
Abstract
Boronic acids, R–B(OH)2, play an important role in synthetic, biological, medicinal, and materials chemistry. This investigation compares the structure and bonding surrounding the boron atoms in the simple aliphatic boronic acids, R–B(OH)2 (R=H; NH2, OH, and F), and the analogous borinic acids, R–BH(OH). Geometry optimizations were performed using second-order Møller–Plesset perturbation theory (MP2) with the Dunning–Woon aug-cc-pVTZ, aug-cc-pVQZ, and aug-cc-pV5Z basis sets; single-point CCSD(FC)/aug-cc-pVTZ//MP2(FC)/aug-cc-pVTZ level calculations were used to generate a QCI density for natural bond orbital analyses of the bonding. The optimized boron–oxygen bond lengths for the X–B–Ot–H trans-branch of the endo-exo form of the boronic acids and for the X–B–O–H cis-branch of the boronic and borinic acids (X=N, O, and F, respectively) decrease as the electronegativity of X increases. The boron–oxygen bond lengths are generally longer in the endo-exo or anti forms of the boronic acids than in the corresponding borinic acids. NBO analyses suggest the boron–oxygen bond in H2BOH is a double bond; the boron–oxygen bonding in the remaining boronic and borinic acids in this study has a significant contribution from dative pπ–pπ bonding. Values for Δ\({\text{H}}_{298}^{0}\) for the highly balanced reaction, R–B(OH)2 + R–BH2 → 2 R–BH(OH), suggest that the bonding surrounding the boron atom is stronger in the borinic acid than in the corresponding boronic acid.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Boronic acids, RB(OH)2, are a versatile class of compounds with applications in synthetic, biological, medicinal, and materials chemistry [1–32] and are widely used in the field of molecular recognition as chemical sensors for 1,2- and 1,3-diols. Boronic acid host compounds readily form boronate esters with saccharides in one of the few processes where a covalent bond can be rapidly made and subsequently broken [33]. When ortho-aminomethyl phenylboronic acids are rationally designed, the resulting boronate esters fluoresce due to subtle changes in Lewis acid–base chemistry of the proximal amine and the boron atom. A modification in the chemical structure adjacent to the boronic acid moiety can change the saccharide selectivity, pH range of operation, and fluorescence reporting abilities of the host compound.
Like their boronic acid counterparts, borinic acids, R1R2B(OH), have a variety of applications, e.g., in catalysis [34–36] and medicine [37–41], although their utility in molecular recognition is only beginning to be exploited [35, 36, 42–45]. Chudzinski et al. [42] recently highlighted this “neglected class of organoboron compounds” for diol recognition and detailed the effectiveness of diphenylborinic acid to preferentially bind the diol compounds catechol, L-lactic acid, and Alizarin Red S. Indeed, association constants for diphenylborinic acids bound to these particular substrates are nearly an order of magnitude larger than for similar boronic acids. Chudzinski et al. [42] speculated that this increase in affinity toward catechol, L-lactic acid, and Alizarin Red S could be a steric effect. However, there have been no systematic investigations of the structural and/or bonding differences in corresponding boronic and borinic acids that might help identify those features that affect their preference toward specific substrates.
The nature of the bonding surrounding the boron atom in both boronic and borinic acids is distinctive as a result of the empty p-orbitals on the trivalent boron atoms. Donor–acceptor interactions involving lone-pair orbitals on the hydroxyl oxygen atoms and these empty p-orbitals play a significant role in the nature of the boron–oxygen bonding. Lone-pair orbitals available from simple aliphatic substituents provide competitive donor–acceptor interactions that also impact the boron–oxygen bonding. Boronic acids differ from borinic acids by the presence of two boron–oxygen bonds and an intramolecular B–O–H…O hydrogen-bonding interactions inherent in the lower-energy endo-exo conformers of boronic acids [46–48]. These differences are expected to result in distinct steric and electronic properties of corresponding boronic and borinic acids that will influence their effectiveness as diol receptors to specific substrates. In this investigation, we compare the structure and bonding surrounding the trivalent boron atoms in the simple aliphatic boronic acids, R–B(OH)2 (R=H; NH2, OH, and F) and their related borinic acids, R–BH(OH), as a first step toward understanding their distinguishing properties.
Computational methods
Equilibrium geometries of all the molecules involved in this article were obtained using second-order Møller–Plesset perturbation theory with the frozen-core option, MP2(FC) [49], which neglects core–electron correlation; the Dunning–Woon aug-cc-pVTZ basis set was employed for all the geometry optimizations and the more complete aug-cc-pVQZ and aug-cc-pV5Z basis sets in selected optimizations [50–53]. Frequency analyses were performed analytically for all the compounds in this investigation at the MP2(FC)/aug-cc-pVTZ level to confirm that the geometry optimized structures were local minima on the potential energy surfaces (PESs). The GAUSSIAN 03 [54] and GAUSSIAN 09 [55] suits of programs were used for all the calculations. The geometry optimizations were followed by single-point calculations at the CCSD(FC)/aug-cc-pVTZ//MP2(FC)/aug-cc-pVTZ level to generate the QCI density for analyses of the bonding. Atomic charges were obtained from natural population analyses (NPA), and the bonding was analyzed with the aid of natural bond orbitals (NBOs). NBO version 3.1 embedded in GAUSSIAN 03 [54] and GAUSSIAN 09 [55] was used for all these calculations. The RESONANCE and E2PERT keywords were employed; the E2PERT command enables second-order perturbative estimates of donor–acceptor (bond–antibond) stabilization interaction energies, E(2), to be calculated in the NBO basis set [56–59]. Since NBO analyses using the (non-variational) MP2 density can be problematic, we consistently employed results from the HF/aug-cc-pVTZ and QCI densities [60]). NBOPro version 6.0 was used for visualization of natural bonding orbitals [61]. Atoms in molecules (AIM) calculations [62] using the MP2(FC)/aug-cc-pVDZ//MP2(FC)/aug-cc-pVDZ density were performed in selected cases to provide bond orders and atomic charge comparisons; the implementation in G03 [54] by Ciolslowski et al. [63, 64] was used for these calculations.
Results and discussion
Boron–oxygen bonding
R–B(OH)2 (R=H; NH2, OH, and F)
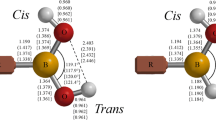
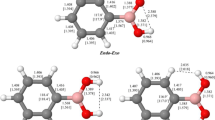
The optimized structures of the lowest-energy endo-exo conformers of R–B(OH)2 (R=H; NH2, OH, and F) are all planar at the MP2(FC)/aug-cc-pVTZ level [46–48, 65–70]. Selected structural parameters are shown in Fig. 1, and additional values are listed in Table 1a; the corresponding parameters from MP2(FC)/aug-cc-pVQZ and MP2(FC)/aug-cc-pV5Z optimizations are also given in Table 1a. Bond lengths for the higher-energy anti conformers are given in the footnotes to Table 1a.

Selected distances (Å) and angles (°) for the endo-exo conformers of the boronic acids H–B(OH)2, (H2N–B(OH)2), [HO–B(OH)2], and {F–B(OH)2} and for the cis conformers of the corresponding borinic acids calculated at the MP2(FC)/aug-cc-pVTZ//MP2(FC)/aug-cc-pVTZ level
Experimental structural data available for simple aliphatic boronic acids are rather limited. In 1981, Boggs and Cordell [65] presented approximate r0 structures of HB(OH)2 and FB(OH)2 based on earlier microwave studies in the gas phase reported by Kawashima et al. [71, 72]; the boron–oxygen distances were given as 1.353 and 1.366 Å for HB(OH)2 and as 1.360 and 1.365 Å for FB(OH)2. An experimental solid-phase boron–oxygen distance of 1.368 Å for B(OH)3 was given by Gajhede et al. [73]. Our calculated B–O distances, see Table 1a, are in reasonable agreement with these experimental values. More recently, Cyrański et al. [74] reported the results of an X-ray crystallographic study of n-butylboronic acid; the endo-exo monomers are arranged in centrosymmetric dimers in the crystal structure. The observed boron–oxygen distances are 1.3672(22) and 1.3795(20) Å, and the O–B–O angle is 116.048(135)° [75]; the corresponding values calculated at the MP2(FC)/aug-cc-pVTZ level for the gas-phase monomeric structure, 1.372 Å, 1.379 Å, and 117.4°, are in acceptable agreement with the crystallographic values, providing confidence in this computational level to generate sensible geometries for simple aliphatic boronic acids.
The MP2(FC)/aug-cc-pVTZ optimized boron–oxygen bond lengths, 1.386, 1.374, and 1.369 Å for the X–B–O–H cis-branch, and 1.379, 1.374, and 1.361 Å for the X–B–Ot–H trans-branch, X=N, O, and F, respectively) clearly decrease as the electronegativity of X increases [76, 77]. Improving the basis set to aug-cc-pVQZ or aug-cc-pV5Z results in a decrease in all the boron–oxygen distances by ca. 0.005 Å, see Table 1a, but does not alter the overall trend.
NBO analyses using either the HF or QCI densities with the aug-cc-pVTZ basis set identify only two boron–oxygen natural bonding orbitals for the endo-exo (and anti) conformers of H–B(OH)2, see Fig. 2, along with a total of four lone-pair orbitals, two on each oxygen atom. A similar description of the boron–oxygen bonding is also found for NH2–B(OH)2, OH–B(OH)2, and F–B(OH)2. However, second-order perturbative estimates of the donor–acceptor interaction energies, E(2) [56–59], available using the HF/aug-cc-pVTZ density in the NBO basis, indicate that significant stabilization is provided for each of these boron–oxygen bonds as a result of n O → n*B interactions involving the p-orbitals perpendicular to the planes of the molecules; the E(2) values for the cis and trans interactions of the endo-exo (anti) conformers are as follows: H, 65.0 and 66.2 kcal/mol (anti 68.5 kcal/mol); NH2, 55.6 and 58.8 kcal/mol (anti 58.3 kcal/mol); OH, 61.2 and 61.2 kcal/mol (anti 60.5 and 63.9 kcal/mol); and F: 63.3 and 66.6 kcal/mol (anti 66.4 kcal/mol). Clearly, the E(2) interaction energies increase in going from NH2 to OH and F as the electronegativity increases from N to F. Thus, NBO analyses suggest a model in which the boron–oxygen bonds in these simple aliphatic boronic acids are significantly stabilized by dative pπ–pπ interactions. This finding is in accord with the classification scheme for boron–oxygen bonding proposed by Straub [78] some 20 years ago. Based on a variety of experimental bond lengths, he proposed that boron–oxygen bonds with lengths in the range 1.36–1.37 Å have partial double bond character, BO. Indeed, the calculated boron–oxygen bond lengths for these simple boronic acids are generally in this Straub range, see Table 1a.

The two boron–oxygen natural bonding orbitals of the endo-exo conformer of H–B(OH)2, identified by the NBO analysis using HF densities with the aug-cc-pVTZ basis set
This NBO description of the boron–oxygen bonding in H–B(OH)2, HO–B(OH)2, and F–B(OH)2 is rather different from that proposed by Mierzwa et al. [79] based on a topological analyses of the electron localization functions (ELFs). These authors found that the boron–oxygen bonds in H–B(OH)2, HO–B(OH)2, and F–B(OH)2 are each described by one V(B,O) attractor with average basin populations very close to 2.0e (2.05e, 1.99e, and 2.01e, respectively), indicative of single bonds. Furthermore, they found that the non-bonding electron density for each oxygen atom is characterized by a single V(O) basin with populations about 4.0e, i.e., two lone pairs of electrons on each oxygen atom. Mierzwa et al. [79] concluded that there is “no indication of dative pπ–pπ bonding” and that the partial double bonds “proposed by Straub are actually single B–O bonds” [78, 79].
The composition of the natural atomic hybrid orbitals of the boron–oxygen NBOs is given in Table 1SA of the Supplementary Materials. The p-character of the atomic hybrids on the boron atoms for the two distinct groups of boron–oxygen bonds for H2N–B(OH)2, HO–B(OH)2, and F–B(OH)2 using the QCI density (cis: sp 2.11, sp 2.00, and sp 1.91; trans: sp 2.14, sp 2.00, and sp 1.89) decreases, while the p-character of the atomic hybrids on the corresponding oxygen atoms (cis: sp 1.41, sp 1.47, and sp 1.52; trans: sp 1.39, sp 1.47, and sp 1.51) increases, as the electronegativity increases from N to F, in accord with Bent’s rule [80]. As would be expected, the NPA charge on the boron atoms for R–B(OH)2 become more positive (QCI: +1.13e, 1.21e, 1.29e) as the electronegativity of the substituent increases, see Table 2a, while the occupancies of the boron lone-pair orbitals decrease (QCI: 0.392e, 0.371e, and 0.353e for R=NH2, OH, and F) [80]. AIM charges based on the MP2(FC)/aug-cc-pVDZ density display the same trend.
R–BH(OH) (R=H; NH2, OH, and F)
Optimized structures of the corresponding borinic acids, R–BH(OH) for (R=H; NH2, OH, and F), are also planar at the MP2(FC)/aug-cc-pVTZ level, and selected structural parameters are shown in Fig. 1, and additional values are listed in Table 1b. For comparing these R–BH(OH) structures, we specifically utilized conformers where the X–B–O–H dihedral angles (X=H; N, O, F) are cis, see Fig. 1. Although these acids lack the intramolecular B–O–H…O interactions inherent in the endo-exo conformers of the corresponding boronic acids, they retain possible intramolecular interactions of the form B–O–H…R. (The conformers of the borinic acids with the X–B–O–H dihedral angles in a trans arrangement were also optimized; bond lengths are given in the footnotes to Table 1. For X=N the trans conformer is ~1.8 kcal/mol lower in energy than the cis conformer, whereas for X=F the trans form is ~0.4 kcal/mol higher in energy.)
There is a paucity of experimental structural data for simple borinic acids. Borinic acid itself, H2BOH, is unstable, but its microwave structure has been reported by Kawashima et al. [77]: B–O = 1.352(4) Å, O–H = 0.967(14) Å, B–H = 1.200 Å (assumed), <B–O–H = 112.0(17)°, cis < H–B–O = 121.8(8)°, and trans < H–B–O = 117.2(8)°. Our calculated MP2(FC)/aug-cc-pVTZ structural parameters are in reasonable accord with these microwave values, see Fig. 1 and Table 1b, as well as with previous ab initio results [81–83]; improving the basis set to aug-cc-pVQZ and aug-cc-pV5Z does decrease the boron–oxygen bond length from 1.360 to 1.356 and 1.354 Å, respectively, in better agreement with the microwave value. Kawashima et al. [71] also observed F–BH(OH) by microwave spectroscopy in the hydrolysis of BF3 and diborane. They identified the observed conformer as trans, although they conjectured that the cis conformer would be lower in energy, consistent with what we found at the MP2(FC)/aug-cc-pVTZ level. No structural parameters were reported by Kawashima et al. [71].
The calculated boron–oxygen distance in H2B(OH), 1.360 Å at the MP2(FC)/aug-cc-pVTZ level, is 0.014 Å shorter than the corresponding distance in the H–B–O–H cis-branch of the endo-exo form of H–B(OH)2, 1.374, and 0.006 Å longer than either boron–oxygen distance in the anti form, see Table 1. In contrast to what we found for H–B(OH)2, NBO analyses using either the HF or QCI densities classify this boron–oxygen bond in H2B(OH) as a double bond, see Fig. 3. The calculated AIM boron–oxygen bond order for H2B(OH) using the MP2(FC)/aug-cc-pVDZ density is also greater than that of either boron–oxygen bonds in H–B(OH)2. Indeed, the boron–oxygen bonds in a variety of second-row borinic acids R–BH(OH) (R=Li, HBe, H2B, and H3C), where there are no competing lone pairs available for dative bonding to the boron atom, are all calculated to be slightly shorter than in their boronic acid analogs at the MP2(FC)/aug-cc-pVTZ level, and they are consistently classified as double bonds in NBO analyses using either the HF or QCI densities. Although Straub [78] did not consider H2B(OH) directly, he did classify the boron–oxygen bond in H2B(OCH3) as a double bond. The experimental gas-phase boron–oxygen bond length in H2B(OCH3) is 1.352 Å [84, 85], and the MP2(FC)/aug-cc-pVTZ geometry optimized value is almost the same, 1.351 Å. Mierzwa et al. [79] also performed a topological analysis of the ELF for H2B(OCH3) and found that the electron population of the V(B,O) basin was 2.22e and the electron population of the lone pair V(O) was only 3.29e, values indicative of some multiple bond character, but certainly not a double bond.

(left) top and (right) side views of the σ- and π-bonding orbitals of H2BOH. The NBO analysis using HF densities with the aug-cc-pVTZ basis set classified this bond as a double bond
It is important to note that a definitive boron–oxygen double bond has been somewhat elusive. In 2005, Vidovic et al. [86] prepared and characterized the first Lewis acid-coordinated oxoborane in which the boron–oxygen bond retained significant double bond character. The X-ray boron–oxygen distance in the >B=O…AlCl3 moiety was 1.304(2) Å, much shorter than the boron–oxygen bond lengths in H2B(OCH3) and (CH3)2BOB(CH3)2, 1.352 and 1.359 Å, that Straub [78] classified as having B=O bonds, but much longer than the experimental B≡O triple bonds for HB≡O, 1.201 Å [87], or the MP2(FC)/aug-cc-pV5Z optimized length, 1.209 Å. Subsequently, geometry optimizations and NBO analyses showed that the core borocycle (–HN=CH–CH=CH–NH–B–)O at the heart of the Vidovic et al. [86] structure also had a B=O double bond in the gas phase; at the MP2(FC)/aug-cc-pVTZ level, the boron–oxygen distance in (–HN=CH–CH=CH–NH–B–)O is 1.270 Å [88]. Thus, it appears that a B=O double bond can be significantly shorter than the boron–oxygen bond found in H2B(OH) and that the Straub classification of B=O double bond lengths being ~1.35 Å may be closer to the upper limit for the boron–oxygen double bond length.
For the borinic acids H2N–BH(OH), HO–BH(OH), and F–BH(OH), where competitive donor–acceptor interactions involving the lone pairs on N, O and F are possible, the calculated boron–oxygen distances are ~0.01 Å shorter than those in the cis-branch of the corresponding boronic acids, compare Table 1a, b. In these three cases, however, NBO analyses using QCI densities classify the boron–oxygen bonds as single bonds, similar to what we found for the corresponding boronic acids. NBO analysis with the HF density also predicts the boron–oxygen bond in H2N–BH(OH) to be a single bond. However, the second-order stabilization energy associated with the nO → n*B interaction is 61.5 kcal/mol, some 6 kcal/mol greater than the corresponding value in the analogous boronic acid, H2N–B(OH)2. The HF density for F–BH(OH) characterizes the boron–oxygen bond as a double bond. Consistent with these NBO findings, AIM calculations predict the boron–oxygen bond orders for the borinic acids to be greater than those for either of the boron–oxygen bond orders in the analogous boronic acids.
The p-character of the atomic hybrid orbitals on the boron atoms associated with the boron–oxygen bonds in H2N–BH(OH), HO–BH(OH), and F–BH(OH) is greater than that for either of the boron–oxygen bonds in the corresponding boronic acids, compare Tables 1SA and 1SB. Consistent with what we observed for the boronic acids, the p-character on the boron atom decreases (QCI density: sp 2.31, sp 2.24, and sp 2.17), while the p-character on the oxygen atom increases (QCI density: sp 1.27, sp 1.30, and sp 1.33) as the electronegativity of the substituent increases.
The NPA and AIM charges on the boron atoms for the borinic acids are significantly less positive than for the corresponding boronic acids, see Table 2. The NPA occupation of the boron lone-pair orbital on the borinic acid is lower than that on the corresponding boronic acid, e.g., 0.267e for F–BH(OH) compared to 0.353e for F–B(OH)2.
Boron–nitrogen bonding
H2N–B(OH)2
The calculated boron–nitrogen bond length in H2N–B(OH)2 is 1.417 Å at the MP2(FC)/aug-cc-pVTZ level; the authors are unaware of any experimental value for this bond length. For comparison, we note that the corresponding bond lengths at this computational level for HN≡BH, H2N=BH2, and H3N → BH3 are 1.246, 1.395, and 1.652 Å, respectively; the experimental values for H2N=BH2 and H3N → BH3 are 1.391 Å [89, 90] and 1.672 Å [91] (for some interesting crystal structure comparisons, see [92]).
Straub’s [78] bond-length classification scheme for boron–nitrogen bonds is: B≡N, 1.22–1.26 Å; B=N, 1.34–1.41 Å; B–N, 1.65–1.67 Å; Paetzold [93] suggested similar values: B≡N, 1.26 Å; B=N, 1.41; B–N, 1.58 Å. For HN≡BH and H2N=BH2, the MP2(FC)/aug-cc-pVTZ boron–nitrogen bond lengths are clearly within Straub’s triple and double bond-length ranges, respectively, and the results of our NBO analyses using HF and QCI densities support these classifications [94–96]. Berski et al. [94] performed a topological ELF analysis of H2N=BH2 and found two bonding attractors V(B,N) localized above and below the symmetry plane, consistent with the usual Lewis formula; the total population of the two basins was nearly 4.0e consistent with a boron–nitrogen double bond.
For H2N–B(OH)2, the calculated MP2(FC)/aug-cc-pVTZ bond length is just outside the double bond range suggested by Straub [78], although the MP2(FC)/aug-cc-pVQZ and MP2(FC)/aug-cc-pV5Z lengths are slightly shorter, see Table 1a. NBO analyses using the QCI or HF densities identify only one boron–nitrogen natural bond orbital in H2N–B(OH)2. However, the second-order stabilization energy available using the HF density for the nN → n*B interaction is 76.8 kcal/mol, some 20 kcal/mol larger than that for either of the nO → n*B interactions, 55.6 and 58.8 kcal/mol, suggesting significant boron–nitrogen double bond character. Consistent with this finding, the calculated AIM boron–nitrogen bond order is larger than either of the two boron–oxygen bond orders.
The AIM and NPA charges clearly indicate that the nitrogen atom is more negatively charged than either of the oxygen atoms, see Table 2a. The natural atomic hybrids that compose the natural bond orbitals show that the boron–oxygen bonds involve greater p-character than the boron–nitrogen bond, see Table 1S.
H2N–BH(OH)
The calculated boron–nitrogen bond length for the borinic acid H2N–BH(OH) is 1.412 Å at the MP2(FC)/aug-cc-pVTZ level, ca. 0.005 Å shorter than the boron–nitrogen bond in the corresponding boronic acid; similar differences are at found at the higher levels, see Table 1a, b. To the author’s knowledge, there are no experimental structural parameters available for this acid. The calculated length of the boron–nitrogen bond is essentially at the upper limit of Straub’s suggested length for a double bond 1.34–1.41 Å [78]. NBO analyses using the QCI density identify the boron–nitrogen bond as a double bond, whereas the HF density classifies it as a single bond, albeit with a large nN → n*B second-order interaction energy of 85.8 kcal/mol, ~9 kcal/mol greater than for H2N–B(OH)2. Consistent with this finding, the calculated AIM boron–nitrogen bond order in H2N–BH(OH) is larger than the corresponding bond order in H2N–B(OH)2 at the MP2(FC)/aug-cc-pVDZ level.
The charge on the boron atom is less positive and the charge on the nitrogen atom is less negative in H2N–BH(OH) than in H2N–B(OH)2, see Table 2. As noted above for H2N–B(OH)2, the natural atomic hybrids that compose the natural bond orbitals show that the boron–oxygen bonds involve greater p-character than the boron–nitrogen bond, see Table 1S. The p-character of the natural atomic hybrids on the boron atom for the boron–nitrogen bond in H2N–BH(OH) (B:sp 1.97; N:sp 1.20) is greater than it is in H2N–B(OH)2 (B:sp 1.77; N:sp 1.31), whereas the reverse holds for the p-character of the nitrogen atom.
Boron–fluorine bonding
F–B(OH)2
The calculated boron–fluorine bond length in F–B(OH)2 is 1.338 Å at the MP2/aug-cc-pVTZ level and only slightly shorter at the MP2(FC)/aug-cc-pVQZ and MP2(FC)/aug-cc-pV5Z levels, 1.334 and 1.333 Å, see Table 1a, somewhat longer than the estimated experimental value, ~1.323 Å, which is based on results from F2B(OH) [65, 97]. In view of the lack of an experimental bond length for the boron–fluorine bond in F–B(OH)2, we calculated the length of a variety of boron–fluorine bonds in several related small molecules. The MP2/aug-cc-pVTZ level boron–fluorine bond lengths for BF(1Sg), H2BF, HBF2, and BF3 are 1.271, 1.326, 1.320, and 1.317 Å, respectively. These calculated lengths are consistently somewhat longer than the gas-phase experimental values: 1.263 Å [98], 1.316 Å [99], 1.311 Å [99], and 1.309 Å [100]. Using the more complete aug-cc-pVQZ and aug-cc-pV5Z basis sets, the calculated lengths of these bonds are shorter (BF: 1.265 and 1.2644 Å; H2BF 1.321 and 1.320 Å; HBF2: 1.316 and 1.315 Å; BF3: 1.313 and 1.312 Å), but remain slightly longer than the corresponding experimental values. For F–B(OH)2, the MP2(FC)/aug-cc-pVQZ and MP2(FC)/aug-cc-pV5Z fluorine–boron bond lengths are 1.334 and 1.333 Å, suggesting that the estimated experimental value of 1.323 Å may be somewhat short.
Straub’s [78] bond-length classification for boron–fluorine bonds is: B≡F, 1.21–1.26 Å; BF, 1.31–1.33 Å; B–F, 1.37–1.40 Å. As Straub clearly noted, there were good structural data for only one molecule with a B=F double bond, i.e., H2BF; interestingly, the experimental fluorine–boron bond length in this case, 1.316 Å [99], is in the range Straub classified as having only partial double bond character. NBO calculations on H2BF using either the HF or QCI densities identify two fluorine–boron bonds (σ and a π(dative)), along with and two lone pairs on the F atom. In contrast, corresponding NBO analyses for F2BH, where the experimental boron–fluorine bond length is 1.311 Å [99], find only two single boron–fluorine σ bonds and a total of six lone pairs on the F atoms. However, the second-order stabilization energy available using the HF density for the nF → n*B interaction is substantial, 54.9 kcal/mol, indicative of a partial boron–fluorine double bond, BF, consistent with Straub’s classification [78]. The experimental boron–fluorine bond length for BF3, 1.309 Å [100], is shorter than the corresponding bond lengths in either H2BF or HBF2. NBO analyses find a total of three sigma bonds, and the second-order stabilization energy available using the HF density for each nF → n*B interaction is 53.1 kcal/mol, similar to the value found for F2BH, suggesting partial double bonds, consistent with the Straub classification [78]. NBO analyses identify one boron–fluorine σ bond in F–B(OH)2, but the second-order nF → n*B interaction energy, 47.2 kcal/mol using the HF density, is only slightly smaller than the corresponding energies for BF3 or H2BF, indicative of a partial double bond.
Both AIM and NPA charges indicate that the charge on the fluorine atom is only about 55–60 % of the charge on the oxygen atoms, see Table 2. It is also interesting to note that the p-character of the atomic hybrids on the boron atom for the B–F bond sp 2.21 has greater p-character than for the B–O bonds, sp 1.89 and sp 1.91, see Table 1S.
F–BH(OH)
The calculated length of the boron–fluorine bond in F–BH(OH), 1.339 Å, is essentially the same as the corresponding bond length in F–B(OH)2, compare Table 1a, b. Although Kawashima et al. [71] detected F–BH(OH) in microwave experiments, no specific structural parameters were reported. Interestingly, a semi-experimental equilibrium structure of F2BOH has been generated in an extraordinary paper by Breidung et al. [101]. Our MP2(FC)/aug-cc-pVTZ optimized structure for this molecule is in acceptable agreement with their semi-experimental structure, e.g., our boron–oxygen distance is 1.355 Å compared to 1.3459(4) Å, and our boron–fluorine cis and trans distances are 1.333 and 1.323 Å compared to 1.322(6) and 1.316(6) Å.
NBO analyses using either the HF or QCI densities identify the boron–fluorine bond in F–BH(OH) as a single bond. Since the HF density identifies the boron–oxygen bond in F–BH(OH) as a double bond (see above), the largest second-order stabilization energy in this case involves delocalization from a fluorine lone-pair orbital into a boron–oxygen antibonding orbital; the value of E(2) for this nF → π*B-O interaction is 43.6 kcal. The boron–fluorine and boron–oxygen AIM bond orders are larger in F–BH(OH) than in F–B(OH)2.
Concluding remarks
Our systematic comparison of the boron–oxygen NBOs in R–B(OH)2 and R–BH(OH) (R=H; NH2, OH, and F) finds large nO → n*B second-order stabilization energies, indicating that the boron–oxygen bonds in these simple aliphatic boronic and borinic acids have a significant contribution from dative pπ–pπ bonding; indeed, π- and σ-bonds are identified in the NBO analyses of H2B(OH). Our analyses also show that the p-character of boron hybrid orbitals associated with the B–X (X=N, O, F) bonds is larger for the borinic acid than for the corresponding boronic acid. The results of geometry optimizations reveal that the boron–oxygen bond length in each borinic acid is shorter than either of the boron–oxygen bond lengths in the corresponding boronic acid. Furthermore, the boron–oxygen bond lengths in the X–B–Ot–H trans-branch of the endo-exo form of the boronic acids and for the X–B–O–H cis-branch of the boronic and borinic acids (X=N, O, and F, respectively) decrease as the electronegativity of X increases.
Further insight into the bonding in the corresponding boronic and borinic acids can be gained by evaluating the thermodynamics for the reaction R–B(OH)2 + R–BH2 → 2 R–BH(OH), in which the formal bonding in reactants and products is balanced in the spirit of homodesmotic reactions [102–105]. Using the endo-exo form for the boronic acids and the cis form of the borinic acids, the values of \(\Delta{\text{H}}_{298}^{0}\) for this reaction are −1.1, +1.6, −4.1 and −7.1 kcal/mol for R=H, NH2, OH, and F, respectively; using the anti form for the boronic acids, which eliminates the B–Ot–H…O intramolecular hydrogen bond, yields −2.1, −1.3, −8.1, and −7.7 kcal/mol. The exothermicity of these reactions suggests somewhat stronger bonding surrounding the boron atom in the borinic acids than in the boronic acids, consistent with the structural features noted above.
Our investigation highlights the significant differences in the strength and composition of the bonding and in the detailed structural features of simple aliphatic boronic and borinic acids that will affect their relative capabilities to bind to specific substrates. Future studies will include bulkier substituents and model substrates.
References
Arimori S, Bell ML, Oh CS, Frimat KA, James TD (2002) Modular fluorescence sensors for saccharides. J Chem Soc Perkin Trans 1(6):803–808
Arzt M, Seidler C, Ng DY, Weil T (2014) Reversible click reactions with boronic acids to build supramolecular architectures in water. Chem Asian J 9(8):1994–2003
Baker SJ, Tomsho JW, Benkovic SJ (2011) Boron-containing inhibitors of synthetases. Chem Soc Rev 40(8):4279–4285
Bull SD, Davidson MG, den Elsen Van, Jean MH, Fossey JS, Jenkins ATA, Jiang Y et al (2012) Exploiting the reversible covalent bonding of boronic acids: recognition, sensing, and assembly. Acc Chem Res 46(2):312–326
Cambre JN, Sumerlin BS (2011) Biomedical applications of boronic acid polymers. Polymer 52(21):4631–4643
Dembitsky VM, Quntar AA, Srebnik M (2004) Recent advances in the medicinal chemistry of α-Aminoboronic acids, amine-carboxyboranes and their derivatives. Mini Rev Med Chem 4(9):1001–1018
Deng CC, Brooks WL, Abboud KA, Sumerlin BS (2015) Boronic acid-based hydrogels undergo self-healing at neutral and acidic pH. ACS Macro Lett 4(2):220–224
Dienstmaier JF, Gigler AM, Goetz AJ, Knochel P, Bein T, Lyapin A et al (2011) Synthesis of well-ordered COF monolayers: surface growth of nanocrystalline precursors versus direct on-surface polycondensation. ACS Nano 5(12):9737–9745
Fossey JS, D’Hooge F, van den Elsen Jean MH, Pereira Morais MP, Pascu SI, Bull SD et al (2012) The development of boronic acids as sensors and separation tools. Chem Rec 12(5):464–478
Fu H, Fang H, Sun J, Wang H, Liu A, Sun J et al (2014) Boronic acid-based enzyme inhibitors: a review of recent progress. Curr Med Chem 21(28):3271–3280
Fujita N, Shinkai S, James TD (2008) Boronic acids in molecular self-assembly. Chem Asian J 3(7):1076–1091
Galbraith E, James TD (2010) Boron based anion receptors as sensors. Chem Soc Rev 39(10):3831–3842
Guan Y, Zhang Y (2013) Boronic acid-containing hydrogels: synthesis and their applications. Chem Soc Rev 42(20):8106–8121
Guo Z, Shin I, Yoon J (2012) Recognition and sensing of various species using boronic acid derivatives. Chem Commun 48(48):5956–5967
Hall DG (2006) Boronic acids: preparation, applications in organic synthesis and medicine, 1st edn. Wiley, New Jersey
Huang S, Jia M, Xie Y, Wang J, Xu W, Fang H (2012) The progress of selective fluorescent chemosensors by boronic acid. Curr Med Chem 19(16):2621–2637
James TD, Shinkai S (2002) Artificial receptors as chemosensors for carbohydrates. In: Penadés S (ed) Host-Guest Chemistry. Springer, Berlin Heidelberg, pp 159–200
James TD, Phillips MD, Shinkai S (2006) Boronic acids in saccharide recognition. Royal Society of Chemistry, Cambridge
Korich AL, Iovine PM (2010) Boroxine chemistry and applications: a perspective. Dalton Trans 39(6):1423–1431
Lacina K, Skládal P, James TD (2014) Boronic acids for sensing and other applications-a mini-review of papers published in 2013. Chem Cent J 8(1):60
Loveless D, Holtsclaw J, Weaver J, Ogle J, Saini R (2014) Multifunctional boronic acid crosslinker for fracturing fluids. IPTC 2014: International petroleum technology conference 2014
Martin AR, Vasseur J, Smietana M (2013) Boron and nucleic acid chemistries: merging the best of both worlds. Chem Soc Rev 42(13):5684–5713
Matsumoto A, Miyahara Y (2014) Current development status and perspectives of self-regulated insulin delivery systems: a review. Electron Commun Jpn 97(12):57–61
Nevalainen V (1996) HB, (OH)2 and H2 CO as probes for a study on binding of dialkoxyboranes and ketones to oxazaborolidines capable of catalyzing the enantioselective reduction of ketones. Tetrahedron Asymmetry 7(9):2655–2664
Nishiyabu R, Kubo Y, James TD, Fossey JS (2011) Boronic acid building blocks: tools for self assembly. Chem Commun 47(4):1124–1150
Nishiyabu R, Kubo Y, James TD, Fossey JS (2011) Boronic acid building blocks: tools for sensing and separation. Chem Commun 47(4):1106–1123
Petasis NA (2007) Expanding roles for organoboron compounds–Versatile and valuable molecules for synthetic, biological and medicinal chemistry. Aust J Chem 60(11):795–798
Scorei R (2012) Is boron a prebiotic element? A mini-review of the essentiality of boron for the appearance of life on Earth. Origins of Life and Evolution of Biospheres 42(1):3–17
Smoum R, Rubinstein A, Dembitsky VM, Srebnik M (2012) Boron containing compounds as protease inhibitors. Chem Rev 112(7):4156–4220
Touchet S, Carreaux F, Carboni B, Bouillon A, Boucher J (2011) Aminoboronic acids and esters: from synthetic challenges to the discovery of unique classes of enzyme inhibitors. Chem Soc Rev 40(7):3895–3914
Trippier PC, McGuigan C (2010) Boronic acids in medicinal chemistry: anticancer, antibacterial and antiviral applications. Med Chem Commun 1(3):183–198
Wang X, Xia N, Liu L (2013) Boronic acid-based approach for separation and immobilization of glycoproteins and its application in sensing. Int J Mol Sci 14(10):20890–20912
Collins BE, Metola P, Anslyn EV (2013) On the rate of boronate ester formation in ortho-aminomethyl-functionalised phenyl boronic acids. Supramol Chem 25(2):79–86
Lee D, Taylor MS (2011) Borinic acid-catalyzed regioselective acylation of carbohydrate derivatives. J Am Chem Soc 133(11):3724–3727
Chan L, Taylor MS (2011) Regioselective alkylation of carbohydrate derivatives catalyzed by a diarylborinic acid derivative. Org Lett 13(12):3090–3093
Lee D, Williamson CL, Chan L, Taylor MS (2012) Regioselective, borinic acid-catalyzed monoacylation, sulfonylation and alkylation of diols and carbohydrates: expansion of substrate scope and mechanistic studies. J Am Chem Soc 134(19):8260–8267
Bailey P, Cousins G, Snow G, White A (1980) Boron-containing antibacterial agents: effects on growth and morphology of bacteria under various culture conditions. Antimicrob Agents Chemother 17(4):549–553
Baker SJ, Akama T, Zhang Y, Sauro V, Pandit C, Singh R et al (2006) Identification of a novel boron-containing antibacterial agent (AN0128) with anti-inflammatory activity, for the potential treatment of cutaneous diseases. Bioorg Med Chem Lett 16(23):5963–5967
Benkovic S, Liu C, Tomsho JW (2015) Inventors; boronic and borinic acid compound as inhibitors of sulfenic acid-containing proteins. Patent 20,150,140,635, May 21, 2015
Lee V, Benkovic SJ (2004) Inventors; antibiotics containing borinic acid complexes and methods of use. Patent 20,040,224,923, Nov 11, 2004
Simpelkamp J, Jones JB (1992) Borinic acid inhibitors as probes of the factors involved in binding at the active sites of subtilisin carlsberg and α-chymotrypsin. Bioorg Med Chem Lett 2(11):1391–1394
Chudzinski MG, Chi Y, Taylor MS (2011) Borinic acids: a neglected class of organoboron compounds for recognition of diols in aqueous solution. Aust J Chem 64(11):1466–1469
Gouliaras C, Lee D, Chan L, Taylor MS (2011) Regioselective activation of glycosyl acceptors by a diarylborinic acid-derived catalyst. J Am Chem Soc 133(35):13926–13929
Lee D, Taylor MS (2013) Regioselective silylation of pyranosides using a boronic acid/Lewis base co-catalyst system. Org Biomol Chem 11(33):5409–5412
Dimitrijević E, Taylor MS (2013) 9-Hetero-10-boraanthracene-derived borinic acid catalysts for regioselective activation of polyols. Chem Sci 4(8):3298–3303
Bhat KL, Markham GD, Larkin JD, Bock CW (2011) Thermodynamics of boroxine formation from the aliphatic boronic acid monomers R–B (OH)2 (R=H, H3C, H2N, HO, and F): a computational investigation. J Phys Chem A 115(26):7785–7793
Bock CW, Larkin JD (2012) Heats of formation for the boronic acids R–B (OH)2 and boroxines R3 B3 O3 (R=H, Li, HBe, H2 B, H3C, H2N, HO, F, and Cl) calculated at the G2, G3, and G4 levels of theory. Comput Theor Chem 986:35–42
Rao NZ, Larkin JD, Bock CW (2015) A computational investigation of monosubstituted boroxines (RH2B3O3): structure and formation. Struct Chem 26:1151–1162
Møller C, Plesset MS (1934) Note on an approximation treatment for many-electron systems. Phys Rev 46(7):618
Dunning TH Jr (1989) Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys 90(2):1007–1023
Kendall RA, Dunning TH Jr, Harrison RJ (1992) Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J Chem Phys. 96(9):6796–6806
Peterson KA, Woon DE, Dunning TH Jr (1994) Benchmark calculations with correlated molecular wave functions. IV. The classical barrier height of the H H2 → H2 H reaction. J Chem Phys 100(10):7410–7415
Woon DE, Dunning TH Jr (1993) Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J Chem Phys. 98(2):1358–1371
Frisch M, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, et al (2008) Gaussian 03, revision C. 02
Frisch M, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, et al (2009) Gaussian 09, revision A. 1. Gaussian Inc., Wallingford, CT
Carpenter J, Weinhold F (1988) Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure. J Mol Struct (Thoechem) 169:41–62
Foster J, Weinhold F (1980) Natural hybrid orbitals. J Am Chem Soc 102(24):7211–7218
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83(2):735–746
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem Rev 88(6):899–926
Weinhold F, Glendening ED (2001) NBO 5.0 Program Manual: Natural Bond Orbital Analysis Programs. Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin, Madison, WI 53706
Glendening E, Badenhoop J, Reed A, et al (2013) NBO 6.0. Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin, Madison, WI
Bader RF (1990) Atoms in molecules. John Wiley & Sons, Ltd
Cioslowski J, Mixon ST (1991) Covalent bond orders in the topological theory of atoms in molecules. J Am Chem Soc 113(11):4142–4145
Cioslowski J, Nanayakkara A (1994) A new robust algorithm for fully automated determination of attractor interaction lines in molecules. Chem Phys Lett 219(1):151–154
Boggs JE, Cordell FR (1981) Accurate ab initio gradient calculation of the structures and conformations of some boric and fluoroboric acids. Basis-set effects on angles around oxygen. J Mol Struct (Thoechem) 76(4):329–347
Duan X, Linder D, Page M, Soto M (1999) Structures and thermochemistry of BH l F m (OH) n and several XYBO compounds at the G-2 level of theory. J Mol Struct (Thoechem) 465(2):231–242
Larkin JD, Bhat KL, Markham GD, Brooks BR, Schaefer HF, Bock CW (2006) Structure of the boronic acid dimer and the relative stabilities of its conformers. J Phys Chem A 110(36):10633–10642
Larkin JD, Bhat KL, Markham GD, Brooks BR, Lai JH, Bock CW (2007) A computational investigation of the geometrical structure and protodeboronation of boroglycine, H2 N-CH2-B (OH) 2. J Phys Chem A 111(28):6489–6500
So S (1982) A theoretical study of the conformations of BF (OH)2. J Mol Struct (Thoechem) 89(3):255–258
Stefani D, Pashalidis I, Nicolaides AV (2008) A computational study of the conformations of the boric acid (B(OH)3), its conjugate base ((HO)2BO−) and borate anion. J Mol Struct (Thoechem) 853(1):33–38
Kawashima Y, Takeo H, Matsumura C (1978) Microwave spectroscopic detection of BFHOH and BH(OH)2. Chem Phys Lett 57(1):145–147
Kawashima Y, Takeo H, Matsumura C (1979) Microwave spectrum of fluorodihydroxy borane, BF(OH)2. J Mol Spectrosc 78(3):493–505
Gajhede M, Larsen S, Rettrup S (1986) Electron density of orthoboric acid determined by X-ray diffraction at 105 K and ab initio calculations. Acta Crystallogr B 42(6):545–552
Cyrański MK, Jezierska A, Klimentowska P, Panek JJ, Żukowska GZ, Sporzyński A (2008) Structural and spectroscopic properties of an aliphatic boronic acid studied by combination of experimental and theoretical methods. J Chem Phys. 128(12):124512
Private Communication with A. Sporzynski
Wells PR (1968) Group electronegativities. Prog Phys Org Chem 6:111–145
Kawashima Y, Takeo H, Matsumura C (1981) Microwave spectrum of borinic acid BH2OH. J Chem Phys 74(10):5430–5435
Straub DK (1995) Lewis structures of boron compounds involving multiple bonding. J Chem Educ 72(6):494
Mierzwa G, Gordon AJ, Latajka Z, Berski S (2015) On the multiple B O bonding using the topological analysis of electron localisation function (ELF). Comput Theor Chem 1053:130–141
Bent HA (1961) An appraisal of valence-bond structures and hybridization in compounds of the first-row elements. Chem Rev 61(3):275–311
Dill J (1975) Schleyer PvR, Pople J. Molecular orbital theory of the electron structure of organic compounds. XXIV. Geometries and energies of small boron compounds. Comparisons with carbocations. J Am Chem Soc 97(12):3402–3409
Cordell FR, Boggs JE (1980) The barriers to methyl group rotation in s-cis-and s-trans-methyl nitrite. J Mol Struct 64:57–65
Gropen O, Johansen R (1975) Barrier to internal rotation and π-bonding In hydroxyborane, H2BOH, studied by ab in1tio calculations. J Mol Struct 25(1):161–167
Carpenter JD, Ault BS (1992) Matrix isolation study of the mechanism of the reaction of diborane with ammonia: pyrolysis of the H3 B NH3 adduct. Chem Phys Lett 197(1):171–174
Kawashima Y, Takeo H, Matsumura C (1986) Microwave spectrum, structure, dipole moment, quadrupole coupling constants, and barrier to internal rotation of methoxyborane, CH3 OBH2. J Mol Spectrosc 116(1):23–32
Vidovic D, Moore JA, Jones JN, Cowley AH (2005) Synthesis and characterization of a coordinated oxoborane: Lewis acid stabilization of a boron–oxygen double bond. J Am Chem Soc 127(13):4566–4567
Kawashima Y, Endo Y, Hirota E (1989) Microwave spectrum, molecular structure, and force field of HBO. J Mol Spectrosc 133(1):116–127
Larkin JD, Bhat KL, Markham GD, James TD, Brooks BR, Bock CW (2008) A computational characterization of boron–oxygen multiple bonding in HN CH–CH CH–NH–BO. J Phys Chem A 112(36):8446–8454
Ortiz J (1989) An electron propagator study of bonding in aminoborane. Chem Phys Lett 156(5):489–493
Sugie M, Takeo H, Matsumura C (1987) Microwave spectrum and molecular structure of aminoborane, BH2 NH2. J Mol Spectrosc 123(2):286–292
Thorne L, Suenram R, Lovas F (1983) Microwave spectrum, torsional barrier, and structure of BH3NH3. J Chem Phys 78(1):167–171
Klooster WT, Koetzle TF, Siegbahn PE, Richardson TB, Crabtree RH (1999) Study of the NH HB dihydrogen bond including the crystal structure of BH3NH3 by neutron diffraction. J Am Chem Soc 121(27):6337–6343
Iminoboranes Paetzold P (1987) Adv Inorgan Chem Radiochem 31:123–170
Berski S, Latajka Z, Gordon AJ (2011) On the multiple B–N bonding in boron compounds using the topological analysis of electron localization function (ELF). New J Chem 35(1):89–96
Leroy G, Sana M, Wilante C (1993) Evaluation of the bond energy terms for the various types of boron–nitrogen bonds. Theoret Chim Acta 85(1–3):155–166
Niedenzu K, Dawson JW (1965) Boron nitride. In: Niedenzu K, Dawson JW (eds) Boron–nitrogen compounds, vol. 6. Springer, Berlin Heidelberg, pp 147–153
Takeo H, Curl R (1972) Microwave spectrum of BF2OH. J Chem Phys 56:4314–4317
Cazzoli G, Cludi L, Degli Esposti C, Dore L (1989) The millimeter and submillimeter-wave spectrum of boron monofluoride: equilibrium structure. J Mol Spectrosc 134(1):159–167
Takeo H, Sugie M, Matsumura C (1993) Microwave spectroscopic detection of fluoroborane, BH2 F. J Mol Spectrosc 158(1):201–207
Cox AP, Hubbard SD, Waterfield S (1986) The microwave spectrum and structure of CH3 BBr2. J Mol Spectrosc 118(2):459–470
Breidung J, Demaison J, D’Eu J, Margulès L, Collet D, Mkadmi E et al (2004) Ground-state constants, ab initio anharmonic force field, and equilibrium structure of F2 BOH. J Mol Spectrosc 228(1):7–22
George P, Trachtman M, Bock CW, Brett AM (1975) An alternative approach to the problem of assessing stabilization energies in cyclic conjugated hydrocarbons. Theoret Chim Acta 38(2):121–129
George P, Trachtman M, Bock CW, Brett AM (1976) An alternative approach to the problem of assessing destabilization energies (strain energies) in cyclic hydrocarbons. Tetrahedron 32(3):317–323
George P, Trachtman M, Bock CW, Brett AM (1976) Homodesmotic reactions for the assessment of stabilization energies in benzenoid and other conjugated cyclic hydrocarbons. J Chem Soc Perkin Trans 2(11):1222–1227
Wheeler SE, Houk KN, Schleyer PvR, Allen WD (2009) A hierarchy of homodesmotic reactions for thermochemistry. J Am Chem Soc 131(7):2547–2560
Acknowledgments
This research was supported in part by the National Science Foundation through XSEDE resources provided by the XSEDE Science Gateways program. The PQS Cluster Facility at Philadelphia University was also used for the calculations described in this manuscript. J.D.L. would like to thank the National Institutes of Health for research support (Grant: 5K22HL113045-04).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rao, N.Z., Larkin, J.D. & Bock, C.W. A comparison of the structure and bonding in the aliphatic boronic R–B(OH)2 and borinic R–BH(OH) acids (R=H; NH2, OH, and F): a computational investigation. Struct Chem 27, 1081–1091 (2016). https://doi.org/10.1007/s11224-015-0730-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-015-0730-5