
Abstract
Recently, adenosine A2A receptor antagonists have been identified as an interesting drug target for the treatment of Parkinson’s disease (PD). Radiolabelled molecular imaging technologies such as positron emission tomography (PET) have emerged in the research field of medicinal chemistry as a diagnostic tool for PD. In the current study, we have performed quantitative structure–activity relationship (QSAR) analysis of 35 xanthine ligand PET tracers as A2AR (adenosine receptors) antagonists in order to determine their structural features required to have binding affinity and selectivity towards A2AR. The division of the dataset into training and test sets was done using a random method, while the feature selection for the binding affinity was done using Genetic Algorithm (GA). The best model with five descriptors was obtained using the spline option in the GA run. QSAR models with four descriptors were also developed for A2AR selectivity, where significant descriptors were selected from the large pool of descriptors using stepwise regression method followed by Best Subset Selection (BSS) method. Furthermore, to improve the quality of the external predictions, we used the “Intelligent Consensus Predictor” tool (http://teqip.jdvu.ac.in/QSAR_Tools/DTCLab/). Both the models showed robustness in terms of statistical parameters. Molecular docking studies have been carried out to understand the molecular interactions between the ligand and receptor, and the results are then correlated with the structural features obtained from the QSAR models. Furthermore, the information derived from the newly found descriptors gives an insight for the development of new candidate PET tracers for the use in PD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder of the central nervous system characterized by muscle rigidity, bradykinesia, and tremor. The disease affects older people, and it is known that 2–3% of the population ≥ 65 years of age are more prone towards this disease [1]. It is also associated with loss of dopaminergic neurons in the substantia nigra, lewy body generation, and abnormal clustering of α-synuclein protein, which is directly connected to expectancy of long life. Hence, effective research for neurodegenerative disease treatment is one of the vital clinical needs of today’s life. The current therapy of PD includes restoration of dopamine with levodopa in the striatum of the brain. However, to maintain the therapeutic level, the dosage has to be increased which does not prevent the underlying neuronal loss [2, 3]. On the other hand, such long-term treatment in addition may cause adverse effects which include levodopa-induced dyskinesia and behavioral disturbances in the individuals [4, 5].
Adenosine enzyme inhibitors can be considered an alternative medication in the treatment of PD having less degree of adverse effects. Adenosine is an endogenous modulator of different physiological functions in the peripheral tissues in addition to the central nervous system (CNS). It is a purine nucleoside having four varieties of subtypes consisting of A1, A2A, A2B, and A3. A2A receptors are highly expressed in striatum (dopamine-rich areas of the CNS) where it is almost co-located with dopamine D2 receptor on GABAergic striatopallidal neurons [6]. A2A antagonistically interferes with the D2 receptor and as a result decreases the affinity of the D2 receptor for dopamine upon stimulation and show opposite effect on motor function [7]. Thus, adenosine A2A receptor blockade may show motoric improvement as proven by many animal models [8,9,10,11].
Positron emission tomography (PET) [12] and single-photon emission computed tomography (SPECT) [13] are non-invasive methodologies which make use of the dynamic distribution of the radiotracers and provide 3D map of the brain quantifying the biological processes. These imaging agents help in the detection and quantification of dopamine and adenosine receptors in the brain thereby creating a path for early detection of the disease. PET studies are superior to SPECT in terms of accurate results and in determining the temporal measurements of radioactivity with their regional distributions. Agonists and antagonists containing positron-emitting radioisotopes can be introduced in vivo to get 3D image of the receptors which have been helpful in CNS diagnosis. The PET tracers can be used as in vivo–imaging agents in order to improve the pharmacokinetics, physicochemical properties, and mapping of the receptor as per interest. As search of new compounds with desired activity is time-consuming and expensive, pharmaceutical companies have a great interest upon theoretical approaches to design compounds with desired activity.
Quantitative structure–activity relationships (QSAR) have gained a lot of attention in molecular modeling field and are beneficial due to less involvement of human resource and cost-effectiveness [14, 15]. It attempts to develop a correlation between the chemical structures with a well-defined activity. It expresses chemical structures and physiological property in the numerical form and develops a mathematical correlation between them. Furthermore, this relationship can be used to predict the biological response of other existing chemical structures. QSAR-based studies have shown useful applications in drug discovery, molecular modeling, pharmaceutical toxicity modeling, pharmacokinetics/toxicokinetics, data mining, environmental toxicity (ecotoxicity), chemical or drug property modeling, food science, agricultural sciences, pesticide toxicity, fragrance, nanoscience (Nano-QSAR), and many other fields [16,17,18,19,20,21,22,23,24]. QSAR is also used to predict the absorption, distribution, metabolism, excretion, and toxicological (ADMET) of drug like compounds [25, 26]. QSAR has widespread applications in drug design, medicinal chemistry, and predictive toxicology. It has also become an effective tool in understanding and determining the major biochemical features associated with the Parkinson’s disease [27, 28].
In the present study, we have tried to develop QSAR models with PET tracers of xanthine ligands as A2AR (adenosine receptors) antagonist using only 2D descriptors to explore the structural features required for binding affinity towards A2AR and selectivity of the tracers between A2B and A2A receptors.

Observed vs predicted A2AR binding affinity scatter plot
Materials and methods
Dataset
The experimental binding affinity and selectivity data of 35 xanthine ligand–based PET tracers were taken from a previously published literature [29] and applied for QSAR modeling to determine the essential structural features needed for binding affinity and explore the structural requirements necessary to be present in the antagonists for selectivity towards A2A adenosine receptors. The experimental values of selectivity and binding affinity (Ki) ranged from 0.1–20 nM and 7.84–16,500 nM respectively, and the details are provided in Supplementary Material I (Table S1). The experimental values were converted into negative logarithm scale during modeling and were used as independent values. No compounds with binding affinity data were removed during modeling but some compounds (14, 32, 33, and 34) with no experimental selectivity values were eliminated during modeling. Here, the binding affinity and selectivity were separately used as endpoints or independent variables in modeling. The compounds for both the dataset were represented in the MarvinSketch software [30] with proper aromatization and addition of hydrogen bond as necessary.
Molecular descriptors
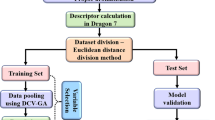
In the present study, QSAR models were developed using a selected class of two-dimensional molecular descriptors involving E-state indices, connectivity, constitutional, functional, 2D atom pairs, ring, atom-centered fragments, molecular property descriptors, and extended topochemical atom (ETA) indices. The ETA descriptors were calculated using the PaDel-Descriptor software [31], whereas the non-ETA descriptors were calculated using the Dragon 7 software [32]. Intercorrelated (|r| > .95), constant (variance < 0.0001), and other incompetent and redundant data was removed using an in-house software available at http://dtclab.webs.com/software-tools before model development.
Dataset division
Dataset division is a crucial part of QSAR modeling in order to develop a properly validated and robust model. Rational data division ensures an unbiased external validation along with uniform data distribution [33]. The division of the dataset into training set (~ 70%) and test set (~ 30%) was performed employing random dataset division method [34] for both binding affinity and selectivity end points. The training set was used for model development, and the test set was used for model validation.
Variable selection and model development
Prior to model development, we have performed variable selection strategies such as Genetic Algorithm (GA) [35, 36] and stepwise regression [35, 37] for binding affinity and selectivity, respectively, to extract the important and influential descriptors and created a reduced pool of descriptors. After obtaining the important descriptors, we went for model development. The best model with five descriptors was obtained using the spline option in the GA run on Discovery Studio version 4.1 for the binding affinity. On the other hand, for A2AR selectivity, four models with four descriptors were selected from the Best Subset Selection (BSS) method based on MAE criteria [38]. Furthermore, to improve the quality of the external prediction via “intelligent” selection of multiple models, we have applied an “Intelligent consensus predictor” tool [39] developed in our laboratory [40].
Statistical validation metrics
The statistical quality of the models developed in the present study was rigorously examined using multiple approaches to check the robustness and predictivity of the developed models. All the models were validated both externally and internally. Various parameters like determination coefficient R2, explained variance R2a, variance ratio (F), and standard error of estimate (s) were computed. Internal predictivity parameters such as predicted residual sum of squares (PRESS) and leave-one-out cross-validated correlation coefficient (Q2LOO) were also calculated along with external predictivity parameters like R2pred or \( {Q}_{F1}^2 \), \( {Q}_{F2}^2 \), and concordance correlation coefficient (CCC) [41]. It has been reported that consensus models are better in performance in comparison with an individual model [41]. Therefore, we have also performed “Intelligent Consensus Prediction (ICP)” using multiple models to see whether the quality of predictions can be increased through an intelligent selection.
Applicability domain
Applicability domain (AD) [42] is a theoretical region in the chemical space developed based on modeled descriptors and modeled response of the training set, where the developed model could make predictions basing on some logical reliability. Here, we have checked AD using standardization approach using the tool developed in our laboratory [40].
Molecular docking
Molecular docking analysis has been implemented in the present work that helps in understanding the intermolecular interactions taking place between the PET tracer antagonists and the A2A receptor. The protein structure for adenosine A2A receptor is retrieved from the protein data bank with PDB ID:3UZA [43]. The X-ray crystal structure of the protein consists of a bound ligand T4G commonly known as 6-(2,6-dimethylpyridin-4-yl)-5-phenyl-1,2,4-triazin-3-amine (formula: C16H15N5). Before docking the target PET tracers, protein preparation was done by cleaning the protein for any missing residues, explicit hydrogen addition, and generation of the docking site. The generation of active docking site was done in the BIOVIA Discovery Studio platform from the ligand-binding domain of the bound ligand T4G by the selection of the ligand and generating the site “from current selection” program in receptor-ligand interaction module of the software. After the generation of the active ligand-binding domain, the bound ligand was removed for new molecule docking. For ligand preparation, the PET tracers were put through small molecule module in the Discovery Studio platform where a series of ligand conformers were generated. Each of these generated conformers was then used in the CDOCKER module energy for molecular docking involving CHARMm interaction [44]. The CDOCKER interaction energy parameter (kJ/mol) was checked for all the receptor ligand complexes, and the top scoring (most negative, thus favorable to binding) poses were kept.
Results and discussion
Based on the binding affinity and selectivity endpoints of 35 xanthine PET tracer antagonists of adenosine A2A receptor, we have developed one model for the binding affinity (Q2 = 0.85, R2 = 0.90, Q2F1 = 0.80) and 4 models (Q2 = 0.80–0.87, R2 = 0.87–0.91, Q2F1 = 0.84–0.85) for selectivity. All the models were externally and internally validated which showed model robustness and good predictivity in terms of the statistical results. We have also checked the rm2 parameters for both internal sets (\( \overline{r_{m(loo)}^2},\varDelta {r}_{m(loo)}^2 \)) and external sets (\( {r}_{m(test)}^2 \) and\( \varDelta {r}_{m(test)}^2 \)), and the statistical results were above the critical point justifying the reliability of the models. To improve the quality of the external prediction for selectivity, we also performed “Intelligent Consensus Prediction” of the multiple MLR models using the ICP tool [39], and found that the consensus predictions were better than the individual MLR model–derived predictions. The winner model was consensus model 0 (CM0).
Modeling binding affinity of PET tracers towards adenosine (A2A) receptor
The model for binding affinity consists of five descriptors: C-025, F09 [N-O], nBnz, NRS, and nCIR which significantly influence the binding of the antagonists to the adenosine (A2A) receptor. The 5 descriptor MLR model (Eq. 1) developed using Genetic Function Algorithm (GFA) could predict 85.0% variance of the training set and 80.0% of the test set. The values of all descriptors appearing in the model for training and test set compounds are given in Supplementary Material II (Excel file) and the scatter plot of the observed vs. predicted binding affinity is shown in Fig. 1.
Essential features required for binding and receptor interaction
The descriptors obtained in the QSAR model (Table 1) give an insight regarding the mechanism of interaction occurring during binding of the xanthine PET tracer antagonists to adenosine A2A receptor. Unsaturation and aromaticity play a dominating role in regulating the receptor binding affinity which is evident from the occurrence of descriptors such as C-025, nBnz, NRS, and nCIR. Descriptors like nBnz and nCIR have positive influences on the adenosine A2A receptor binding (Fig. 2). But on the other hand, descriptors like C-025 and NRS have negative effects on the binding affinity of the PET tracers (Fig. 3). The occurrence of these similar types of descriptors with opposite influence is contradictory and leads to a conclusion that aromaticity provided by benzene nucleus (as seen in compounds like A-32 and A-23) is more important for binding. On the other hand, the presence of heterocyclic aromatic rings and fused-ring systems decrease the overall binding affinity of the radiotracer molecule (found in compounds A-1, A-2, and A-20).

Features increasing the binding affinity (pKi) value

Features decreasing the binding affinity (pKi) value
The 2D atom pair descriptor F09 [N-O] gives information about the electronegativity of the compounds, and the positive coefficient of the descriptor suggests that higher occurrence of nitrogen and oxygen at topological distance 9 would enhance binding affinity of the compounds as seen in compounds A-4 and A-32. It is found that the presence of electronegative atoms in the compounds or chemical structures can influence the binding to the receptor through hydrogen bonding [45].
Molecular docking
Molecular docking helped in understanding the optimized conformation of the complex between the imaging agent and A2A receptor and gave evidences related to the orientation of the imaging agents at the binding zone of the receptor. The major goal was to understand the molecular interactions taking place during radiotracer binding and correlate these findings with QSAR analysis. The docking analysis showed the predominance of different types of π bonding interactions and hydrogen-bonding interactions. In higher active compounds (Fig. 4) like A-4, A-8, and A-25 (pA2AR(BA) = 0.699, 1.000, and 0.398 respectively), the interaction forces include mainly hydrogen-bonding interactions (conventional hydrogen bond and carbon-hydrogen bond interaction), π interactions (π-cation, π-donor hydrogen, π-π stacked, π-π T-shaped, and π-alkyl). Other interactions include halogen and alkyl interaction in compound A-4 and salt bridge formation in compound A-8. Higher number of interacting residues supports the fact that these compounds have higher binding affinity. Compounds having binding affinity in the medium range (Fig. 5) like compound numbers A-14 and A-27 (pA2AR(BA) = − 0.301 and − 0.255 respectively) make less number of interactions with the adenosine receptor, but the type of interactions remains similar, i.e., π interactions and hydrogen-bonding interactions. The lowest active compounds (Fig. 5) like compound numbers A-20 and A-35 (pA2AR(BA) = − 1.301and − 1.204 respectively) show the least number of interactions. All the details of binding including interacting residues and type of binding interactions are given in Table 2.

Docking interactions for compounds having higher binding affinity (pKi)

Docking interactions for compounds having medium (A-14) and low (A-35) binding affinity (pKi)
Relationship with QSAR models
The docking study shows different types of π interactions occurring between the PET radiotracer molecules and adenosine A2A receptor. This observation supports the occurrence of nBnz and nCIR descriptors obtained in the QSAR models. The presence of aromatic rings like benzene can enhance binding with the receptor through aromatic π-π stacking interaction with the phenyl/imidazole residue of the receptor [46]. The interaction of these antagonists through π-π stacking interaction eventually blocks the receptor in the indirect pathway thus blocking the activity of GABA-mediated influence in the globus pallidus pars externa (GPe). This helps the PD patients to gain the motor function again by regaining the balance between direct and indirect pathway. Nitrogen and oxygen are capable of hydrogen bond formation and various types of hydrogen bonding as observed in both higher active and lower active compounds, and this can be also correlated to the F09[N-O] descriptor which gives an idea about the electronegativity of the molecule.
Modeling selectivity of PET tracers towards adenosine (A2A) receptor
In the current work, we have developed four MLR models to understand the selectivity of the PET tracer molecules towards adenosine A2A receptor. A single QSAR model may not be efficient enough for the prediction of activity since the property of molecules cannot be understood by a limited number of features. The use of multiple models for prediction using consensus approach helps in reducing model uncertainty by enhancing the prediction quality of the external set and also in reducing the prediction errors [38]. The four MLR models are given below:
Model 1
Model 2
Model 3
Model 4
The significant descriptors obtained from the four MLR models (M1–M4) contributing to A2A receptor selectivity are C-040, C-027, F09 [N-O], ETA_Beta_s, nCIC, T (F..Cl), SsssN, and F07[C-C]. All the descriptors positively contribute to the A2A receptor selectivity, except C-040, as identified from the regression coefficients of the descriptors and summarized in Table 3. We have also checked the applicability domain of the developed MLR models. The models showed good predictive ability as per the statistical results. The details of the descriptors, their contribution, and frequency of appearance in all the four models are explained elaborately in Table 3. The values of all descriptors appearing in the models for training and test set compounds are given in the Supplementary Material II (Excel file) and the scatter plots of the observed vs. predicted selectivity values are given in Figure 6.

Observed vs predicted A2AR selectivity plots for all four MLR models
Mechanistic interpretation
All the descriptors obtained in the four models and their frequency give an idea about their importance in modeling the selectivity of the PET tracers towards adenosine A2A receptor. The descriptors like C-027, F09[N-O], SsssN, T(F..Cl), and ETA_Beta_s appearing in the models give information about the electronic feature of the compounds and are essential when the selectivity of receptor is considered (Fig. 7). Electronegativity is a chemical property that describes the tendency of an atom to draw electron towards itself. If a compound contains higher number of electronegative atoms in its structure, then the selectivity of the A2A receptor for that compound also increases.

Features affecting the adenosine A2A selectivity
The presence of atom-centered fragments like C-027 (R--CH--X) in compounds like A-23 and A-25 increase the antagonist selectivity of the PET compounds. Since ‘X’ represents any electronegative atom like O, N, S, P, Se, and halogens, the presence of heteroatoms increases the selectivity of the compounds towards A2A receptor. The descriptor F09[N-O] explains the frequency of presence of nitrogen and oxygen at the topological distance 9, and its positive regression coefficient indicates its influential activity on the antagonistic behavior of the imaging agents (as seen in compounds A-4 and A-27). Another similar kind of descriptor is T (F..Cl), explaining the information about sum of topological distances between F and Cl atoms in the chemical structure. These descriptors give information about the electronegative atoms, i.e., nitrogen and oxygen in F09[N-O] and fluorine and chlorine in T(F..Cl). ETA_Beta_s (Σβs) is an extended topochemical atom (ETA) descriptor, which can be represented as sum of βs values of all non-hydrogen vertices divided by 2. The term ′βs′ can be denoted as
Here, x represents contribution of sigma bonds and σ signifies parameters related to sigma bonds. During the computation of β values, the sigma bond value for two similar types of electronegative atoms should be considered 0.5, and dissimilar electronegative atoms should be considered 0.75. This suggests that compounds bearing dissimilar heteroatoms will have greater selectivity to A2A receptor as seen in compounds A-25, A-23, and A-4. Sigma bonds connected with different heteroatoms will have higher descriptor values indicating that the presence of dissimilar heteroatoms is more favorable for selectivity than similar heteroatoms. E-state descriptor SsssN (> N—) encodes the intrinsic electronic state of the nitrogen atom as perturbed by the electronic influence of other molecules with the context of topological character within the molecule. The electronegative contribution of nitrogen is well-depicted in this descriptor, and the positive regression coefficient shows that an increase in the number of tertiary nitrogen benefits in receptor selectivity as seen in compounds A-30 and A-4.
Other descriptors which significantly contribute to A2A receptor selectivity are nCIC, F07[C-C], and C-040. These descriptors give information about the number of rings present, type of bonds, and size of the antagonists showing selectivity towards the receptor. The number of rings (cyclomatic number) in the structure is indicated by nCIC descriptor. The positive regression coefficient of the descriptor suggests that the presence of high number of rings increases the selectivity towards the A2A receptor as observed in compounds A-25 and A-4. F07[C-C], a 2D atom pair stands for frequency of C–C fragment at the topological distance 7. It provides information about the size (chain length) of the molecule. This means that with an increase in the number of this fragment, i.e., carbon chain, the selectivity towards the A2A receptor increases (as in compounds A-4 and A-25). The atom-centered fragment descriptor, C-040 (Table 3) gives information about the number of carbon atoms that are attached to heteroatoms by single/double or triple bonds in the straight chain length. The negative regression coefficient suggests that an increase in the number of such fragments decreases the selectivity of the compound towards the A2A receptor as seen in compounds A-6, A-7, and A-35. As this fragment suggests high number of double and triple bonds attached with the carbon, it can be concluded that unsaturation in the straight chain of the antagonists is unfavorable for the receptor selectivity.
Intelligent consensus predictions
For further refinement of the predictions obtained from the individual models, we have applied intelligent consensus modeling methods. Consensus modeling helps in enhancing the prediction performance of the models and also reduces the test set errors. It was observed that consensus prediction of the test set compounds (Table 4) is better in terms of both MAE-based criteria and predicted R2 parameter. Four different consensus approaches were used employing “Intelligent Consensus Prediction” tool [39]: CM0 (simple average of predictions), CM1 (average of predictions from the ‘qualified’ individual models), CM2 (weighted average predictions (WAPs) from ‘qualified’ individual models), and CM3 (best selection of predictions (compound-wise) from ‘qualified’ individual models). From the four consensus model obtained, CM0 was found to be the best.
Applicability domain
Applicability domain (AD) is an important tool for reliable application of QSAR models. It can be considered a “theoretical region in chemical space defined by the respective model descriptors and responses in which the predictions are reliable” [42, 47]. We have checked the AD of all the models using standardization approach [48] to check whether any molecule in the test set lies outside the AD of a model. From the domain of applicability analysis, it was found that there were no test set compounds outside the AD, and no compound in the training set came as an outlier (see Supplementary II Excel file).
Comparison with a previously published model
A direct comparison between the current and a previously published model [29] is infeasible due to the differences in the composition of training and test sets. However, the current model can be considered more advantageous since it has been developed using simple and easily interpretable two-dimensional descriptors which does not require any conformational analysis or energy minimization before their calculation.
Conclusion
Parkinson’s disease is a neurodegenerative disease affecting the elderly person around the world. An important target for its treatment is blocking adenosine A2A receptor which is co-located with the D2 receptor and is pharmacologically opposite in motor function. Many studies hint that blocking A2A receptor would be a beneficial strategy in the treatment of PD. Thus, this work endeavors exploring QSAR analysis to correlate the chemical structures with their biological activity with the aim to filter the essential chemical features of an antagonist for selectivity and binding affinity to A2A receptor. The computational approach used in this work consists firstly the calculation of the molecular descriptors, and secondly, correlating these descriptors with the binding affinity and selectivity using different chemometric tools such as Genetic Function Algorithm (GFA), Best Subset Selection (BSS) method, and Intelligent consensus predictor (ICP) tools. The statistical quality of the models was checked using traditional metrics both internally and externally. We have also discussed about the contributions of the descriptors in the light of known binding mechanisms such as π-π stacking interaction, hydrophobic interaction, and hydrogen bonding with the different protein residues present in the receptor binding sites. From the insights obtained from such mechanism, we found that electronegative atoms and presence of aromatic ring like benzene are favorable for enhancing the binding affinity to the A2A receptor. Furthermore, the docking studies supported the conclusions derived from the QSAR studies. In conclusion, the study highlights the pharmacophoric features mainly responsible for antagonizing adenosine receptors that can be further modified for better binding and selectivity to A2A receptor. In case of selectivity also, electronegativity and aromaticity of the compounds play essential and influential roles. The simple two-dimensional (2D) descriptors appearing in all the models are easier to compute requiring no conformation analysis or energy minimization process. Thus, this information would help in the future development and synthesis of newer PET tracer targeted towards adenosine receptor.
References
Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, Lang AE (2017) Parkinson disease. Nat Rev Dis Primers 3:1–21
Voss T, Ravina B (2008) Neuroprotection in Parkinson’s disease: myth or reality? Curr Neurol Neurosci Rep 8:304–309
Ahmed SS, Ahameethunisa A, Santosh W (2010) QSAR and pharmacophore modeling of 4-arylthieno [3, 2-d] pyrimidine derivatives against adenosine receptor of Parkinson’s disease. J Theor Comput Chem 9:975–991
Chen JJ, Swope DM (2007) Pharmacotherapy for Parkinson’s disease. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 27:161S–173S
Jankovic J, Stacy M (2007) Medical management of levodopa-associated motor complications in patients with Parkinson’s disease. CNS Drugs 21:677–692
Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53:527–552
Fuxe K, Ferré S, Genedani S, Franco R, Agnati LF (2007) Adenosine receptor–dopamine receptor interactions in the basal ganglia and their relevance for brain function. Physiol Behav 92:210–217
Chen JF, Xu K, Petzer JP, Staal R, Xu YH, Beilstein M, Sonsalla PK, Castagnoli K, Castagnoli N, Schwarzschild MA (2001) Neuroprotection by caffeine and A2A adenosine receptor inactivation in a model of Parkinson’s disease. J Neurosci 21:RC143–RC143
Grondin R, Bedard PJ, Tahar AH, Gregoire L, Mori A, Kase H (1999) Antiparkinsonian effect of a new selective adenosine A2A receptor antagonist in MPTP-treated monkeys. Neurology 52:1673–1673
Ongini E, Monopoli A, Impagnatiello F, Fredduzzi S, Schwarzschild M, Chen JF (2001) Dual actions of A2A adenosine receptor antagonists on motor dysfunction and neurodegenerative processes. Drug Dev Res 52:379–386
Ikeda K, Kurokawa M, Aoyama S, Kuwana Y (2002) Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J Neurochem 80:262–270
Pike VW (2009) PET radiotracers: crossing the blood–brain barrier and surviving metabolism. Trends Pharmacol Sci 30:431–440
Rahmim A, Zaidi H (2008) PET versus SPECT: strengths, limitations and challenges. Nucl Med Commun 29:193–207
Roy K (2018) Quantitative structure-activity relationships (QSARs): a few validation methods and software tools developed at the DTC laboratory. J Indian Chem Soc 95:1497–1502
Gramatica P (2020) Principles of QSAR modeling: comments and suggestions from personal experience. IJQSPR 5:1–37. https://doi.org/10.4018/IJQSPR.20200701.oa1
Puzyn T, Leszczynski J, Cronin MT, eds. (2010) Recent advances in QSAR studies: methods and applications, Vol. 8 Springer Science & Business Media, Berlin, Germany
Gao DW, Wang P, Yang L, Peng YZ, Liang H (2002) Study on the screening of molecular structure parameter in QSAR model. J Environ Sci Heal A 37:601–609
Tropsha A (2004) Application of predictive QSAR models to database mining. Chemoinformatics Drug Discov 23:437–455
Roy K (2020) Ecotoxicological QSARs. Springer, New York
Kar S, Roy K, Leszczynski J (2017) In: Roy K. (eds) On applications of QSARs in food and agricultural sciences: history and critical review of recent developments. Advances in QSAR Modeling, Springer, Cham, Switzerland
Ojha PK, Roy K (2018) Chemometric modeling of odor threshold property of diverse aroma components of wine. RSC Adv 8:4750–4760
Tantra R, Oksel C, Puzyn T, Wang J, Robinson KN, Wang XZ, Ma CY, Wilkins T (2015) Nano (Q) SAR: Challenges, pitfalls and perspectives. Nanotoxicology 9:636–642
Mikolajczyk A, Gajewicz A, Mulkiewicz E, Rasulev B, Marchelek M, Diak M, Hirano S, Zaleska-Medynska A, Puzyn T (2018) Nano-QSAR modeling for ecosafe design of heterogeneous TiO 2-based nano-photocatalysts. Environ Sci Nano 5:1150–1160
Mikolajczyk A, Sizochenko N, Mulkiewicz E, Malankowska A, Rasulev B, Puzyn T (2019) A chemoinformatics approach for the characterization of hybrid nanomaterials: safer and efficient design perspective. Nanoscale 11:11808–11818
Hoekman D (1996) Exploring QSAR fundamentals and applications in chemistry and biology, volume 1. hydrophobic, electronic and steric constants, Volume 2 J. Am. Chem. Soc. 1995, 117, 9782. J Am Chem Soc 118:10678-10678
Klein C, Kaiser D, Kopp S, Chiba P, Ecker GF (2002) Similarity based SAR (SIBAR) as tool for early ADME profiling. J Comput Aided Mol Des 16:785–793
Sebastián-Pérez V, Martínez MJ, Gil C, Campillo NE, Martínez A, Ponzoni I (2019) QSAR Modelling to identify LRRK2 inhibitors for Parkinson’s disease. J Integr Bioinform 16
Khanfar MA, Al-Qtaishat S, Habash M, Taha MO (2016) Discovery of potent adenosine A2a antagonists as potential anti-Parkinson disease agents. Non-linear QSAR analyses integrated with pharmacophore modeling. Chem Biol Interact 254:93–101
Tamiji Z, Salahinejad M, Niazi A (2018) Molecular modeling of potential PET imaging agents for adenosine receptor in Parkinson’s disease. Struct Chem 29:467–479
MarvinSketch software, https://www.chemaxon.com. Accessed on 05 Jan 2020
Yap CW (2011) PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem 32:1466–1474
Dragon version 7, Kodesrl, Milan, Italy, 2016; software available at http://www.talete.mi.it/index.htm. Accessed 07 Jan 2020
Golbraikh A, Shen M, Xiao Z, Xiao Y-D, Lee K-H, Tropsha A (2003) Rational selection of training and test sets for the development of validated QSAR models. J Comput Aided Mol Des 17:241–253
Golbraikh A, Tropsha A (2000) Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. Mol Divers 5:231–243
Khan PM, Roy K (2018) Current approaches for choosing feature selection and learning algorithms in quantitative structure–activity relationships (QSAR). Expert Opin Drug Discovery 13:1075–1089
Devillers J (1996) Genetic algorithms in molecular modeling. Academic Press, Cornwall, Great Britain
Pope PT, Webster JT (1972) The use of an F-statistic in stepwise regression procedures. Technometrics 14:327–340
Roy K, Das RN, Ambure P, Aher RB (2016) Be aware of error measures. Further studies on validation of predictive QSAR models. Chemom Intell Lab Syst 152:18–33
Roy K, Ambure P, Kar S, Ojha PK (2018) Is it possible to improve the quality of predictions from an “intelligent” use of multiple QSAR/QSPR/QSTR models? J Chemom 32:e2992
DTC Lab QSAR Tools http://teqip.jdvu.ac.in/QSAR_Tools/DTCLab
Roy K, Mitra I (2011) On various metrics used for validation of predictive QSAR models with applications in virtual screening and focused library design. Comb Chem High Throughput Screen 14:450–474
Gadaleta D, Mangiatordi GF, Catto M, Carotti A, Nicolotti O (2016) Applicability domain for QSAR models: where theory meets reality. IJQSPR 1:45–63
Congreve M, Andrews SP, Doré AS, Hollenstein K, Hurrell E, Langmead CJ, Mason JS, Ng IW, Tehan B, Zhukov A, Weir M (2012) Discovery of 1, 2, 4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J Med Chem 55:1898–1903
Wu G, Robertson DH, Brooks Iii CL, Vieth M (2003) Detailed analysis of grid-based molecular docking: a case study of CDOCKER—a CHARMm-based MD docking algorithm. J Comput Chem 24:1549–1562
Pan AC, Borhani DW, Dror RO, Shaw DE (2013) Molecular determinants of drug–receptor binding kinetics. Drug Discov Today 18:667–673
Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322:1211–1217
Yun YH, Wu DM, Li GY, Zhang QY, Yang X, Li QF, Cao DS, Xu QS (2017) A strategy on the definition of applicability domain of model based on population analysis. Chemom Intell Lab Syst 170:77–83
Roy K, Kar S, Ambure P (2015) On a simple approach for determining applicability domain of QSAR models. Chemom Intell Lab Syst 145:22–29
Funding
PD thanks Indian Council of Medical Research, New Delhi, for awarding with a Senior Research Fellowship. JR received financial assistance from the Department of Atomic Energy—Board of Research in Nuclear Sciences (DAE-BRNS) (ref. 36(3)/14/08/2017-BRNS). KR thanks DAE-BRNS for a major research project (ref. 36(3)/14/08/2017-BRNS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary I
contains structures and experimental A2AR binding affinity [pA2AR(BA)] and A2AR selectivity values. (DOCX 1269 kb)
Supplementary II
contains descriptor values and AD information of all compounds for different models. (XLSX 23 kb)
Rights and permissions
About this article

Cite this article
De, P., Roy, J., Bhattacharyya, D. et al. Chemometric modeling of PET imaging agents for diagnosis of Parkinson’s disease: a QSAR approach. Struct Chem 31, 1969–1981 (2020). https://doi.org/10.1007/s11224-020-01560-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01560-6