
Abstract
Theoretical studies on Lewis acid-base behavior of hypervalent halogen fluorides, F3X and F5X (X = Cl, Br, I) have been instrumental in guiding this work. We have also examined whether the hole-lump concept explains the formation of the F5X∙∙∙CO complexes. Calculations of proton affinities (PA) and gas-phase basicity (GB) on hypervalent halogen fluorides show that F3X and F5X molecules can act as Lewis bases in gas phase. Moreover, theoretical calculations indicate that F3X and F5X molecules can act as Lewis acids forming stable complexes with a Lewis base as CO. The quantum theory of atoms in molecules (QTAIM) shows that the electrostatic interaction between the lone pair of the Lewis base (CO) and nucleus of the hypervalent halogen atom (X) plays a key role in stabilizing and determining the optimal geometry of the F5X∙∙∙CO complexes, as in conventional XBs. The localized molecular orbital energy decomposition analysis (LMOEDA) reveals that electrostatic component plays an important role in the stability of the FnX···CO complexes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Various hypervalent halogen fluorides are known. These have been conveniently employed to introduce a few fluorine atoms into the desired place in the substrate [1]. Hypervalent derivatives of the iodine are now routinely used in organic synthesis, probably due to its oxidizing properties, their benign environmental character and commercial availability [2]. Akiba has summarized some general aspects about structure and bonding of hypervalent halogen compounds [3].
Recently, the geometric features of some interactions given by di-, tri-, and tetravalent iodine atoms have been discussed [4]. According to Catalano et al. the fingerprint of XBs is the directionality of the attractive interaction with Lewis base because of anisotropic distribution of the electron charge density around the halogen-atom bridge (Lewis acid).
Moreover, it is well known that monovalent halogen atoms can function as Lewis bases [5,6,7,8] (equatorial region). As an example, a search in the Cambridge Structural Database (CSD) for C–X∙∙∙M+ interactions (with M+ = Li+, Na+, K+, Rb+, Cs+ and X = Cl, Br, I) reveals that the median value of the C–X∙∙∙ M+ angle is 103.08°. That is to say, cations enter the most negative region of the halogen atom (equatorial region to the C–X bond).4 In addition, a monovalent halogen atom can also act as Lewis acid and form attractive interactions with Lewis bases [8,9,10,11], these are called halogen bond (XB). All these interactions are understood when the anisotropic distribution of the electron density of the monovalent halogen atoms is taken into account.
Landrun et al. have reported an analysis of intermolecular interactions between hypervalent molecules Ph2IX and F3X (X = Cl, Br, I) dimers, using a combination of Density Functional Theory (DFT) calculations and qualitative arguments [12]. These authors concluded that the secondary bonding between these species can be understood using the language of donor-acceptor interactions. There is also a strong electrostatic contribution to the secondary bonding. The calculated strengths of these halogen-halogen secondary interactions are all less than 10 kcal mol−1.
Wang has found in the CSD that hypervalent halogen centers behave as acceptors of electron density (Lewis acids) [13]. In addition, Wang showed, through accurate computational results, that the halogen bond involving hypervalent halogen may be weaker than the corresponding halogen bond involving monovalent halogen even in the case that the hypervalent halogen is more positively charged than the monovalent halogen. According to this author the AIM analysis shows that there is no difference between the halogen bond involving hypervalent halogen and the halogen bond involving monovalent halogen [13]. We must make it clear that in any of these research papers, any halogenated compounds having a pentavalent halogen centers were considered.
Grabowski [14] has studied the acidic characteristics of some hypervalent halogen fluorides. In this work, he explains the geometry of the complexes FnBr∙∙∙B (n = 3, 5 and B: Lewis base) by means of the location of the positive maxima of the electrostatic potential on the bromine atom surface. Through this analysis, this author concluded that a hypervalent bromine atom should always act as the Lewis acid center, while a monovalent halogen atom may act as the Lewis acid and a Lewis base simultaneously. Grabowski also showed that the location of the positive maxima of the molecular electrostatic potential on the surface of both monovalent and polyvalent halogen compounds, predicts the geometry of the studied complexes [14].
Moreover, the Valence Shell Electron Pair Repulsion (VSEPR) theory [15,16,17] predicts the presence of one and two lone pairs in hypervalent halogen fluorides F5X and F3X (X = Cl, Br, I) respectively. It seems, on the one hand, as previously stated, that F3X compounds can act as Lewis base [12], but: Can the F5X compounds act as Lewis base?
On the other hand, the positive σ-hole concept explains many of the characteristics of the conventional XBs [9,10,11]. According to this concept established by Politzer et al., when a monovalent halogen atom forms a covalent bond, some of its electronic charge is transferred towards the bond region, causing the electronic charge to be diminished in its outer region (along the extension of the covalent bond) but increased in its equatorial region [9,10,11]. That is to say, the positive region on X corresponds to the electronically-depleted outer lobe of the half-filled p-type orbital of X that is involved in forming the R–X covalent bond [10, 11, 18]. In the hypervalent halogen fluorides F5X, as it is mentioned above, there is a lone pair on its outermost portions, centered along the extension of the Fe–X bond. Therefore, we can ask ourselves: rigorously, does the positive σ-hole concept explain the formation of the complexes F5X∙∙∙B (X = Cl, Br, I and B: Lewis base)? or is it only valid for XBs that involve monovalent halogens? Is the nature of these X∙∙∙B interactions the same as in the conventional XBs?
Recently, the characteristics of the conventional XBs through Quantum Theory of Atoms in Molecules (QTAIM) have been explained by means of the hole-lump concept [7, 19,20,21,22,23]. It seems that this concept can explain those interactions halogen bonding in which the positive σ-hole concept cannot do it [19, 20]. According to our interpretation of the hole-lump concept the formation of a XB is mainly due to the electrostatic interaction between the charge density provided by the Lewis base and the nucleus of the halogen-atom bridge (Lewis acid) [7, 20, 22, 23].
In this work we firstly investigate the Lewis acid-base behavior of F3X and F5X (X = Cl, Br, I) molecules. Secondly, we investigate whether the hole-lump concept explains the formation of the F5X∙∙∙CO complexes.
Computational details
Monomers and complexes were optimized without any constraint at the MP2/6–611++G(2d,2p) level of theory using the Gaussian 09 suite of programs [24]. All stationary points were confirmed to be true minima by the absence of any imaginary frequencies. For the study of Lewis base behavior of F3X and F5X molecules, we have calculated the proton affinities (PA) and gas-phase basicity (GB). These calculations were performed with G4 [25] method, and we also performed calculations at the MP2/6–311++G(2d,2p) level of theory. The study of Lewis acid behavior of F3X and F5X molecules was performed in FnX∙∙∙CO (n = 1, 3, 5 and X = Cl, Br, I) complexes. The binding energies of these complexes were obtained using the supermolecular approach, and the basis set superposition error (BSSE) was corrected by the counterpoise procedure of Boys and Bernardi [26]. The QTAIM analysis was performed with the AIMAll [27] software.
The energies of interaction have been decomposed following the localized molecular orbital energy decomposition analysis (LMOEDA) [28] formalism, according to the equation below:
where E elect is the electrostatic component, E ex-rep is the exchange-repulsion component resulting from the Pauli exclusion principle, and E pol and E disp correspond to polarization and dispersion terms, respectively. These calculations have been carried out with the GAMESS program (version 2013-R1) [29] at the same calculation level as the optimization.
Results and discussion
Monomers
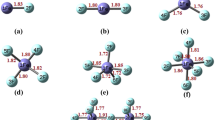
The L(r) = −∇2ρ(r) function is of particular interest to the present study due to the fact that its topology shows the regions of the space where the electron density is locally concentrated [basic region – L(r) > 0] or depleted [acidic region – L(r) < 0] [30, 31]. The valence shell of an atom is divided into an inner region where L(r) > 0 and an outer one where L(r) < 0. We will use this function to locate the preferential acidic and basic sites of the interhalogen compounds studied here. According to Bader et al. the topology of L(r) function has been shown to provide information about the spatial localization of electronic charge. The topology of this function provides a physical basis for the VSEPR theory [15,16,17]. The L(r) function exhibits maxima which indicate the presence of localized concentrations of electronic charge (lump) and shows the regions of the charge depletion (hole), both in the valence shell of an atom. Figure 1 shows at the envelope graph at L(r) = 1.0 au of the F3Br and F5Br molecules. The regions of the valence shell of the bromine atom in which there is a higher probability of finding an opposite spin electron pair (lone pairs of the Lewis model) are indicated (lump). In Fig. 1, regions where there is depletion of the electron charge density (hole) are observed. In other words, the L(r) function reveals that hypervalent halogen fluorides can act as Lewis acids or bases. In fig. S1 of the supporting information molecular graph, the critical points (3,-3) (nonbonded maxima) of the L(r) function and the bond angles, Fe–X–CP [CP: critical point (3,-3) of the L(r) function] are reported.

Envelops at L(r) = 1.0 au., of F3Br and F5Br compounds. Blue arrows indicate electronic depletion regions (Lewis acidic sites) and purple arrows indicate electronic concentration (lone pairs of the Lewis model)
These results are in partial agreement with those reported by Grabowski [14] in which it is stated that “For BrF3 and BrF5, as well as for their analogs analyzed here, the positive electrostatic potential is observed for the whole hemispheres of bromine centers. It may mean that multivalent bromine atom should always act as the Lewis acidic center. A distinct situation is usually observed for the monovalent halogen atom which possesses a dual character since it may act as the Lewis acid and a Lewis base simultaneously”. However, as discussed below, hypervalent halogen fluorides can act as Lewis bases.
Lewis base behavior
Regularly, the gas-phase basicity (GB) and the proton affinity (PA) are used to characterize the ability of a molecule to accept a proton when in the gas phase. The GB phase basicity is the negative of the free energy change associated with the reaction. The most frequently used index, the PA, is the negative of the enthalpy change at standard conditions. Computational approaches can provide reliable values for GB and PA, which is important since they are hard to determine experimentally. In this work we have investigated these magnitudes and results have been gathered in Table 1.
G4 estimates of ClF molecule are in very good agreement with the existing data of this molecule [32], while MP2 calculation is slightly lower.
According to the values of PA and GB calculated at MP2 level, the ability of these molecules to accept a proton when in the gas phase decreases in the order FX > F3X > F5X (for the same X) and increases in the order FnCl < FnBr < FnI (for n = 1, 3). These results are according with the strong electron-withdrawing effect of the fluorine atom and with the polarizability of the X halogen atoms respectively. That is to say, when the electron-withdrawing effect on X atom increases its ability to accept a proton decreases, and the higher the polarizability of X atom, the greater its ability to accept a proton. However, the same trend is not observed at G4 level.
In general, values PA and GB are relatively low compared with ammonium (PA = 853.6 kJ mol−1 and GB = 819.0 kJ mol−1). However, they are on the same order of magnitude of that CO (PA = 426.3 kJ mol−1 and GB = 402.2 kJ mol−1) and HF (PA = 484.0 kJ mol−1 and GB = 456.7 kJ mol−1), and they are higher than F2 molecule (PA = 332.0 kJ mol−1 and GB = 305.5 kJ mol−1) [33].
Interatomic distances and local topological parameters can be interpreted as a measure of the strength of the chemical bonds. Table 2 shows these parameters obtained at the MP2/6-311++G(2d,2p) level of theory. It can be observed that the interatomic distances X–H(+) are relatively short, d[Cl–H(+)] ≈ 1.31 Å, d[Br–H(+)] ≈ 1.45 Å and d[I–H(+)] ≈ 1.62 Å. But these distances are longer than the respective hydrogen halides [d(Cl–H) = 1.275 Å, d(Br–H) = 1.414 Å and d(I–H) = 1.609 Å respectively]. Therefore, we can establish that the X–H(+) bonds in these species are weaker than the respective hydrogen halides.
The equilibrium angles, Fe–X–H (for the some X), in FX–H(+), F3X–H(+), F5X–H(+) species, are near to 95°, 100° and 180° respectively. These values agree with the prediction made by the L(r) topology (see fig. S1 of the supporting information).
It is well known that local topological parameters of the AIM theory allows to characterize the interatomic interactions [30, 31, 34]. That is to say, the values of ρ(r b), ∇2ρ(r b) and H(r b) reveal the nature of the interactions. When ρ(r b) is relatively large, ∇2ρ(r b) < 0 and H(r b) < 0 then we have shared interactions (covalent bond), while when ρ(r b) is relatively lower, ∇2ρ(r b) > 0 and H(r b) > 0 then we have closed-shell interactions. Calculated local topological properties at the X–H(+) BCPs, shown in Table 2, present values typical of shared interactions. It is observed that, for the same pair of interacting atoms, the magnitudes ρ(r b), ∇2ρ(r b) and H(r b) increase in the order FX–H(+) < F3X–H(+) < F5X–H(+). That is to say, the covalent character of the interactions X–H(+) increases in this order.
It is important to note that geometric analysis gives different results to local topological parameters of charge density at the intermolecular BCP.
Lewis acidic behavior
Table 3 reports the values of the main parameters that describe the geometry of the studied systems. It can be seen that in all the cases, X···C intermolecular distances are substantially shorter than the sum of the van der Waals radii [35] of the X and C atoms. This is due to the mutual penetration of X and C atoms electronic densities. In addition, X···C intermolecular distances for the same X halogen decrease with the increase of the binding energy. The strength of the interactions FnX···CO (n = 1, 3, 5 and X = Cl, Br, I), for the same n, varies with the X halogen, increasing in the order FnCl···CO < FnBr···CO < FnI···CO. These remarks agree with the polarizability of the halogen atoms, which increases in the order Cl < Br < I.
The strength of the XBs depends on the electron-withdrawing ability of the group to which the halogen is attached. If the substituents of hypervalent halogen are strong electron-withdrawing groups such as fluorine atoms, the hypervalent halogen must be more positively charged than the corresponding monovalent halogen. Consequently, it is reasonable to think that the strength of hypervalent halogen-bonded interactions increases with the number of F atoms added. However, the findings of the current study do not support this idea. In Table 3 it is observed that the strength of the interactions, for the same X, increases in the order F5X···CO < F3X···CO < FX···CO.
Moreover, it is well established that the bond angle in the conventional XBs is always close to 180°, in the FX···CO and F3X···CO complexes this is true. But, in the F5X···CO complexes, the bond angles Fe–X···C are less than 180°. In addition, it is observed that these bond angles decrease in the order F5Cl···CO > F5Br···CO > F5I···CO, that is to say, in order inverse to the increase of the strength of the F5X···CO interactions.
QTAIM analysis is a powerful tool to investigate the electronic properties of the molecular system. In the present study, this theory was systematically applied to obtain a deep insight into different factors defining these interactions. Furthermore, this methodology has been successfully applied in the properties study of a variety of conventional XBs [7, 19,20,21,22,23].
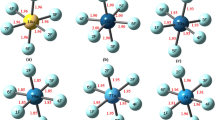
Table 4 reports the selected topological properties of the complexes studied here. The topological analysis of the electron density shows the presence of a BCP and an atomic interaction line of maximum electron density connecting the bridge atom (X) of the Lewis acid FnX with the carbon atom of the CO molecule (see Fig. 2), confirming that between these two atoms there is a bonded interaction. The topological characteristics at the intermolecular BCP show typical properties of closed-shell interactions. The electron density values range from 0.0100 to 0.0504 au and the Laplacian values range from 0.0299 to 0.1160 au, which correlates fairly well with the values reported for similar interactions [13, 14]. According to the topological analysis of electron density distribution in the QTAIM [30, 31], the electron density at the BCP, ρ(r b), is used to describe the strength of a bond. In general, the larger the value of ρ(r b), the stronger the bond is. In the complexes studied here it is observed that, for the same X, ρ(r b) increases following this order: F5X∙∙∙CO < F3X∙∙∙CO < FX∙∙∙CO. In other words, ρ(r b) increases in the same order that binding energies.

Envelops at L(r) = 0.0125 au. Note that in FBr···CO and F3Br···CO complexes, the molecules are oriented so that the lumps in the VSCC of the carbon atom are aligned with the hole in the VSCC of the bromine atom. However, in F5Br···CO complex, the lump in the VSCC of the carbon atom interacts with a belt of electronic charge depletion located around of a cap of charge concentration. In addition, the bond paths of ρ(r) are shown
The integration of electron density on the atomic basins, provides a useful tool for analyzing atomic charges, q(Ω), quadrupole moments, Q(Ω) and the electrostatic interaction energy between the total charge distribution of A atom and the nucleus of B atom, V e-n(A,B). These values are reported in Table 4.
The integrated atomic charges show that interactions occur between positively charged atoms, Xδ+···δ+C. The product of the positive charges of these interacting atoms (for the same X) is increased in the order FX∙∙∙CO < F3X∙∙∙CO < F5X∙∙∙CO. In this order repulsive electrostatic interactions increase and, therefore, the stability of the complexes decreases.
The atomic quadrupole moment in a particular direction (i.e. along the z-axis Q zz) is a measure of the deviation of electron density from spherical symmetry. That is to say, a spherical electron density distribution results in a value of zero for Q zz. This component measures how the electron density is elongated or compressed along the z-axis relative to a direction perpendicular to z. Thus, if Q zz is negative, the electron density is concentrated along this axis and, consequently, the electron distribution is prolate with respect to the z-axis. Similarly, when the component Q zz is positive, the electron distribution is oblate [30, 31]. This property of Q zz allows us to quantify the deformation experienced by the electron density on the atomic basin of interacting atoms. In Table 4, the atomic quadrupole moment values calculated on the basin halogen atom, Q zz(X), measures in the direction of the Fe–X bond are reported. The results show that in the FX···CO and F3X···CO complexes Q zz(X) > 0, which indicates that in this direction an electron charge density depletion is present. However, Q zz(X) < 0 for F5X···CO complexes, which indicates that in this direction an electron charge density accumulation is present.
When comparing Q zz(X) in the FnX∙∙∙CO complexes (for the same X), it is observed that increases in the order F5X∙∙∙CO < F3X∙∙∙CO < FX∙∙∙CO and comparing Q zz(X) in the FnX∙∙∙CO complexes (for n = 1, 3), it is observed that augments in magnitude following the order FnCl∙∙∙CO < FnBr∙∙∙CO < FnI∙∙∙CO. In general Q zz(X) augments follow the same order than that of binding energies.
In a previous work it was shown that the formation of the conventional XBs (D − X∙∙∙B) is due to the electrostatic interaction between the charge density provided by the Lewis base and the nucleus of the halogen atom [7, 20, 22, 23, 36]. The halogen atom has an electronic depletion region localized at the outset region in the direction of the D–X bond. This depletion determines the geometry and strength of the conventional XBs. Figure 2 undoubtedly shows that the formation of the FX···CO and F3X···CO complexes results from the interaction between a region of the space where the electron density is locally concentrated [lump − L(r) > 0] and the nucleus of the X atom through the region of the space where the electron density is locally depleted [hole − L(r) < 0]. In FX···CO and F3X···CO complexes it is clearly observed that these molecules are oriented so that the lump in the valence shell charge concentration (VSCC) of the carbon atom (Lewis base) is aligned with the hole in the VSCC of the halogen atom (Lewis acid). However, on the F5X···CO complexes the halogen atom X has a cap of electronic charge density concentration on its outermost portions, centered along the extension of the Fe–X bond. This small region of charge concentration causes an interelectronic repulsion with a lone pair of the Lewis base. This repulsion is responsible that the equilibrium angle Fe–X···C is other than 180°. In addition, the L(r) function shows clearly that, in the valence shell of the halogen atom X on the F5X···CO complexes, there is a region of electronic charge depletion around the cap of charge concentration capable of undergoing a nucleophilic attack. It is this region of electronic charge depletion which produces the formation and determines the geometry of the F5X···CO complexes. According to the preceding discussion, it seems that the electrostatic interaction between the lone pair of Lewis base (CO) and the nucleus of halogen atom plays a key role in stabilizing and determining the optimal geometry of these hypervalent halogen bonding interactions, like in the conventional XBs [7, 20, 23, 36] and in the double hole-lump interaction between halogen atoms [22].
In Fig. 2 it is also observed that in the F3Br···CO complex, the two lone pairs of the bromine atom, LP(Br), are confronted with the lone pair of the Lewis base, LP(C). In addition, the two bonding pairs F–Cl, BP(F–Br), are also faced to the lone pair of the Lewis base. In the F5Cl···CO complex it is observed one lone pair of the bromine atom and four bonding pairs F–Cl which are confronted with the lone pair of the Lewis base. That is, despite that the hypervalent halogen has a higher positive charge than the monovalent halogen, the halogen bonding interaction involving hypervalent halogen are weaker than the corresponding XBs involving monovalent halogen. This can be explained by the strong interelectronic repulsions of the lone pair of the Lewis base [LP(C)] with the lone pair/s [LP(X)] and the bonding pairs F–X [BP(F–X)] of the hypervalent halogen atom.
These repulsive interactions LP(Br)↔LP(C) and BP(F–Br)↔LP(C) make that Br···C intermolecular distances increases in the order FBr···CO < F3Br···CO < F5Br···CO.
According to Politzer et al. a halogen bond is a highly directional, electrostatically-driven noncovalent interaction between a region of positive electrostatic potential (positive σ-hole) on the outer side of the halogen X in a molecule D–X and a negative site B, such as a lone pair of a Lewis base or the p-electrons of an unsaturated system [9,10,11, 18]. There is no doubt that this is true to the monovalent halogens attached to a D group with medium or high electron-withdrawing power. However, as we have seen, in hypervalent halogen bonding interactions F5X···CO the halogen atom X (Cl, Br or I) has a cap of electronic charge density concentration on its outermost portions, centered along the extension of the Fe–X bond. That is to say, in the region where there would be the positive σ-hole proposed by Politzer. Therefore, we consider that the concept positive σ-hole used to explain the formation and directionality of halogen bonding should continue to be reviewed in other systems.
Moreover, the positive σ-hole concept is not suitable in some special situations, for example in the CH3Cl molecule [19]. The chlorine atom in this molecule does not have a positive electrostatic potential on its surface and consequently should not be able to form XBs. However, Riley et al. [37] showed that this molecule is able to form a stable XB with OCH2 molecule. Recently, a similar situation has been observed in the framework of the pnicogen bond [38]. According to Eskandari et al. “the lump-hole concept is more useful than the σ-hole in which the electrostatic part of potential is only considered. It is shown that the existence of hole in the VSCC of pnicogen atom is responsible for the formation and (in the absence of other interactions) the geometry of pnicogen bonded complexes” [38]. For this reason we consider it more appropriate to analyze, in the context of the QTAIM, the electrostatic interaction energy between the electron cloud of the carbon atom and the nucleus of the halogen atom, |V e-n(C,X)| (these values are shown in Table 4). It is observed that |V e-n(C,X)| (for a same X) increases with the module of the binding energy. These findings allow us to establish that, the electrostatic interaction between the lone pair of C atom and the nucleus of X atom plays a key role in stabilizing these halogen bonding interactions.
The electron-nuclear attractive contribution to virial field measured at the X···C BCP, |V e-n(r b)|, is a measure of the electrostatic force exerted by the nuclei X and C on the electronic cloud of the intermolecular region. Figure 3 shows a good quadratic correlation between |V e-n(r b)| and the binding energies. Therefore, the electrostatic interaction between the electron cloud of the intermolecular region and the nucleus of the halogen and carbon atoms play a key role in stabilizing these complexes.

Correlation between |V e-n(r b)| and the binding energies
Finally, we have studied the interaction energy components derived from the LMOEDA method. This partition scheme is a useful tool for a quantitative interpretation of the strength of the intermolecular interactions. In Table 5, the interaction energy components are reported.
It is observed that in all the complexes the most important stabilization term is electrostatic (between 41 and 47% of the stabilizing terms) followed by polarization/dispersion (in smaller proportions). In the complexes with greater interaction energies (FX∙∙∙CO) it is observed that E pol > E disp, while the in the hypervalent halogen-bonded interactions there is larger contribution of the dispersion. This indicates that in FnX∙∙∙CO (n = 3, 5) complexes the orbital interactions, related to polarization term [28], are less important than in the conventional XBs.
Conclusions
In this work, a systematic theoretical study about Lewis acid-base behavior of hypervalent halogen fluorides in gas phase was carried out on a series of binary complexes. The Lewis base behavior was studied in systems FnX–H(+) (n = 1, 3, 5 and X = Cl, Br, I), while the Lewis acidic behavior was studied in complexes FnX∙∙∙CO (n = 1, 3, 5 and X = Cl, Br, I).
The topological analysis of the L(r) function reveals that the hypervalent halogen fluorides, FnX (n = 3, 5 and X = Cl, Br, I) can act as either Lewis bases or Lewis acids. The ability of these molecules, according to proton affinities (PA) and gas-phase basicity (GB), to accept a proton decreases in the order FX > F3X > F5X (for the same X) and increases in the order FnCl < FnBr < FnI (for n = 1, 3).
The analysis of the geometric parameters of the FnX···CO (n = 1, 3, 5 and X = Cl, Br, I) interactions shows that, the X···C intermolecular distances are substantially shorter than the sum of the van der Waals radii of the X and C atoms. The bond angles Fe–X···C are close to 180° in the FnX···CO (n = 1, 3) complexes, while in the F5X···CO complexes, the bond angles Fe–X···C are considerably less than 180°.
QTAIM reveals that the FnX···CO interactions are the result of the electrostatic interaction between the charge density provided by the lone pair of the Lewis base [LP(C)] and the nucleus of hypervalent halogen atom, like in the conventional XBs.
LMOEDA reveals that electrostatic component plays an important role in the stability of the FnX···CO complexes. In addition, in the hypervalent halogen-bonded interactions FnX∙∙∙CO (n = 3, 5), the orbital interactions are less important than in the conventional XBs (FX∙∙∙CO).
References
Hara S, Atta-ur-Rahman (ed) (2006) Advances in organic synthesis: modern organofluorine chemistry-synthetic aspects. Bentham Science Publisher Ltd.
Zhdankin VV, Stang PJ (2008) Chemistry of polyvalent iodine. Chem Rev 108:5299–5358
Akiba K (1998) Chemistry of hypervalent compounds. VCH Publishers, New York
Catalano L, Cavallo G, Metrangolo P, Resnati G, Terraneo G (2016) Halogen bonding in hypervalent iodine compounds. Top Curr Chem 373:289–309
Nakamoto K, Margoshes M, Rundle RE (1995) Stretching frequencies as a function of distances in hydrogen bonds. J Am Chem Soc 77:6480–6486
Schleyer PVR, West R (1959) Comparison of covalently bonded electronegative atoms as proton acceptor groups in hydrogen bonding. J Am Chem Soc 81:3164–3165
Duarte DJR, Sosa GL, Peruchena NM, Alkorta I (2016) Halogen bonding. The role of the polarizability of the electron-pair donor. Phys Chem Chem Phys 18:7300–7309
Nelyubina YV, Antipin MY, Dunin DS, Kotov VY, Lyssenko K (2010) Unexpected “amphoteric” character of the halogen bond: the charge density study of the co-crystal of N-methylpyrazine iodide with I2. Chem Commun 46:5325–5327
Politzer P, Lane P, Concha MC, Ma Y, Murray JS (2007) An overview of halogen bonding. J Mol Model 13:305–311
Politzer P, Murray JS, Clark T (2010) Halogen bonding: an electrostatically-driven highly directional noncovalent interaction. Phys Chem Chem Phys 12:7748–7757
Politzer P, Murray JS, Clark T (2013) Halogen bonding and other σ-hole interactions: a perspective. Phys Chem Chem Phys 15:11178–11189
Landrum GA, Goldberg N, Hoffman R, Myniaev RM (1998) Intermolecular interactions between hypervalent molecules: Ph2IX and XF3 (X = Cl , Br , I) dimers. New J Chem 22:883–890
Wang W (2011) Halogen bond involving hypervalent halogen: CSD search and theoretical study. J Phys Chem A 115:9294–9299
Grabowski SJ (2014) Halogen bond with the multivalent halogen acting as the Lewis acid center. Chem Phys Lett 605–606:131–136
Bader RFW, Johnson S, Tang TH, Popelier PL (1996) The electron pair. J Phys Chem 100:15398–15415
Fradera X, Austen MA, Bader RFW (1999) The Lewis model and beyond. J Phys Chem A 103:304–314
Gillespie RJ (2008) Fifty years of the VSEPR model. Coord Chem Rev 252:1315–1327
Clark T (2013) σ-Holes. Wiley Interdisciplinary Reviews: Comput Mol Sci 3:13–20
Eskandari K, Zariny H (2010) Halogen bonding: a lump-hole interaction. Chem Phys Lett 492:9–13
Duarte DJR, Sosa GL, Peruchena NM (2013) Nature of halogen bonding. A study based on the topological analysis of the Laplacian of the electron charge density and an energy decomposition analysis. J Mol Model 19:2035–2041
Eskandari K, Lesani M (2015) Does fluorine participate in halogen bonding? Chem Eur J 21:4739–4746
Duarte DJR, Peruchena NM, Alkorta I (2015) Double hole–lump interaction between halogen atoms. J Phys Chem A 119:3746–3752
Duarte DJR, Angelina EL, Peruchena NM (2012) On the strength of the halogen bonds: mutual penetration, atomic quadrupole moment and Laplacian distribution of the charge density analyses. Comput Theor Chem 998:164–172
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery Jr JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nak-ajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghava-chari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, PMW G, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 09, Revision A.01. Gaussian, Inc, Wallingford
Curtiss LA, Redfern PC, Raghavachari K (2007) Gaussian-4 theory. J Chem Phys 126:084108-1–084108-12
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Keith TA (2011) AIMAll (Version 11.12.19); TK Gristmill Software, Overland Park KS, aim.tkgristmill.com
Su P, Li H (2009) Energy decomposition analysis of covalent bonds and intermolecular interactions. J Chem Phys 131:014102
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, et al (1993) General atomic and molecular electronic structure system. J Comput Chem 14:1347–1363
Bader RFW (1990) Atoms in molecules. A Quantum Theory; Clarendon, Oxford
Popelier P (ed) (2000) Atoms in molecules: an introduction. Prentice-Hall, Harlow
Cacace F, De Petris G, Pepi F, Rossi M, Sgamellotti A (1998) Elemental chlorine and chlorine fluoride: theoretical and experimental proton affinity and the gas phase Chemistry of Cl2H+ and FClH+ ions. J Phys Chem A 102:10560–10567
Hunter EPL, Lias SG (1998) Evaluated gas phase basicities and proton affinities of molecules: an update. J Phys Chem Ref Data 27:413–656
Matta CF, Boyd RJ (2007) The quantum theory of atoms in molecules: from solid state to DNA and drug design. Weinheim, Wiley-VCH
Bondi A (1964) Van der Waals Volumes and Radii. J Phys Chem 68:441–451
Duarte DJR, Vallejos MM, Peruchena NM (2010) Topological analysis of aromatic halogen/hydrogen bonds by electron charge density and electrostatic potentials. J Mol Model 16:737–748
Riley KE, Hobza P (2008) Investigations into the nature of halogen bonding including symmetry adapted perturbation theory analyses. J Chem Theory Comput 4:232–242
Eskandari K, Mahmoodabadi N (2013) Pnicogen bonds: a theoretical study based on the Laplacian of electron density. J Phys Chem A 117:13018–13024
Acknowledgements
G.J. Buralli, D.J.R. Duarte and N.M. Peruchena acknowledge SEGCYT UNNE and CONICET for the financial support. NMP is a career research of CONICET, Argentina.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
ESM 1
(DOC 1691 kb).
Rights and permissions
About this article

Cite this article
Buralli, G.J., Duarte, D.J.R., Sosa, G.L. et al. Lewis acid-base behavior of hypervalent halogen fluorides in gas phase. Struct Chem 28, 1823–1830 (2017). https://doi.org/10.1007/s11224-017-0966-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-017-0966-3