4+2] cycloaddition reactions indicated that the [2+4] reaction of 1a+5 is in terms of the kinetic stability and the major product is 7a-A, but 6a-p is also present. On the other hand, the [4+2] cycloaddition reaction of 1a+5 is in terms of the thermodynamic stability, which is in agreement with the experimental results (at 130°C), and the reaction becomes reversible with the only 6a-p product.
Moreover, the thermodynamic parameters for these cycloaddition reactions are calculated and given in Table 2 and Table S2 in the supporting information. Accordingly, the activation enthalpies of the [2+4] pathways are the lower than other pathways, in line with the calculated trends of the calculated activation energies.
The geometries of all TSs in these cycloaddition reactions are shown in Fig. 2 and Fig. S2 in the supporting information. An analysis of the lengths of the two forming bonds and BO values at the TSs of the processes reveals that the most asynchronicity is observed for the TSs associated with the formation of the [4+2] cycloadducts. Moreover, the bond order values indicate that the formation of the C1-C9 bonds at the most favorable TSs, TS4a-Ax and TS4b-Ae, is more advanced than the C2-C12 bond, which may be due to the steric effect of SO2Ph and SPh groups.
Finally, in order to evaluate the polar nature of the reactions of 1 with 5, the GEDT at the more favorable TSs was analyzed. In the [4+2] cyclization reactions of 1 with 5, the GEDT values at the most favorable TS3a-px and TS3b-px, which fluxes from 5 to 1, are 0.192 and 0.174 e, respectively. Also, in the [2+4] cyclization reactions of 1 with 5, the GEDT values at the most favorable TS4a-Ae and TS4b-Ax are 0.205 and 0.215 e, respectively. These values indicate that the exo and A pathways are the most polar in character and are in agreement with the computed low activation energies. Also, a comparison between the GEDT values was associated with the reactive pathways of 1 with 2, and 5 indicates that the polarity of the [2+4] cycloaddition reactions of sulfonyl alkenes 1with 1,3-butadiene 2 is the highest.
Analysis of the global reactivity indices
Numerous studies devoted to DA reactions have shown that analysis of the reactivity indices defined within condensed DFT (CDFT) [33,34,35] is a powerful tool to understand organic chemical reactivity. Consequently, in order to characterize the reactivity of 2-sulfonyl-1,3-butadiene, 1a, and 2-sulfonyl-3-sulfanyl-1,3-butadiene, 1b, with 2,3-dimethyl butadiene, 2, and cyclopentadiene, 5, in the cycloaddition reactions, the global reactivity indices are analyzed and reported in Table 3. The global reactivity indices include electronic chemical potential, μ; chemical hardness, η; global electrophilicity, ω; and global nucleophilicity, N.
As can be seen from Table 3, the electronic chemical potentials of (Z)-1a, μ=−4.27 eV; (E)-1a, μ=−4.21 eV; (Z)-1b, μ=−3.69 eV; and (E)-1b, μ=−3.76 eV are lower than (Z)-2, μ=−3.12 eV; (E)-2, μ=−3.30eV; and 5, μ=−3.01 eV. Therefore, the GEDT for these cycloaddition reactions will take place from the studied alkenes, 2 and 5, to the 2-sulfonyl diene, 1.
According to the global electrophilicity, ω, and global nucleophilicity, N, 2-phenylsulfonyl dienes, (Z)-1a (ω=1.65, N=2.09 eV) and (E)-1a (ω=1.66, N=2.24 eV), are classified as strong electrophiles and moderate nucleophiles based on the electrophilicity [36] and nucleophilicity scale [37]. Also, 2-phenylsulfonyl-3-phenylsulfanyl diene, (Z)-1b with ω=1.44 and N=3.07 eV and (E)-1b with ω=1.54 and N=3.07 eV, are classified as moderate electrophiles and strong nucleophiles. The higher nucleophilicity index of 1b relative to 1a can be due to the presence of the sulfanyl group (SPh) in 1b. Moreover, (Z)-2, (E)-2, and 5 with marginal electrophilicity values, ω=0.81, 0.96, and 0.83 eV, and high nucleophilicity values, N=2.98, 3.00 and 3.37 eV, respectively, are classified as marginal electrophiles and strong nucleophiles in the electrophilicity and nucleophilicity scales.
Moreover, from the CDFT analysis performed in this section, we can conclude that the highly electrophilic character of 1a and 1b and the high nucleophilic activation of 2 and 5 point to polar character and, consequently, low activation energy for these cycloaddition reactions.
Prediction of the chemoselectivity of the studied cycloaddition reactions using Parr functions and local reactivity indices
The regio- and chemoselectivity of these reactions has also studied through the DFT-based reactivity descriptors, such as Parr functions, local electrophilicity, and nucleophilicity indices [29]. For the polar reactions, the most favorable reactive pathways involve the initial interaction between the most electrophilic center with the most nucleophilic center of the corresponding reactants [27, 28]. Domingo et al. proposed the electrophilic, Pk+, and the nucleophilic, Pk–, Parr functions, which are obtained from the ASD distribution in the radical anions and the radical cations of the reactants [27, 28, 31].
Therefore, a simple analysis of the Parr functions and ASD allows us to characterize the most electrophilic and the most nucleophilic centers in the reactants and to study the chemoselectivity of the studied reactions. Accordingly, the electrophilic, Pk+, Parr functions of 1a and 1b, based on the ASD in the radical anion, and the nucleophilic, Pk–, Parr functions of butadiene 2 and cyclopentadiene 5, based on the ASD in the radical cation, are analyzed (see Fig. 3 and Table 4). While the electrophilic Parr functions of 1a and 1b are mainly concentrated at C1 (Parr functions of 0.41–0.45), C4 also present considerable electrophilic Parr functions of 0.150, respectively. The electrophilic Parr functions of C2 are also higher than C3. Therefore, the C1 and C2 of the C=C-SO2Ph framework have been more electrophilically activated than the C3 and C4 of the C=C-R framework. These behaviors indicated that the reactivity of the C1=C2-SO2Ph framework of 1 toward diene 2 is higher than the C=C-R framework which can explain the observed chemoselectivity of these reactions.

Maps of ASD of the radical anion and the local electrophilic Parr function of 1a and 1b and ASD of the radical cation and the local nucleophilic Parr function of 2 and 5
The nucleophilicity Parr function and analysis of the ASD of the radical cations of (Z)-2 and (E)-2 indicated that spin density is located mainly at C5 with \( {P}_{C5}^{-} \)=0.45 and 0.49, respectively, which will be preferred position for a nucleophilic attack to the C1 atom of 1. Therefore, the most favorable electrophile–nucleophile interaction along the nucleophilic attack of 2 on 1a and 1b will take place between the C1 atom of 1a,b and the C5 atom of 2 leading to the formation of the most stable regioisomer of 3-px.
For the cycloaddition reactions of 1a and 1b with 5, the most favorable interaction occurs between the C1 atom of 1 and C9 (or C12) atom of 5 (PC9–=0.47, NC9=1.58), which indicates that the C1-C9 bond formation is more advanced than the C4-C9 one. This behavior is similar to that found in analysis of the ASD of the radical cations of 5. Analysis of the Parr functions and ASD for the cycloaddition reactions of 1+5 can explain the source of the regio- and chemoselectivities observed.
ELF analysis of the [2+4] cycloaddition reaction between 2-phenylsulfonyl-1,3-butadiene and cyclopentadiene
An appropriate tool to study the molecular mechanism of reactions is the ELF analysis along the reaction path [13,14,15]. The maximum probability of finding electron pairs, classified as core and valence basins, can result from the ELF analysis. Monosynaptic and disynaptic basins characterize valence basins which involve single and bonding pairs, respectively [38, 39]. The mechanism of the [2+4] cycloaddition reaction between 1a and 5 and C-C bond formations along this reaction have studied using the ELF analysis of the selected structures in the IRC curve of TS4a-Ae at MPWB1K/cc-pVDZ level of theory [40]. The related ELF valence attractors of the most important points together with their populations are shown in Fig. 4.


Schematic representation of the ELF attractors of selected points of IRC path of the most favored pathway of CA reaction of 2-phenylsulfonyl-1,3-butadienes 1a and cyclopentadiene 5
ELF topological analysis of the first point of IRC map show two disynaptic basins V(C3, C4) and V′(C3, C4) in 1a fragment with a total population of 3.4e, and a disynaptic basin V (C10, C11) integrating 2.27e as well as two pairs of disynaptic basins V(C9, C10) and V′(C9, C10) and V(C11, C12) and V´(C11, C12) in 5 fragment with a total population of 3.28e. However, at the C4-C9 distance of 2.09Å (and C3-C12, 2.53 Å), three disynaptic basins V(C3, C4), V(C9, C10), and V(C11, C12) have been observed related to the double bonds of 1a and 5. As shown in Fig. 4, the population associated with V(C3, C4), V(C9, C10), and V(C11, C12) in TS4-Ae decrease, and three monosynaptic basins V(C4), V(C9), and V(C3) emerge with the population of 0.23e, 0.33e, and 0.40e, respectively. In Fig. 4 for points after TS4a-Ae and at a C4-C9 distance of 1.93 Å, two monosynaptic basins V(C4) and V(C9) merge to a disynaptic basin V(C4, C9) with the population of 0.91e where the first single bond has formed. At this point, the population of V(C10, C11) and V(C3) increased to 2.87e and 0.54e, respectively.
At C3–C12 distance of 2.28 Å, the second monosynaptic basin required to form another single bond, V(C12), has appeared with a population of 0.20e. At this point, the population of V(C3), V(C4,C9), and V(C10, C11) increase to 0.76e, 1.41e, and 3.15e, whereas the population of V(C3,C4), V(C9,C10), and V(C11, C12) decrease to 2.26e, 2.20e, and 2.44e. Finally, bicyclic compound has formed by merging two pseudoradical centers V(C3) and V(C12) to create disynaptic basin V(C3,C12) with population of 1.14e at C3–C12 distance of 2.17 Å. However, the population of V(C3, C12) and V(C4,C9) increase to 1.83e and 1.77e at 7-Ae. According to ELF results, the reaction has a one-step two-stage mechanism [40].
Conclusion
The CDA reaction of sulfonyl diene 1a with alkenes 2 and 5 has been carried out in order to explain the experimental outcomes observed by Chou et al. through DFT calculations at the MPWB1K/cc-pVDZ computational level. In addition the cycloaddition reaction of sulfonyl diene 1b has been also analyzed in order to investigate the role of the SPh of 1b in these reactions.
It is known that in the cycloaddition reactions of unsymmetrical 1 with the studied alkenes, up to 16 competitive reaction paths are feasible. The chemo-, regio-, and stereoisomeric reaction pathways involving the two C-C double bonds of sulfonyl dienes 1 have studied. The chemoselectivity results revealed that the significant difference in electron deficiency makes the double bond attached to the sulfonyl group a more reactive dienophile than the other double bond.
Analysis of the relative energies and thermodynamic parameters indicates that these cycloaddition reactions are completely chemo- and stereoselective and take place via a polar and asynchronous process. In the studied CDA reactions of 1 with 2 and 5, the most stable pathways are related to the [2+4] reactions in terms of the kinetic stability. The substituent effect on the regioselectivity indicated that the phenyl sulfanyl group (SPh) is a much more powerful directing element than the phenyl sulfonyl group (SO2Ph) for the control of the regioselectivity of reactions.
Analysis of the reactivity indices shows that 2-sulfonyl dienes 1a and 1b present a strong electrophilic character and studied alkenes 2 and 5 have a strong nucleophilic character, explaining the polar character of these cycloaddition reactions, which is established by analysis of the GEDT computed at the TS of the reactions. The ELF analysis revealed that the [2+4] cycloaddition reaction between 1a and 5 proceeds through a two-stage one-step mechanism.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Sauer J, Sustman R (1980) Mechanistic aspects of Diels-Alder reactions: a critical survey. Angew Chem Int Ed Engl 19:779–807
Boger DL, Mullican MD (1984) Inverse electron demand Diels-Alder reactions of 3-carbomethoxy-2-pyrones controlled introduction of oxygenated aromatics: benzene, phenol, catechol, resorcinol, and pyrogallol annulation regiospecific total synthesis of sendaverine and a preparation of 6, 7-benzomorphans. J Org Chem 49:4033–4044
Boger DL, Panek JS (1985) Inverse electron demand Diels-Alder reactions of heterocyclic azadienes: formal total synthesis of streptonigrin. J Am Chem Soc 107:5745–5754
Andell O, Bäckvall JE (1985) Sulfonylmercuration of conjugated dienes A facile route to allyl-and dienyl-sulfones. Tetrahedron Lett 26:4555–4558
Hardinger SA, Fuchs PL (1987) Synthesis of vinyl sulfones 23 Addition of organometallic reagents to cyclooctenyl phenyl sulfones. J Org Chem 52:2739–2749
Bäckvall JE, Plobeck N, Juntunen SK (1989) Facile cycloaddition of 2-phenylsulfonyl 1, 3-dienes to indoles. Tetrahedron Lett 30:2589–2592
Padwa A, Norman BH (1988) Intramolecular cycloaddition reactions of oximes with vinyl sulfones. Tetrahedron Iett 29:2417–2419
Bäckvall JE, Juntunen SK (1987) 2-(Phenylsulfonyl)-1, 3-dienes as versatile synthons in organic transformations Multicoupling reagents and Diels-Alder dienes with a dual electron demand. J Am Chem Soc 109:6396–6403 and references therein
Cuvigny T, Hervé du Penhoat C, Julia M (1986) Syntheses with sulfones XLVI: stereoselective preparation of 2-benzenesulfonyl-1, 3-dienes and 2-benzenesulfonyl-1, 4-dienes. Tetrahedron 42:5329–5336
Inomata K, Kinoshita H, Takemoto H, Murata Y, Kotake H (1978) Preparation and Diels-Alder reactions of 3-substituted 3-sulfolenes. Bull Chem Soc Jpn 51:3341–3344
Bäckvall JE, Juntunen KS (1988) 2-(Phenylsulfonyl)-1, 3-dienes as versatile synthons in organic transformations Functionalizations via epoxidation reactions. J Org Chem 53:2398–2400
Chou T-S, Hung S-C (1988) Selective cross Diels-Alder reactions of 2-(phenylsulfonyl) 1, 3-dienes. J Org Chem 53:3020–3027
Andrés J, González-Navarrete P, Safont VS (2014) Unraveling reaction mechanisms by means of quantum chemical topology analysis. Int J Quantum Chem 114:1239–1252
Andres J, Berski S, Domingo LR, Polo V, Silvi B (2011) Describing the molecular mechanism of organic reactions by using topological analysis of electronic localization function. Curr Org Chem 15:3566–3575
Polo V, Andres J, Berski S, Domingo LR, Silvi B (2008) Understanding reaction mechanisms in organic chemistry from catastrophe theory applied to the electron localization function topology. J Phys Chem A 112:7128–7136
Zhao Y, Truhlar GD (2004) Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: the MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J Phys Chem A 108:6908–6918
Gonzalez C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113:6378–6396
Cances E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107:3032–3041
Hehre WJ, Radom L, Schleyer PvR, Pople JA (1986) Ab initio molecular orbital theory Wiley New York
Domingo LR (2014) A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4:32415–32428
Domingo LR, Rios-Gutiérrez M, Pérez P (2016) A new model for C–C bond formation processes derived from the Molecular Electron-Density. Tetrahedron 72:1524–1532
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735–746
Parr RG, Liu LV, Szentpaly S (1999) Electrophilicity index. J Am Chem Soc 121:1922–1924
Parr RG, Yang W (1989) Density functional theory of atoms and molecules. Oxford University Press, Oxford
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516
Domingo LR, Chamorro E, Pérez P (2008) Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions A theoretical study. J Org Chem 73:4615–4624
Domingo LR, Pérez LP (2011) The nucleophilicity N index in organic chemistry. Org Biomol Chem 9:7168–7175
Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140:1133–1138
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery, JAJr, Start-mann RE, Burant,JC, Daprich S, Millam JM, Daniels AD,. Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochtersk Ji, Petersson GA, Ayala Y, Ui QC, Morokuma K, Malick DK, Rubuck AD, Raghavachari K, Foresman JB, Cioslowski J, Oritz JV, Stefanov BB, . Liu G, Liashenko A, Piskorz P, Komaromi I, Comperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Gonzalez C, Challa-combe M, Gill MW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople J.A, Gaussian 09, revision A.02 Gaussian, Inc, Wallingford CT, 2009
Domingo LR, Pérez P, Sáez JA (2013) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3:1486–1494
Noury S, Krokidis X, Fuster F, Silvi B (1999) Computational tools for the electron localization function topological analysis. Comput Chem 23:597–604
Parr RG, Yang W (1995) Density-functional theory of the electronic structure of molecules. Annu Rev Phys Chem 46:701–728
Chermette H (1999) Chemical reactivity indexes in density functional theory. J Comput Chem 20:129–154
Ess DH, Jones GO, Houk KN (2006) Conceptual, qualitative, and quantitative theories of 1, 3-dipolar and diels–alder cycloadditions used in synthesis. Adv Synth Catal 348:2337–2361
Domingo LR, Aurell MJ, Pérez PJ, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 58:4417–4423
Jaramillo P, Domingo LR, Chamorro E, Pérez PJ (2008) A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J Mol Struct 865:68–72
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371:683–686
Silvi B (2002) The synaptic order: a key concept to understand multicenter bonding. J Mol Struct 614:3–10
Domingo LR, Saéz JA, Zaragozá RJ, Arnó M (2008) Understanding the participation of quadricyclane as nucleophile in polar [2σ+2σ+2π] cycloadditions toward electrophilic π molecules. J Org Chem 73:8791–8799
Acknowledgements
The authors wish to acknowledge Dr. Louise S. Price, University College London, UK, for reading the manuscript and providing valuable suggestions.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Calculations and optimization of structures, data collection, and analysis were performed by Soheyla Heydari, Mina Haghdadi, and Mahshid Hamzehloueian. Mina Haghdadi and Hassan Ghasemnejad Bosra wrote the first draft of the manuscript, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
The paper is not currently being considered for publication elsewhere. The paper reflects the authors’ own research and analysis in a truthful and complete manner. No data, text, or theories by others are presented as if they were the author’s own.
Consent to participate
All authors consent to participate in the research project, and the following has been explained to us: the research may not be of direct benefit to us. My participation is voluntary.
Consent for publication
All authors approved the version to be published. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of interest
The authors declare no competing interests.
Additional information
Ethical responsibilities of authors:
We warranty that this manuscript is original, has been written by the stated authors, and has not been published elsewhere; the manuscript has not been submitted to more than one journal for simultaneous consideration. We wish to confirm that it has not been published previously (partly or in full). This study is not split up into several parts. We confirm that no data have been fabricated or manipulated. No data, text, or theories by others are presented as if they were the authors own. This manuscript contains no libelous or other unlawful statements and does not contain any materials that violate any personal or proprietary rights of any other person or entity.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 12654 kb)
Rights and permissions
About this article

Cite this article
Heydari, S., Haghdadi, M., Hamzehloueian, M. et al. An investigation of the regio-, chemo-, and stereoselectivity of cycloaddition reactions of 2-phenylsulfonyl-1,3-butadiene and its 3-phenylsulfanyl derivative: a DFT study. Struct Chem 32, 1819–1831 (2021). https://doi.org/10.1007/s11224-021-01758-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-021-01758-2