
Abstract
The adenosine receptors have appeared as potent and selective drug target in various diseases especially for central nervous system diseases. Adenosine receptor A2A antagonists have been known as potential treatment for Parkinson’s disease (PD). Radiolabeled A2AR antagonists can be used as positron emission tomography (PET) tracers and diagnostic tools for PD. In the present investigation, we perform the quantitative structure–activity relationship (QSAR) analysis and docking studies of a series of PET tracers as ligands and Adenosine receptors (A2AR) binding affinity, to elucidate the structural properties required for A2AR antagonist in treatment for PD. Several variable-selection methods were used to choose the descriptors that would lead to good QSAR model. Among several models developed, the best model was a five-variable multiple linear regression (MLR) equation with statistical parameters of squared correlation coefficient R 2 = 0.90 ± 0.01 and cross-validated correlation coefficient Q 2 = 0.84 ± 0.02. The QSAR models were also constructed for A2AR selectivity to tracer ligands, that yielded a four-variable model with R 2 = 0.94 ± 0.01 and Q 2 = 0.89 ± 0.02. The most important variables contributed in models construction involved: partial charge, hydrophobic atoms, rotatable bonds, polar van der Waals surface area, potential energy, and conformation-dependent charge descriptors. Finally, molecular docking analysis was carried out to better understand the interactions between ligands and Adenosine receptors. The importance of π-π stacking interactions between aromatic moiety of the ligands and triazine core of A2AR antagonist was confirmed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In recent years, the adenosine receptors have appeared as potential drug targets [1, 2]. Adenosine, a purine nucleoside, is an endogenous modulator of a number of physiological functions in the central nervous system (CNS) as well as in peripheral tissues [3,4,5]. These receptors have been extensively characterized and divided into four different subtypes including: A1, A2A, A2B, and A3 [4]. It acts at specific membrane G-protein receptors positively (A2A, A2B) or negatively (A1, A3) linked to adenylatecyclase [4]. Among these four subtype adenosine receptors in the CNS, the adenosine A2A receptors (AR) are densely distributed in the central nervous system (striatum, nucleus accumbens, and olfactory tubercles) which controls intracellular AMP (Adenosine monophosphate) levels and play an important role in the regulation of mood and motor function [6]. Adenosine A2A receptor antagonists have developed as potential treatment for Parkinson’s disease in the past decade [7, 8].
Human Parkinson’s disease (PD) is a very serious neurological disorder, and current manner of treatment fail to achieve long-term control. Since adenosine receptors antagonists have been shown to restore the deficits arise from decline of the striatonigral dopamine system, which is compromised by the loss of striatal neurons in this disease, A2A antagonism provides a possible treatment for PD [9]. Dopamine receptors, as a subclass of G protein-coupled receptors, are important in the vertebrate central nervous system. The existence of multiple types of receptors for dopamine was first proposed in 1976 [10]. There are at least five subtypes of dopamine receptors, D1, D2, D3, D4, and D5.Excitation of the A2A receptor was found to reduce the binding desire of dopamine D2 receptors for dopamine and to counter the actions of both D1 and D2 receptors on behavior, gene expression, and secondary messenger systems [11, 12]. Consequently, blockade of the adenosine receptor could compensate for the lack of dopamine D2receptor-mediated control of striato-Gpe neurons [12]. At the moment, a number of pharmacological models that can recapitulate many of the symptoms displayed in Parkinsonian patients such as bradykinesia, rigidity, and tremor, and A2A receptor antagonists appeared to have a useful effect in many of these models [13,14,15,16,17,18].
Positron emission tomography (PET) [19, 20] is the most advanced methods for non-invasive medical imaging modality that provides 3D maps of the brain. PET studies show better accuracy and resolution in quantification of regional distribution and temporal measurements of radioactivity and thus are superior to all other imaging modalities including single photon emission computed tomography (SPECT) [21, 22]. Both agonist and antagonist ligands containing positron emitting radioisotopes have been introduced for 3-dimensional in vivo imaging of the receptors [23]. Such ligands for PET might prove useful for eventual diagnostic use in the CNS. Ligands for in vivo PET imaging of A1, A2A, and A3ARs have been developed. A2AR antagonist PET tracers are of two types, xanthine and non-xanthine PET tracers [24, 25]. Development of new ligands that may lead to new candidates for PET tracer to improve physicochemical and pharmacokinetics properties and mapping adenosine receptors is of great interest.
The search for new compounds with desired properties requires enormous human resource and cost. That is why the pharmaceutical industry has shown great interest in theoretical approaches that enable the logical design of pharmaceutical agents. Quantitative structure-activity relationships (QSAR) studies, as significant research fields in drug design and medicinal chemistry, have obtained an important place within modern chemistry and therefore, there exists a keen interest on the development of these techniques [26,27,28]. The purpose of QSAR approaches is to develop some quantitative models to predict activity of a compound, and these models can reduce the research time and cost of new drugs.
In this study, we built the QSAR model with molecular descriptors, to explore the correlations of the molecular structure of a series of PET tracers as ligands and for A2A receptor antagonist binding affinity and selectivity of the tracer between A1 and A2A receptors. Molecular docking analysis was also carried out to better understand the interactions between ligands and Adenosine receptors.
Methods
Data set
The data set employed for the QSAR analyses contains 35 Xanthine ligands as A2AR antagonist PET tracers were taken from the literature [29] and applied for QSAR analysis. The structures of all the compounds along with their experimental binding affinity and selectivity are presented in Table 1. The A2AR selectivity (SA2R) and binding affinity (AA2R) values ranged between 0.1–20 and 7.84–16,500 nM, respectively. These values were converted into the corresponding logarithm values and utilized as dependent variables.
Descriptor calculation
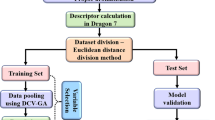
Optimized and energy-minimized structures of the molecules were used to calculate molecular descriptors. The energy minimization of the molecules was done using AM1 (Austin model) Hamiltonian method available in MOPAC module with a convergence criterion of 0.001 kcal/molÅ. Various kinds of molecular descriptors involving: physichochemical, structural, partial charge, topological, and geometrical descriptors were computed using Molecular Operating Environment (MOE, Chemical Computing Group Inc. 2011) package.
As a first step in variables reduction, descriptors exhibiting constant or nearly constant values as well as those with poor correlation to the dependent variables (R 2 < 0.10) were removed.
Descriptor selection
Feature selection is of considerable importance in QSAR modeling to reduce the computational complexity, improving the prediction performance of constructed models and providing a better understanding of the underlying process [30]. Three feature selection procedures including: genetic algorithm (GA), enhanced replacement method (ERM), genetic function approximation (GFA), and stepwise multiple linear regression (stepwise-MLR) were applied to choose the best subset of descriptors out of a large pool of descriptors.
Genetic algorithms
Genetic algorithms (GA), explained by Holland [31], mimic natural evolution and selection. In biological systems, genetic data that distinguishes the individuality of an organism is stored in chromosomes. Chromosomes are replicated and passed onto the next generation with selection criteria depending on fitness. Genetic information can however be changed through genetic operations such as crossover and mutation.
Genetic function approximation (GFA)
GFA is a helpful technique for searching in a large parameter space when the data is small. This technique can choose descriptors automatically and optimize parameters, expand a population of models simultaneously, and avoid local optima. Models are improved by performing a crossover operation to recombine terms of better scoring models. GFA technique is a method using the idea of natural selection and evolution in higher dimensional space to choose optimal descriptor combinations able of explaining bioactivity variation among training compounds from a large pool of possible descriptor combinations [32, 33].
The best subset selection version 1.2 program available at https://teqip.jdvu.ac.in/QSAR_Tools/ was used to carry out the GFA variable selection.
Enhanced replacement method
Enhanced replacement method (ERM) suggested by Mercader et al. [34] is an improved version of replacement method (RM) [35, 36]. This technique approaches the minimum of S by judiciously taking into account the relative errors of the coefficients of the least-squares model given by a set of d descriptors d = X 1, X 2,..., X d .
In this equation, N is the number of molecules in the set of train, and res i the residual for molecule i, the difference between the experimental activity and predicted activity. More detailed information about these algorithms can be found in reference [34].
Stepwise-multiple linear regressions
Stepwise-MLR, a multiple-term linear equation was constructed step-by-step. The basic approach contains (1) recognized an initial model, (2) iteratively “stepping,” that is, frequently altering the model at the previous step by adding or removing a predictor variable in accordance with the “stepping criteria,” and (3) terminating the research when stepping is no longer possible given the stepping criteria, or when a specified maximum number of steps have been attained. Especially, at each step all variables are reviewed and evaluated to determine which one will contribute most to the equation. That variable is then contained in the model, and the process starts again.
Model construction and validation
The data set was randomly divided into a training set (80%), which was used to adjust the parameters of the models, and a test set (20%) to evaluate the prediction ability of the models obtained.
QSAR models were generated for this series using multiple linear regression (MLR) and partial least squares (PLS) regression methods and those which come out with promising results are discussed here.
For each model cross-validated correlation coefficient (Q 2), correlation coefficients of calibration of train set (R 2 train), correlation coefficient of prediction set (R 2 pred), root mean square error of calibration (RMSEC) and root mean square error of prediction (RMSEP) were calculated. Y-randomization test was also applied to internally validate the models obtained. In Y-randomization test, the model was reconstructed based on randomized values of Y variable. The correlation coefficients of randomization (R 2 Yrand) values imply that acceptable models were obtained for the given data sets by the current modeling method and they did not show any chance correlation [37]. The external validation of final model was also checked by mean absolute error (MAE) based criteria [38]. The applicability domain (AD) of final QSAR models was explored based on standardization approach proposed by Roy et al. [39].
Molecular docking
For the present docking study, AutoDock program, version 4.1.1, was adopted. Discovery Studio Visualizer (Accelrys Software Inc) and Pymol (The PyMOL Molecular Graphics System) programs were employed for docking simulation. Fine 3D structure with a resolution of 3.27 Å of Adenosine A2A receptor was retrieved from the Protein Data Bank (PDB ID, 3UZA).
All of the compounds in the dataset were docked into the binding site of Adenosine receptor to provide the interaction between the ligand and the receptor. Initially, for the purpose of docking studies, the protein was considered without ligand and water molecules. Hydrogen atoms and the active torsions of ligand were assigned using Autodock Tools (ADT). AutoGrid was employed to generate grid maps around the active site. The volume of the grid was set to cover the binding site with a grid-spacing interval of 1.0 Å with dimensions of 40 × 40 × 40 Å. When docking was performed, some residues in the protein active site and all the torsional bonds in the ligand were set free. Lamarckian Genetic Algorithm (LGA) was employed then for conformational search with standard protocol. The final structures were clustered and ranked according to the Autodock scoring function.
Results and discussion
Tables 2 and 3 represent the statistical performance of model obtained based on selected descriptors with different variable selection methods for A2AR binding affinity and selectivity o respectively. GFA, ERM, GA, and stepwise procedures were used for variable selection. MLR and PLS methods were used to build models. The number of descriptors base on the statistical parameters arriving from the models with different number of descriptors. From Tables 2 and 3, it is observed that for all the models the data fit (R 2 = 0.68–0.92) and the predictive capability of models (R 2 pred ≥ 0.68) is acceptable.
The best QSAR model obtained forA2AR binding affinity was ERM-MLR, although the stepwise-MLR model showed similar statistical performance (Table 2). The multiple linear regressions based on ERM-selected descriptors, were performed to set up a statistically reliable model with good predictive ability for A2AR binding affinity including: R 2 train = 0.90 ± 0.01, Q 2 = 0.84 ± 0.02, and R 2 pred = 0.80 ± 0.15.The optimal QSAR model of A2AR binding affinity according to ERM-MLR was
Both of density and Kier1 descriptors, which represent the molecular shapes, showed positive effect impact on binding affinity whereas weinerPol descriptor as a distance-based molecular structure showed negative impact on binding affinity. Q_VSA_POL describes the total polar van der Waals surface area of molecules and showed negative impact on binding affinity. Table 4 gives a brief description of the most important descriptors which selected and involved in all constructed QSAR models for binding affinity. The capacity factor descriptors (vsurf_CW1 and vsurf_CW2) represent hydrophilic regions and computed as the ratio of hydrophilic surface on the overall molecule surface. GCUT_SLOGP descriptors represent the atomic contribution to logP (octanol-water partition coefficient). Most of involved descriptors indicate on the importance of partial charge properties and volume, shape, and subdivided surface area properties of Xanthine ligands, as PET tracer, on binding affinity to A2AR.
Table 3 showed the statistical performance of different QSAR models obtained based on selected descriptor of various variable selections for A2AR selectivity to Xanthine ligands. As could be seen, the best QSAR model obtained for A2AR selectivity was ERM-MLR with five variables. The multiple linear regressions based on ERM-selected descriptors, were performed to set up a statistically reliable model with good predictive ability for A2AR selectivity including R 2 train = 0.94 ± 0.01, Q 2 = 0.89 ± 0.02, and R 2 pred = 0.88 ± 0.01.The optimal QSAR model of A2AR selectivity according to ERM-MLR was
Total negative partial charge (PEOE_PC-) and angle bend potential energy (E_ang) with positive regression coefficients showed positive impact on selectivity of A2AR. The next two variables, namely b_rotR, that calculate fraction of rotatable bonds, and PC-, which describes the negative partial charge, have negative contributions to the A2AR selectivity to Xanthine ligands.
Table 5 represents a brief description of the most important descriptors which selected and involved in all constructed models for A2AR selectivity to Xanthine ligands. The vsurf_DW23 descriptor intents to describe lipophilicity and vsurf_IW1 represent hydrophilic regions of a molecule. As could be obvious from Table 5, the molecular flexibility and electrostatic interactions play important role in the selectivity of adenosine A2A receptor to Xanthine ligands.
Xanthine ligands that show high affinity and selectivity to A2AR antagonists usually contain alkyl/alkynyl moieties at N atoms of Xanthine backbone [40]. The ability of Xanthine ligand to form hydrogen bonding and steric hindrances in molecular structure has considerable impact on A2AR selectivity and A2AR binding affinity.
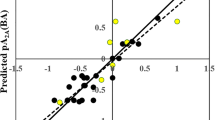
Figure 1 compares the experimental and the predicted values of A2AR selectivity and A2AR binding affinity for ERM-MLR models.

The fitted values resulting from linear models on the x-axis relative to the experimental values on the y-axis for ERM-MLR method for (a) binding affinity and (b) selectivity of PET tracer ligands to A2A antagonist
Table 6 shows the performance parameters of MAE-based criteria for validation tests of A2AR binding affinity and selectivity QSAR models. MAE and MAE + 3δ values, concordance correlation coefficient (CCC), Q 2 F1, and Q 2 F2 [41] were calculated for both QSAR models. These parameters were calculated using “Xternal Validation Plus” tool that is freely available at http://dtclab.webs.com/software-tools. As can be seen from Table 6, both QSAR models show high values for CCC, Q 2 F1, and Q 2 F2 indicating the reliability and high-performance prediction of proposed models to estimate and predict new compounds A2AR selectivity and A2AR binding affinity. If a QSAR model follows the criteria: MAE ≤ 0.1 × data range and MAE ± 3σ ≤ 0.2 × data range, then the model could be consider as good predictor. From Table 6, these criteria are preset in both final QSAR models.
The performance of models obtained from prediction errors was shown in Table 6. The values of variance, Bias2 and mean square error (MSE) parameters confirm no systematic error in quantitative predictions.
The applicability domain (AD), as a tool to check the reliability of prediction power of QSAR models, was calculated using “Applicability domain using standardization approach” freely available at http://dtclab.webs.com/software-tools. The AD analysis of the A2AR binding affinity model reveals that one outlier in train set (compound 2, Table 1). Removing of compound considered as outlier had no significant effect on the predictive power of the model. The AD analysis of the A2AR selectivity model show no outliers in both train and test set.
Molecular docking
Molecular docking studies of these compounds with adenosine A2A receptor represented very good binding interactions and warrants further studies to corroborate their binding with human A2A receptor for the design and development of better treatment for PD. All of the ligands were docked into the active site of A2A receptor to study the possible mode of their interactions. Docking of these ligands into inhibitor binding cavity ofA2AR confirms that these ligands dock in a similar binding modus like native co-crystallized ligand (Fig. 2). Inhibitor binding cavity of A2AR is predetermined by residues I1e-66, Ala-63, Leu-85, Phe-168, Met-177, Leu-249, His-250, Asn-253, Ala-277, Ile-274, T4g-330, and His-278. Analysis of the receptor/ligand complex models generated after successful docking of the compounds was based on parameters such as (1) hydrogen bond interactions, (2) hydrophobic interaction, (3) binding energy, (4) RMSD of active site residues and (5) orientation of the docked compound within the active site (Fig. 3). Lowest RMSD value was 1.37 Å (for compound 14) and RMSD value lower or close to 2 Å was considered as a successful docking [42]. In this research, RMSD values were within 2.0 Å representing our docking methods are valid for the given structures.

The docking of the native co-crystallized ligand with protein binding site

The ligand interactions profile
The most powerful ligand was selected to perform the docking study in the dataset. Figure 3 shows the several of interactions between the ligand and the receptor. An aromatic π-π stacking between aromatic ligand with Phe168 for all ligands was the common interaction pattern. An amino group of the ligand as hydrogen bond donor has main role in the high-affinity ligands and with side chain of the conserved Asn253 are established the powerful polar interactions. Also, a hydrophobic interaction between amino triazine core and the side chain of Asn253 can be observed.
Conclusions
As a first study on QSAR modeling of binding affinity and selectivity of Xanthine PET tracers to A2AR antagonist, different modeling strategies including ERM-MLR, GA-PLS, stepwise-MLR, and GFA-MLR were performed to achieve a reliable and robust model. The QSAR models obtained indicated the impact of molecular properties such as van der Waals surface area combination with the electrostatic property, the number of double bonds and rotatable bonds, partial charge, hydrophilic properties and molecular potential energy in the binding affinity, and selectivity of A2AR antagonist to Xanthine-type PET tracers. The provided model could be a helpful tool in the prediction of the A2AR binding affinity and A2AR selectivity, in a fast and costless manner, for any future studies that may require an estimation of these important characteristics of A2AR antagonist PET tracers.
Docking study reveals that the π-π stacking interactions between aromatic ligand and triazine core was the common interaction pattern in the binding affinity and selectivity of A2AR antagonist to Xanthine-type PET tracers. The importance of hydrogen bonding interactions of ligands and amino group of A2AR antagonist was also confirmed.
References
Müller CE, Scior T (1993) Adenosine receptors and their modulators. Pharm Acta Helv 68(2):77–111
Sachdeva S, Gupta M (2013) Adenosine and its receptors as therapeutic targets: an overview. Saudi Pharm J 21(3):245–253
Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24(1):31–55
Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M (1994) Nomenclature and classification of purinoceptors. Pharmacol Rev 46(2):143–156
Haas HL, Selbach O (2000) Functions of neuronal adenosine receptors. Naunyn-Schmiedeberg’s arch. Pharmacology 362(4–5):375–381
Poulsen S-A, Quinn RJ (1998) Adenosine receptors: new opportunities for future drugs. Bioorg Med Chem 6(6):619–641
Kim DS, Palmiter RD (2003) Adenosine receptor blockade reverses hypophagia and enhances locomotor activity of dopamine-deficient mice. Proc Natl Acad Sci 100(3):1346–1351
Pinna A (2014) Adenosine A2A receptor antagonists in Parkinson’s disease: progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs 28(5):455–474
Jacobson KA, Van Galen PJ, Williams M (1992) Adenosine receptors: pharmacology, structure-activity relationships, and therapeutic potential. J Med Chem 35(3):407–422
Cools AR, Rossum JV (1976) Excitation-mediating and inhibition-mediating dopamine-receptors: a new concept towards a better understanding of electrophysiological, biochemical, pharmacological, functional and clinical data. Psychopharmacology 45(3):243–254
Ongini E, Adami M, Ferri C, Bertorelli R (1997) Adenosine A2A receptors and neuroprotection. Ann N Y Acad Sci 825(1):30–48
Richardson PJ, Kase H, Jenner PG (1997) Adenosine A2A receptor antagonists as new agents for the treatment of Parkinson’s disease. Trends Pharmacol Sci 18(4):338–344
Fenu S, Pinna A, Ongini E, Morelli M (1997) Adenosine a 2A receptor antagonism potentiates L-DOPA-induced turning behaviour and c-fos expression in 6-hydroxydopamine-lesioned rats. Eur J Pharmacol 321(2):143–147
Kanda T, Jackson MJ, Smith LA, Pearce RK, Nakamura J, Kase H, Kuwana Y, Jenner P (1998) Adenosine A2A antagonist: a novel antiparkinsonian agent that does not provoke dyskinesia in parkinsonian monkeys. Ann Neurol 43(4):507–513
Kanda T, Shiozaki S, Shimada J, Suzuki F, Nakamura J (1994) KF17837: a novel selective adenosine A2A receptor antagonist with anticataleptic activity. Eur J Pharmacol 256(3):263–268
Mally J, Stone TW (1996) Potential role of adenosine antagonist therapy in pathological tremor disorders. Pharmacol Ther 72(3):243–250
Shimada J, Koike N, Nonaka H, Shiozaki S, Yanagawa K, Kanda T, Kobayashi H, Ichimura M, Nakamura J, Kase H (1997) Adenosine A 2A antagonists with potent anti-cataleptic activity. Med Chem Lett 7(18):2349–2352
Shiozaki S, Ichikawa S, Nakamura J, Kitamura S, Yamada K, Kuwana Y (1999) Actions of adenosine A2A receptor antagonist KW-6002 on drug-induced catalepsy and hypokinesia caused by reserpine or MPTP. Psychopharmacology 147(1):90–95
Segovia F, Górriz J, Ramírez J, Salas-Gonzalez D, Álvarez I, López M, Chaves R, Initiative AsDN (2012) A comparative study of feature extraction methods for the diagnosis of Alzheimer’s disease using the ADNI database. Neurocomputing 75(1):64–71
Zhang D, Wang Y, Zhou L, Yuan H, Shen D, Initiative AsDN (2011) Multimodal classification of Alzheimer’s disease and mild cognitive impairment. NeuroImage 55(3):856–867
Alavi A, Basu S (2008) Planar and SPECT imaging in the era of PET and PET–CT: can it survive the test of time? Eur J Nucl Med Mol Imaging 35(8):1554–1559
Rahmim A, Zaidi H (2008) PET versus SPECT: strengths, limitations and challenges. Nucl Med Commun 29(3):193–207
Holschbach MH, Olsson RA (2002) Applications of adenosine receptor ligands in medical imaging by positron emission tomography. Curr Pharm Des 8(26):2345–2352
Elsinga PH, Hatano K, Ishiwata K (2006) PET tracers for imaging of the dopaminergic system. Curr Med Chem 13(18):2139–2153
Ishiwata K, Kimura Y, de Vries J, Erik F, Elsinga PH (2007) PET tracers for mapping adenosine receptors as probes for diagnosis of CNS disorders. Cent Nerv Syst Agents Med Chem 7(1):57–77
Ghasemi J, Salahinejad M, Rofouei M (2011) Review of the quantitative structure–activity relationship modelling methods on estimation of formation constants of macrocyclic compounds with different guest molecules. Supramol Chem 23(9):614–629
Salahinejad M, Ghasemi J (2014) 3D-QSAR studies on the toxicity of substituted benzenes to Tetrahymena pyriformis: CoMFA, CoMSIA and VolSurf approaches. Ecotoxicol Environ Saf 105:128–134
Du Q-S, Huang R-B, Chou K-C (2008) Recent advances in QSAR and their applications in predicting the activities of chemical molecules, peptides and proteins for drug design. Curr Protein Pept Sci 9(3):248–259
Khanapur S, van Waarde A, Ishiwata K, Leenders KL, Dierckx RA, Elsinga PH (2014) Adenosine A2A receptor antagonists as positron emission tomography (PET) tracers. Curr Med Chem 21(3):312–328
Guyon I, Elisseeff A (2003) An introduction to variable and feature selection. J Mach Learn Res 3:1157–1182
Holland JH (1992) Adaptation in natural and artificial systems: an introductory analysis with applications to biology, control, and artificial intelligence. MIT Press, Cambridge,
Ayers JT, Clauset A, Schmitt JD, Dwoskin LP, Crooks PA (2005) Molecular modeling of mono-and bis-quaternary ammonium salts as ligands at the α4β2 nicotinic acetylcholine receptor subtype using nonlinear techniques. AAPS J 7(3):E678–E685
Vadlamudi SM, Kulkarni VM (2003) 3D-QSAR of protein tyrosine phosphatase 1B inhibitors by genetic function approximation. Internet Electron J Mol Des 2:000
Mercader AG, Duchowicz PR, Fernández FM, Castro EA (2008) Modified and enhanced replacement method for the selection of molecular descriptors in QSAR and QSPR theories. Chemom Intell Lab Syst 92(2):138–144
Duchowicz PR, Castro EA, Fernández FM (2006) Alternative algorithm for the search of an optimal set of descriptors in QSAR-QSPR studies. MATCH Commun Math Comput Chem 55:179–192
Duchowicz PR, Fernández M, Caballero J, Castro EA, Fernández FM (2006) QSAR for non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg Med Chem 14(17):5876–5889
Rücker C, Rücker G, Meringer M (2007) Y-randomization and its variants in QSPR/QSAR. J Chem Inf Model 47(6):2345–2357
Roy K, Das RN, Ambure P, Aher RB (2016) Be aware of error measures. Further studies on validation of predictive QSAR models. Chemom Intell Lab Syst 152:18–33
Roy K, Kar S, Ambure P (2015) On a simple approach for determining applicability domain of QSAR models. Chemom Intell Lab Syst 145:22–29
Choi H, Lee DS (2015) PET and SPECT of neurobiological systems. J Nucl Med 56(11):1805–1805
Chirico N, Gramatica P (2011) Real external predictivity of QSAR models: how to evaluate it? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J Chem Inf Model 51(9):2320–2335
Stigliani J-L, Bernardes-Génisson V, Bernadou J, Pratviel G (2012) Cross-docking study on InhA inhibitors: a combination of Autodock Vina and PM6-DH2 simulations to retrieve bio-active conformations. Org Biomol Chem 10(31):6341–6349
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Tamiji, Z., Salahinejad, M. & Niazi, A. Molecular modeling of potential PET imaging agents for adenosine receptor in Parkinson’s disease. Struct Chem 29, 467–479 (2018). https://doi.org/10.1007/s11224-017-1044-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-017-1044-6