
Abstract
Boroxines have emerged as an important class of compounds with diverse applications in many fields. However, there are limited experimental and/or computational structural and thermal data available for these compounds or for their corresponding boronic acids. In this investigation, we report structural parameters for a variety of aliphatic monosubstituted boroxines (RH2B3O3) and their enthalpies of formation via the dehydration reactions from the boronic acids (R–B(OH)2), i.e., R–B(OH)2 + 2H–B(OH)2 → RH2B3O3 + 3H2O. Equilibrium geometries of all the boronic acids and monosubstituted boroxines involved in this article were obtained using second-order Møller–Plesset perturbation theory with the Dunning–Woon aug-cc-pVDZ and aug-cc-pVTZ basis sets; heats of formation were calculated at the G3 level of theory. The specific substituents, R, include all the second- and third-period hydrides, as well as a selection of electron-donating/electron-withdrawing groups.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Boroxines are an important class of compounds having numerous applications in chemistry, medicine, and material science [1–4]. Although some experimental/computational structural and thermochemical data for symmetrically trisubstituted boroxines (R3B3O3) are currently available [5–13], there is a paucity of such data for heteroboroxines, particularly when aliphatic substituents are involved [8, 14–17]. Although we are concerned in this article with aliphatic substituents, it is important to note that Tokunaga et al. [17] experimentally established the existence of several heterophenylboroxines in the gas phase, as well as in solution. In our continuing effort to obtain reliable data for a series of fundamental boronic acids and boroxines, and the reactions that lead to their interconversion, we have undertaken a systematic investigation of the structures of a wide variety of monosubstituted boroxines (RH2B3O3) and the thermodynamics of their formation via the dehydration reactions
The substituents R in this investigation include all the second- and third-period hydrides, as well as a selection of electron-donating/electron-withdrawing groups. Since limited experimental and/or computational data are available for many of the corresponding boronic acids in this study, we performed extensive geometry optimizations for many of these acids to determine the global minima on their potential energy surfaces (PESs). Boronic acids serve as important building blocks for the synthesis of a variety of natural products, pharmaceuticals, and materials [1]; the results of the calculations reported in this article, in conjunction with those from our previous studies [5, 13], provide the most comprehensive collection of structural and thermochemical data currently available for aliphatic boronic acids.
Computational methods
Equilibrium geometries of all the boronic acids and monosubstituted boroxines involved in this article were obtained using second-order Møller–Plesset perturbation theory (MP2) [18] with the frozen core (FC) option, which neglects core-electron correlation; the Dunning–Woon aug-cc-pVDZ and aug-cc-pVTZ basis sets [19–22] were employed in these optimizations. The GAUSSIAN 03 [23] and GAUSSIAN 09 [24] suits of programs were used for all the calculations. Frequency analyses were performed analytically for all the boronic acids and boroxines in this investigation at the MP2/aug-cc-pVDZ level to confirm that the optimized structures were local minima on the potential energy surface (PES) and to correct reaction energies to 298.15 K; for the boronic acids and a few of the boroxines, frequencies were also calculated at the MP2/aug-cc-pVTZ level. Heats of formation at 298.15 K were calculated at the G3 level of theory [25, 26]. Atomic charges were obtained from natural population analyses (NPA), and the bonding was analyzed with the aid of natural bond orbitals (NBOs) [27–30].
Boronic acids
There is a lack of reliable experimental data for monomeric boronic acids because it is difficult to obtain pure samples that have the formula R–B(OH)2; these acids often separate from aqueous solutions as hydrates that undergo dehydration to yield boroxines [31].
Structures
For the boronic acids, three different orientations of the hydroxyl groups are typically possible: endo–exo, anti, and syn, see Fig. 1. It should be noted that in some cases the two X–B–O angles (where X denotes the atom of R directly bonded to the B atom) can be quite different, resulting in a bridging-like structure [32]; this is especially true when X carries a partial positive charge, resulting in an X+O− electrostatic interaction.

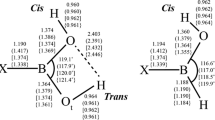
Endo–exo, anti, and syn conformers of the R–B(OH)2 monomers; the distances (Å) and angles (°) are for H–B(OH)2[O–B(OH)2] at the MP2(FC)/aug-cc-pVTZ//MP2(FC)/aug-cc-pVTZ computational level
Calculations in vacuo (and using implicit solvation models) have generally found that the endo–exo form of R–B(OH)2 is lowest in energy for a variety of small aliphatic substituents and that the syn form is highest in energy; energy differences between the endo–exo and anti conformers can be relatively small [5, 13, 32–38]. It should be noted that some exceptions have been reported, e.g., at the MP2/aug-cc-pVTZ level, the anti conformer of Cl–B(OH)2 is slightly lower in energy than the endo–exo conformer [13]. In view of this finding and since we consider more complex substituents in this article, including some charged species, we initially explored the PESs of the boronic acids not described in our previous articles [13] with particular attention to the structure of the –B(OH)2 moiety and the orientation of the substituent R.
Selected geometrical parameters for the endo–exo conformers of the boronic acids used in this article are listed at the MP2/aug-cc-pVDZ and MP2/aug-cc-pVTZ levels in Table 1; in some cases, the corresponding parameters for the anti conformers are given in the footnotes of the table. Enthalpy changes at 298.15 K, \(\Delta {\text{H}}_{298}^{0}\), for the endo–exo → anti conversions are listed in Table 2. For many of these simple acids, the endo–exo → anti conversion is endothermic by a few kcal/mol, in accord with our previous results [13] for the hydride substituents across the second row of the periodic table. This energy difference is in part due to the intramolecular B–O–H…O interaction within the boronic acid moiety in the endo–exo form; H…O distances for the acids in this study range from 2.275 Å for Li–B(OH)2 to 2.593 Å for NH3 +–B(OH)2 [5, 13]. However, interactions between the hydroxyl hydrogen atoms of the –B(OH)2 moiety and the substituent can stabilize the anti form relative to the endo–exo form. For example, consider the acids with R=O−, COO−, NO2, and COOH where there are two possible B–O–H…Osubstituent interactions in the anti form compared to only one in the endo–exo form. For all four of these acids, the anti form is calculated to be lower in energy than the endo–exo form, see Table 2.
For some substituents, the NPA charge on the atom directly connected to the boron atom is extremely positive, a feature that can lead to very asymmetric endo–exo structures. For example, the charge on the Mg atom in HMg–B(OH)2 is +1.267e using the MP2/aug-cc-pVTZ density, and the two Mg–B–O angles are 133.1° and 112.8°; this structural feature separates the positively charged Mg atom from one of the positively charged hydroxyl H atoms while increasing one Mg+O− electrostatic interaction. In the anti conformer of HMg–B(OH)2, this distortion does not occur, leading to a somewhat elevated value of \(\Delta {\text{H}}_{298}^{0}\) for the endo–exo → anti conformer conversion, see Table 2.
It may be noted that the O–B–O angle increases across the second and third periods of the periodic table as the electronegativity of the substituents increases, see Table 1S; the character of the NBOs is clearly in accord with Bent’s rule [39]. From the data in Table 1A.3, A.4, it can be seen that the O–B–O angles are less than 120° for the groups usually thought of as electron donating and greater than 120.0° for those thought of as electron withdrawing.
It is of interest to compare the orientations of the BH2, NH2, AlH2, and PH2 groups in the endo–exo conformers of R–B(OH)2. As might be expected, the BH2 moiety in H2B–B(OH)2 is perpendicular to the –B(OH)2 plane; if H2B–B(OH)2 is constrained to be planar, the resulting structure is a rotational TS. In contrast, H2N–B(OH)2 is planar and NBO analysis using the MP2 density indicates that the nitrogen–boron bond is actually a double bond, consisting of a σ-bond and a π-dative bond; the structure with the NH2 moiety perpendicular to the –B(OH)2 plane is a rotational TS. The endo–exo conformer of H2Al–B(OH)2 is also planar, and a structure in which the AlH2 moiety is perpendicular to the plane is a rotational TS. The PH2 group in H2P–B(OH)2 is in a pyramidal orientation with the two hydrogen atoms on the same side of the –B(OH)2 plane, and NBO analysis shows that the P–B is a single σ-bond; as would be expected, a completely planar form of H2P–B(OH)2 is an inversion TS.
The highest value of \(\Delta {\text{H}}_{298}^{0}\) for the endo–exo → anti conversion for the boronic acids in this investigation is +7.8 kcal/mol when R=H3N+, see Table 2; this is partially the result of substantial electrostatic repulsion involving the five positively charged H atoms in the anti conformer. In most cases, the value of \(\Delta {\text{H}}_{298}^{0}\) for this endo–exo → anti conversion is slightly lower for the corresponding acid in the third period compared to that in the second period, see Tables 2a, b. Values of ΔH† for the endo–exo → anti barrier at the MP2/aug-cc-pVTZ level are also listed in Table 2, and the values range from 6.1 kcal/mol for R=O− to 10.4 kcal/mol for R=NH3 +.
Charges
NPA charges from the MP2/aug-cc-pVTZ density on the boron atoms for the boronic acids in this investigation are listed in Table 3. Although these boron charges are always positive, they range widely from +0.278e for Li–B(OH)2, where the charge on the Li atom is +0.664e, to +1.267e for F–B(OH)2, where the charge on the F atom is −0.480e. Across the second and third rows of the periodic table, the positive charges on the boron atoms in these acids generally increase as the electronegativity of the substituent increases, see Table 1S. The charge of the atom directly connected to the boron atom varies from −1.085e for the nitrogen atom in H2N–B(OH)2 to +1.276e for the Mg atom in HMg–B(OH)2, see Table 3.
Heats of formation
In Table 4, we list heats of formation at 298.15 K, ΔHf(298 K), for selected endo–exo conformers of R–B(OH)2 calculated at the G3 level of theory [25, 26]; this particular extrapolation procedure was chosen based on our previous experience calculating heats of formation for boronic acids and boroxines [13]. It is important to note that the scarcity of high-quality experimental thermodynamic data for boronic acids has excluded these compounds from the various data sets that have been used to assess the Gn methodologies for testing heats of formation; in particular, there are no boron-oxygen bonded species in the G3/05 data set [40].
Nevertheless, there are a few experimental/computational heats of formation available in the literature for comparison. (1) Porter and Gupta [7] determined ΔHf (298 K) for H–B(OH)2 to be −153.8 ± 2.0 kcal/mol, and Gurvich et al. [11] estimated its value as −154.0 ± 2.0 kcal/mol, see Table 2SA; the most reliable calculated value, −154.6 ± 1.5 kcal/mol, is due to Grant and Dixon [41], and the G3 value is −153.9 kcal/mol [13]. (2) The JANAF [8] and Gurvich et al. [11] compilations list ΔHf (298 K) for B(OH)3 as −237.2 ± 0.6 and −240.0 ± 0.6 kcal/mol, respectively; Grant and Dixon [41] calculated ΔHf (298 K) to be −239.8 ± 1.5 kcal/mol, and the G3 value is −238.8 kcal/mol [13]. 3) The heat of formation of F–B(OH)2 was determined as 249.8 ± 2.5 kcal/mol by Porter et al. [42], estimated by Gurvich et al. [11] as −250.9 ± 5.0 kcal/mol and calculated at the G2 level by Duan et al. [37] to be −250.6 kcal/mol; the G3 value is −250.5 kcal/mol. (4) Gurvich et al. [11] reported ΔHf (298 K) for Cl–B(OH)2 as −192.5 ± 5.0 kcal/mol; the G3 value is −193.1 kcal/mol. Thus, heats of formation calculated at the G3 level of theory are in reasonably good agreement with the limited experimental/computational data available from the literature.
The calculated heats of formation for the boronic acids in this investigation vary widely. Based on the fact that ΔHf (298 K) for NH4 + is +154.1 kcal/mol [9, 10], it is not surprising that the highest heat of formation we found, −16.8 kcal/mol, is for R=NH3 +. The most negative heat of formation, −318.8 kcal/mol, is for R=CF3. It should also be pointed out that except for the Group 1 metals Li and Na, the heats of formation become more negative across both periods 2 and 3 of the periodic table, reflecting the increase in the electronegativity of the atom directly connected to the boron atom or of various group electronegativity schemes, see Table 1S; with the exception of the Na and MgH, the Period 2 heats of formation are more negative than the corresponding Period 3 values, following a similar trend in electronegativities.
Boroxines
Structures
H3B3O3 is the only boroxine compound whose gas-phase experimental structure has been reported. Using electron diffraction, Chang et al. [43] found this molecule to be planar (D3h) with r(B–H) = 1.192 ± 0.017 Å, r(B–O) = 1.3758 ± 0.0021 Å, <B–O–B=<O–B–O=120.0 ± 0.04°, and <H–B–O = 120° [11]. The structure of boroxine has also been studied computationally [5, 13, 44]; the calculated structural parameters at the MP2/aug-cc-pVTZ level, see Table 1, are in excellent agreement with these experimental results. It should be pointed out that the ring angles in H3B3O3 calculated at the MP2/aug-cc-pVDZ differ by ~0.6° from the experimental values and those calculated at the MP2/aug-cc-pVTZ level.
In Table 1, we list selected geometrical parameters for a variety of monosubstituted boroxines; the parameters are identified in Fig. 2. Many of the boroxines in this investigation are planar and have C2v symmetry (e.g., R=Li, MgH, NH2, CN, and O−) or involve a distortion from this symmetry that retains the vertical symmetry plane (e.g., R=COO−, vide infra); in these cases, r a = r a′ , β = β’, etc. However, when there is a distortion from C2v symmetry that retains the horizontal symmetry (e.g., R=SH, CHO, and COOH), the Bipso…Opara line is no longer a twofold rotational axis and r a ≠ r a’, β ≠ β’, etc.; the “prime side” of the ring was taken as shown in Fig. 2. For R = XY3, symmetry was not enforced during the calculation, but one of the BOXY torsional angles typically optimized to ~0°, which was used to define the “prime side,” see Fig. 2.

Identification of the B–O bonds and B–O–B and O–B–O angles for the monosubstituted boroxines in Table 1; the ipso angle is labeled as α
Generally, the parameters calculated at the MP2/aug-cc-pVDZ and MP2/aug-cc-pVTZ levels are in reasonable agreement. As expected, the B–O bond lengths in the ring are shorter with the more complete basis set; this amounts to ca. 0.01–0.02 Å. As noted above for H3B3O3, the predicted bond angles in the ring from these two computational levels can differ significantly, although there are some regularities to the differences, e.g., the calculated ipso angles (α; <O–B–O) are consistently smaller at the MP2/aug-cc-pVTZ level, and the opposing angles (δ; <B–O–B) consistently larger; unfortunately, no experimental data are available for comparison. In view of these differences, the comparisons below will be made using data from the MP2/aug-cc-pVTZ level.
Although some of the monosubstituted boroxines we considered do not have C 2v symmetry, the corresponding B–O lengths, the B–O–B angles, and the O–B–O angles on opposite sides of the Bipso…Opara line are nearly equal in most cases, see Table 1; the largest value of |r a − r a′ | is 0.009 Å for R=OH, and the largest value of |β-β’| is 0.2° for R=OH and OCH3. The ipso angles for the substituents we employed in this study vary from 109.8° for the highly electron-donating group R=O− to 128.0° for the highly electron-withdrawing group R=NH3 +; in fact, the ipso angles for the electron-donating groups in Table 1A.3 are all less than 120°, whereas for the electron-withdrawing groups in Table 1A.4 are all greater than 120°. The ipso angle and the opposing angle δ in the boroxine ring generally increase for the hydrides across the second and third periods of the periodic table; the only exception involves the weakly electron-donating CH3 group where the ipso angle is less than that for both BH2 and NH2.
To a large extent, the angular O–B–O deformation, Δα, of the boroxine ring at the ipso position induced by substitution is compensated for B–O–B angular deformations, Δβ and Δβ’, at the ortho positions of the opposite sign, and Δβ ≈ Δβ′ ≈ −Δα/2, similar to what has been observed by Domenicano et al. [45] for monosubstituted benzenes. The O–B–O angular deformations, Δγ and Δγ’, at the meta positions are generally smaller in magnitude than the B–O–B deformations at the ortho positions; Δγ ≈ Δγ’, and they range from +2.9° for R=O− to −2.4° for R=NH3 +. The angular deformation at the para position, Δδ, can be substantial, e.g., +4.3° for R=NH3 +. Although the magnitude of Δδ is consistently smaller than the magnitude of Δα, the signs are the same. It should be noted that the angle δ for the electron-withdrawing groups we employed in this investigation is greater than 120.0°, whereas for the electron-donating groups, it is less than 120.0°.
It is of interest to compare structural parameters for the monosubstituted boroxines with those for the corresponding endo–exo conformers of the monomers. For the second- and third-period hydrides, the ipso angle for the boroxine is consistently greater than the O–B–O angle in the corresponding monomer, and this is also the case for most of the electron-donating groups we considered, see Table 1. In contrast, for the carboxylate anion, the ipso angle for the boroxine with R=COO−, 112.9°, is significantly smaller than the O–B–O angle in the monomer, 119.7°. However, the structure of the lowest energy form for this boroxine is quite distinctive in that the carbon atom and one of the two carboxylate oxygen atoms are nearly equidistant from the ipso boron atom (the B–C and B–O distances are 1.545 and 1.523 Å, respectively), forming a three-membered CBO ring in an orientation perpendicular to the boroxine core; several attempts to find an analogous structure for the corresponding boronic acid were unsuccessful, and the lowest energy form of this acid was planar with the two carbon–oxygen bond lengths the same. For comparison, we note that a planar version of this boroxine at the MP2/aug-cc-pVDZ level is a TS (α = 116.1°) while a version with the COO− group constrained to be perpendicular to the ring is also a TS (α = 116.3°); thus, even for these more conventional type structures, the ipso angle for the boroxine remains smaller than the O–B–O angle in the monomer. It should be noted that the rather large angle in the endo–exo form of the boronic acid in this case is a result of a very strong B–O–H…O interaction; the O–B–O angle in the anti form of the monomer is even larger, 121.5°. For the electron-withdrawing groups NO2, CF3, COOH, and CHO, which involve either strong B–O–H…O or B–O–H…F interactions in the acids, the O–B–O angle in the monomer is greater than the ipso angle for the corresponding boroxine, whereas for CN and CH3O, it is greater for the boroxine.
Charges
NPA charges from the MP2/aug-cc-pVTZ density on the various boron atoms and the atom X of the substituent R that is directly connected to the boron atom of the boroxines in this investigation are listed in Table 3. The charges on the ipso boron atoms are always positive, ranging from +0.312e for R=Li to +1.319e for R=F, reflecting what we observed for the corresponding boronic acids. Across the second and third periods, the charge on the ipso boron atom is generally more positive than it is for the corresponding boronic acid.
Heats of formation
In Table 4, we list heats of formation at 298.15 K for selected monosubstituted boroxines in this study calculated at the G3 level of theory. Unfortunately, there are few experimental values to compare with. As we have noted previously [5, 13], the heat of formation for H3B3O3 listed in the JANAF compilation [8], −291.0 ± 2.0 kcal/mol, in the ATcT (Active Thermochemical Tables) project of Ruscic and coworkers [9, 10, 46], −291.0 ± 10.0, or in the Gurvich et al. Tables [11], −287.7 ± 5.0 kcal/mol, all appear to be too negative. The value calculated from the G3 level of theory, −276.5 kcal/mol, is likely to be much closer to the correct value [13]. There are a few other heats of formation available in the literature for monosubstituted boroxines that we can use for comparison, see Table 2S: (1) The JANAF compilation [5] lists the heat of formation for FH2B3O3 as −382.0 kcal/mol, and Guest et al. [15] in their computer analyses of a variety of boron compounds give the value as −382.8 kcal/mol; however, numerous assumptions were involved in arriving at these numbers. The value calculated at the G3 level of theory is somewhat less negative, −374.2 kcal/mol. (2) Porter and Gupta [16] in their study of the reactions of H3B3O3(g) with HCl(g) determined the heat of formation at 298 K for ClH2B3O3 to be −314.5 ± 4.0 kcal/mol. The G3 value, −315.7 kcal/mol, is in surprisingly good agreement with this result.
Boroxine formation from boronic acid dehydration
In Table 5, we list reaction enthalpies for the dehydration reactions
calculated at the MP2/aug-cc-pVDZ and MP2/aug-cc-pVTZ computational levels. The following points may be noted: (1) The calculated values of \(\Delta {\text{H}}_{298}^{0}\) for the dehydration at the MP2/aug-cc-pVDZ and MP2/aug-cc-pVTZ levels typically differ from each other by less than 1 kcal/mol; (2) with the exception of R=O−, all the dehydration reactions for these aliphatic boronic acids are endothermic; (3) for the second- and third-period hydrides in toto, \(\Delta {\text{H}}_{298}^{0}\) is not correlated with the atomic or group electronegativities, see Table 1S; (4) for the substituents in the second period with lone pairs, i.e., H2N, HO, and F, \(\Delta {\text{H}}_{298}^{0}\)increases as the σ-electron withdrawing capacity increases and the π-electron donating ability of the group decreases based on values of the Hammett constants [47], see Table 1S; (5) the values of \(\Delta {\text{H}}_{298}^{0}\) for the electron-withdrawing substituents are consistently larger than those for the electron-donating.
Concluding remarks
The availability of experimental thermodynamic data for simple aliphatic substituted boronic acids and boroxines is extremely limited, and their accuracy is not always as good as one might hope for. This problem is exacerbated by the paucity of reliable experimental structural and vibrational data on these compounds. Thus, the heats of formation at 298 K listed in the JANAF and Gurvich et al. [11] compilations have typically employed structural parameters, bond energies, and/or vibrational frequencies adopted from a variety of related molecules, making it difficult to assess the reliability of these tabulated values. In this article, we investigated the structures of a variety of monosubstituted boroxines and their corresponding boronic acids, as well as the reactions that lead to their interconversion via dehydration.
We have shown the following:
(1) Although the endo–exo conformer of the boronic acids is usually the lowest energy form, see Table 2, interactions between the hydroxyl hydrogen atoms of the boronic acid moiety and the substituent can significantly stabilize the anti conformer, e.g., R=COO− or NO2, and make it the lowest energy form. The barriers for the endo–exo → anti conversion for the acids in this study range from 6.1 to 10.4 kcal/mol at the MP2/aug-cc-pVTZ level. (2) The O–B–O angle for the acids increases for the second- and third-period hydride substituents as the electronegativity increases; the character of the NBOs is clearly in accord with Bent’s rule [39]. (3) For the second- and third-period hydrides and the electron-donating groups we considered, the ipso angle for the boroxine is greater than the O–B–O angle in the corresponding monomer; for the electron-withdrawing groups, there are cases with the ipso angle greater and lesser than the O–B–O angle in the corresponding monomer. (4) The boroxine with R=COO− has a distinctive structure with the carbon atom and one of the two oxygen atoms nearly equidistant from the ipso boron atom forming a three-membered CBO ring perpendicular to the nominal plane of the boroxine core. (5) The ipso angles in the monosubstituted boroxines vary from 109.8° (R=O−) to 128.0° (R=NH3 +); the ipso angles for the electron-donating groups in Table 1A.3 are less than 120°, whereas the ipso angles for the electron-withdrawing groups in Table 1A.4 are greater than 120°. (6) With the exception of R=O−, the reactions R–B(OH)2 + 2 H–B(OH)2 → RH2B3O3 + 3 H2O are predicted to be endothermic; the values of \(\Delta {\text{H}}_{298}^{0}\) for the electron-donating groups are generally smaller than for the electron-withdrawing groups. (7) For the second-period substituents with lone pairs, H2N, HO, and F, \(\Delta {\text{H}}_{298}^{0}\) increases as the σ-electron withdrawing π-electron donating capacity of the group increases. (8) For many of the boronic acids and boroxines in this article, the heats of formation calculated at the G3 level of theory listed in Table 4 are the first estimates available for this fundamental property of these compounds; the lack of experimental data for comparison makes it difficult to assess the accuracy of these values. This highlights the important work of Grant and Dixon [41], Schlegel and Harris [48], Duan et al. [37], and Karton and Martin [37, 49] in using high-level calculations to improve the accuracy of heats of formation for boron compounds.
References
Hall DG (2005) Boronic acids: preparation and applications in organic synthesis and medicine, 1st edn. Weinheim, Wiley-VCH Verlag
Korich AL, Iovine PM (2010) Dalton Trans 39:1423–1431
Dembitsky VM, Quntar AA, Srebnik M (2004) Mini Rev Med Chem 4:1001–1018
Baker SJ, Tomsho JW, Benkovic SJ (2011) Chem Rev Soc. doi:10.1039/c0cs00131g
Bhat KL, Markham GD, Larkin JD, Bock CW (2011) J Phys Chem A 115:7785–7793
Yao L, Zeng X, Ge M, Wang D (2007) J Mol Struct 841:104–109
Porter RF, Gupta SK (1964) J Phys Chem 68:2732–2733
Chase MW (1998) NIST—JANAF thermochemical tables (Journal of Physical and Chemical Reference Data Monograph No. 9). American Institute of Physics
Ruscic B, Pinson RE, Morton ML, von Laszevski G, Bittner SJ, Nijsure SG, Amin KA, Minkoff M, Wagner AF (2004) J Phys Chem A 108:9979–9997
Ruscic B, Pinson RE, von Laszevski G, Kodeboyina D, Burcat A, Leahy D, Montoya D, Wagner AF (2005) J Phys Conf Ser 16:561–570
Gurvich LV, Veyts IV, Alcock CB (1994) Thermodynamic properties of individual substances, vol 3 (Parts 1 and 2) elements B, Al, Ga, In, Tl, Be, Mg, Ca, Sr, and Ba and their compounds. CRC Press Inc, Boca Raton
Tokunaga Y, Ueno H, Shimomura Y, Seo T (2002) Heterocycles 57:787–790
Bock CW, Larkin JD (2012) Comput Theor Chem 986:35–42
Ghiasi R (2008) J Mol Struct (THEOCHEM) 853:77–81
Guest MF, Pedley JB, Horn M (1969) J Chem Thermodyn 1:345–352
Porter RF, Gupta SK (1964) J Phys Chem 68:280–289
Tokunaga Y, Ueno H, Shimomura Y (2007) Heterocycles 74:219–223
Møller C, Plesset MS (1934) Phys Rev 46:618–622
Dunning TH Jr (1989) J Chem Phys 90:1007–1023
Woon DE, Dunning TH Jr (1993) J Chem Phys 98:1358–1371
Kendall RA, Dunning TH Jr (1992) J Chem Phys 96:6796–6806
Peterson KA, Woon DE, Dunning TH Jr (1994) J Chem Phys 100:7410–7415
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts RE, Stratmann O, Yazyev AJ, Austin R, Cammi C, Pomelli JW, Ochterski R, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2003) G03, R B.02 ed, Gaussian Inc., Wallingford, CT
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts RE, Stratmann O, Yazyev AJ, Austin R, Cammi C, Pomelli JW, Ochterski R, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) G09 Gaussian Inc., Wallingford, CT
Curtiss LA, Raghavachari K, Redfern PC, Rassolov V, Pople JA (1998) J Chem Phys 109:7764–7776
Curtiss LA, Raghavachari K (2002) Theor Chem Acc 108:61–70
Carpenter JE, Weinhold F (1988) J Mol Struct (THEOCHEM) 169:41–62
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926
Foster JP, Weinhold F (1980) J Am Chem Soc 102:7211–7218
Reed AE, Weinstock RB, Weinhold F (1985) J Chem Phys 83:735–746
Snyder HR, Kuck JA, Johnson JR (1938) J Am Chem Soc 60:105–111
Wagner M, van Eikema Homes NJR, Ndth H, Schleyer PVR (1995) Inorg Chem 34:607–614
Larkin JD, Bhat KL, Markham GD, Brooks BR, Schaefer HF, Bock CW III (2006) J Phys Chem A 110:10633–10642
Larkin JD, Bhat KL, Markham GD, Brooks BR, Lai JH, Bock CW (2007) J Phys Chem A 111:6489–6500
Boggs JE, Cordell FR (1981) J Mol Struct (THEOCHEM) 76:329–347
So SP (1982) J Mol Struct (THEOCHEM) 89:255–258
Duan X, Linder DP, Page M, Soto MR (1999) J Mol Struct (THEOCHEM) 465:231–242
Stefani D, Pashalidis I, Nicolaides AV (2008) J Mol Struct (THEOCHEM) 853:33–38
Bent HA (1961) Chem Rev 61:275–311
Curtiss LA, Redfern PC, Raghavachari K (2005) J Chem Phys 123:124107
Grant DJ, Dixon DA (2009) J Phys Chem A 113:777–787
Porter RF, Bidinosti DR, Watterson VF (1962) J Chem Phys 36:2104–2108
Chang CH, Porter RF, Bauer SH (1969) Inorg Chem 8:1689–1693
Beckmann J, Dakternieks D, Duthie A, Lim AEK, Tiekink ERT (2001) J Organomet Chem 633:149–156
Domenicano A, Vaciago A, Coulson C (1975) Acta Cryst Sect B 31:1630–1641
Tasi G, Iszák R, Matisz G, Császár AG, Kállay M, Ruscic B, Stanton JF (2006) ChemPhysChem 7:1664–1667
Hansch C, Leo A (1979) Substituent constants for correlation analysis in chemistry and biology. Wiley-Interscience, New York
Schlegel HB, Harris SJ (1994) J Phys Chem 98:11178–11180
Karton A, Martin JML (2007) J Phys Chem A 111:5936–5944
Acknowledgments
This research was supported in part by the National Science Foundation through XSEDE resources provided by the XSEDE Science Gateways program.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rao, N.Z., Larkin, J.D. & Bock, C.W. A computational investigation of monosubstituted boroxines(RH2B3O3): structure and formation. Struct Chem 26, 1151–1162 (2015). https://doi.org/10.1007/s11224-015-0577-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-015-0577-9