
Abstract
Boronic acids, R–B(OH)2, play an important role in synthetic, biological, medicinal, and materials chemistry. Borinic acids, R–BH(OH), find relevance in similar fields, although their properties, e.g., binding affinity to diols, can vary significantly. Dative boron–nitrogen bonds, N → B, are critical for protecting the boron atom in these acids from nucleophilic attack. In this article, we study the structure, bonding, and formation thermodynamics of the model donor–acceptor complexes H3N → BR(OH)2 and H3N → BRH(OH) (R = H; NH2, OH, and F). Geometry optimizations were performed using second-order Møller–Plesset perturbation theory (MP2) with the Dunning–Woon aug-cc-pVTZ basis set; single-point CCSD(FC)/aug-cc-pVTZ//MP2(FC)/aug-cc-pVTZ level calculations were used to generate a QCI density for analyses of the bonding. Extensive comparisons are made with results from density functional theory (DFT) optimizations with/without empirical dispersion corrections. The addition of an ammonia molecule dative bonded to the boron atom for these boronic and borinic acids results in the elongation of bonds to the boron atom, e.g., the boron–oxygen bond lengths increase in the range from ~ 4.5 to ~ 6.5%. The calculated values of ∆ \( {H}_{298}^0 \) for the ammoniation reactions, R–B(OH)2 + NH3 → H3N → BR(OH)2 are − 0.6, + 4.6, + 0.1, and − 6.0 kcal/mol for R = H, NH2, OH, and F, respectively, at the CCSD(FC)/aug-cc-pVTZ//MP2(FC)/aug-cc-pVTZ level; the corresponding values of ∆\( {H}_{298}^0 \) for the borinic acid reactions are − 8.9, + 2.1, − 1.5, and − 7.9 kcal/mol.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dative boron–nitrogen bonds, N → B, have been of interest to chemists for many years and are well documented in vacuo, in solution, and in the solid state. These Lewis acid–base type bonds are useful structural directing elements in supramolecular chemistry [1, 2] and have found novel applications in tissue engineering and regenerative medicine [3]. One such application involves the use of phenylboronate containing copolymers (PBCCs) currently under development as molecular scaffolds with poly(vinyl alcohols) (PVAs) [4]. A key feature of PBCC-PVAs is their biodegradability, allowing them to be removed easily from wounds after healing. Only a fructose solution is required to disintegrate the polymer by competitive displacement, leaving no residue. Proximal amine groups are crucial for protecting the phenyl boronic acid (PBA) portion of the PBCC while in the aqueous phase. This is achieved through “amine assistance,” i.e., dative bonding, between an amine nitrogen lone pair and a planar boronic acid [4].
The disruption of a N → B dative bond has long been implicated as the mechanism for fluorescence turn-on in arylboronic acid diol sensors [5]. However, recent evidence [6, 7] suggests that a N → B bond plays a lesser role, and an inserted water molecule is more likely to modulate fluorescence by coordinating the nitrogen and boron atoms. As a step toward better understanding N → B coordination, we carried out an investigation comparing the structure and bonding surrounding the boron atoms in the donor–acceptor complexes H3N → BR(OH)2 and H3N → BRH(OH) (R = H; NH2, OH, and F). Thermochemical parameters are also given for the ammoniation processes
and
The ammonia molecule was initially positioned in the optimization process to form a dative bond to the boron atoms in R–B(OH)2 and R–BH(OH), effectively minimizing the π-dative bond character of other bonds to the central boron atom.
Computational methods
Equilibrium geometries of all the molecules involved in this article were obtained using second-order Møller–Plesset perturbation theory (MP2) [8] with the frozen core (FC) option, which neglects core–electron correlation; the Dunning–Woon aug-cc-pVTZ basis set was employed for all the geometry optimizations and the more-complete aug-cc-pVQZ basis set in selected cases [9,10,11,12]. The GAUSSIAN 0313 and GAUSSIAN 0914 suits of programs were used for all the calculations. Frequency analyses were performed analytically for all the compounds in this investigation at the MP2(FC)/aug-cc-pVTZ level to confirm that the optimized structures were local minima on the potential energy surfaces (PESs) and to correct reaction energies to 298.15 K. The geometry optimizations were followed by single-point calculations at the CCSD(FC)/aug-cc-pVTZ//MP2(FC)/aug-cc-pVTZ level to generate the QCI density for analyses of the bonding. Additional single-point calculations at the MP2(FC)/aug-cc-pVQZ//MP2(FC)/aug-cc-pVTZ and CCSDT(FC)/aug-cc-pVTZ//MP2(FC)/aug-cc-pVTZ levels [9,10,11,12,13,14,15] were performed to provide more reliable thermochemical parameters for the ammoniation reactions. In addition to the MP2(FC) optimizations, a variety of density functional theory (DFT) optimizations were also performed, with and without empirical dispersion corrections [16, 17] using the B3LYP [18], B3LYP-GD3BJ [16], CAM-B3LYP [19], B97D3 [16], PBE1PBE [20, 21], PBE1PBE-GD3BJ [16, 20, 21], B2PLYP [22], B2PLYPD3 [16, 23], M062X [24], and M062X-GD3 [16, 24] functionals with the aug-cc-pVTZ basis set [9,10,11,12]. Atomic charges were obtained from natural population analyses (NPA), and the bonding was analyzed with the aid of natural bond orbitals (NBOs). (NBO Version 3.1 embedded in GAUSSIAN 03 and GAUSSIAN 09 was used for all these calculations.) The RESONANCE and E2PERT keywords were employed; the E2PERT command enables second-order perturbative estimates of donor–acceptor (bond-antibond) stabilization interaction energies, E [2], to be calculated in the NBO basis set [25,26,27,28]. Since NBO analyses using the (non-variational) MP2 density can be problematic, we employed results from the HF and QCI densities [19, 29].
Results and discussion
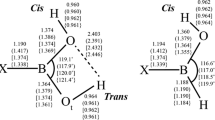
Detailed analyses of the structures and bonding of the isolated boronic and borinic acids, R–B(OH)2 and R–BH(OH) (R = H; NH2, OH, and F), have been given by Rao et al. [30]; a summary of the calculated structural results for the endo–exo conformers of R–B(OH)2 and the cis (relative to R) conformers of R–BH(OH) is shown in Fig. 1. In Table 1S of the Supplementary Materials, selected structural parameters of the isolated boronic and borinic acids are compared to their ammoniated forms.

Selected distances (Å) and angles (°) for the endo–exo conformers of the boronic acids H–B(OH)2, (H2N–B(OH)2), [HO–B(OH)2], and {F–B(OH)2} and for the Cis conformers of the corresponding borinic acids calculated at the MP2(FC)/aug-cc-pVTZ level
A.1 The N → B dative bond in H3N → BR(OH)2 and H3N → BRH(OH)
The lengths of the N → B bonds in H3N → BR(OH)2 and H3N → BRH(OH) (R = H; NH2, OH, and F) are ~ 1.7 Å at the MP2/aug-cc-pVTZ level; see Fig. 2 and Table 1. To provide a measure of the reliability of these calculated boron–nitrogen bond distances, we also calculated the length of the prototype boron–nitrogen dative bond in borazane, H3N → BH3; excellent discussions of the structure of borazane and the energetics of its formation have been given by Demaison et al. [31] and Staubitz et al. [32] A neutron diffraction study at 200 K by Klooster et al. [33] has shown that the H3N → BH3 molecule has a staggered geometry with a boron–nitrogen distance of 1.5 8[2] Å, in reasonable agreement with previous determinations in the solid state, notably the value of 1.56 5[7] Å by Boese et al. [34] The gas-phase boron–nitrogen distance obtained by Thorne et al. [35] using electron diffraction, re = 1.672 2[5] Å and rs = 1.657 6[16] Å, is considerably longer than the solid-state value. At the MP2(FC)/aug-cc-pVTZ [6] level, the boron–nitrogen dative bond length is 1.652 [1.647] Å, in reasonable agreement with the gas-phase experimental value, computational results from other authors, [36, 37] and results from a variety of DFT calculations; see Table 1 (A). It may be noted that various empirical dispersion corrections used in conjunction with the DFT calculations did not alter significantly the calculated length of the B → N bond in ammonia borane.

Structures of a H3N → BR(OH)2 and b H3N → BRH(OH) (R = H; NH2, OH, and F) calculated at the MP2(FC)/aug-cc-pVTZ//MP2(FC)/aug-cc-pVTZ level
It is evident from Table 1 that replacing the borane moiety, BH3, with a boronic acid, R–B(OH)2, or a borinic acid, R–BH(OH) (R = H; NH2, OH, and F), moiety results in a longer N → B bond at every computational level we employed (e.g., at the MP2(FC)[PBE1PBE]{B3LYP}, <B3LYP-GD3BJ>/aug-cc-pVTZ levels, the increases are 3.4%[4.1%]{5.9%} <5.6%> for HB(OH)2 and 2.0%[2.4%]{3.0%} <2.9%> for HBH(OH)). (N → B dative bonds shorter than those calculated for H3N → BH3 can be obtained by replacing hydrogen atoms on the ammonia moiety with hydroxyl groups, e.g., the length of the N → B dative bond in H(HO)2N → BH3 is only 1.5995 Å at the PBE1PBE/aug-cc-pVTZ level and 1.6176 Å at the M062X/aug-cc-pVTZ level.) The N → B dative bond is shorter for the borinic acids than for the corresponding boronic acids, except for R = F. Again, including empirical dispersion in the optimization did not alter significantly the dative bond length.
The NPA charges assigned to the boron atoms, q(B), in H3N → BR(OH)2 are consistently more positive than those assigned to H3N → BRH(OH), similar to that found for the isolated structures, R(OH)2 and R–BH(OH); see Table 2S [30]. Furthermore, the value of q(B) increases in both the isolated and ammoniated species as the electronegativity of X = N, O, F increases. Of course, as a result of the dative bond, the NPA charge assigned to the boron atoms in H3N → BR(OH)2 and H3N → BRH(OH) is less positive than in the isolated structures; see Table 2S. The NBO occupancies of the boron–nitrogen dative bonds are ~ 1.95e using the QCI density.
The p-character of the natural atomic boron hybrid orbitals associated with the boron–nitrogen dative bonds for both H3N → BR(OH)2 and H3N → BRH(OH) decreases slightly as the electronegativity of X (N → O → F) increases (see Table 3S) whereas the p-character of the corresponding natural atomic nitrogen hybrid orbital increases, in accordance with Bent’s rule [38]. The directing of more p-character to the fluorine (and subsequently from the nitrogen to the boron) allows the energy of the bond to increase marginally.
A.2 Distortion of the planar R–B(OH)2 and R–BH(OH) structures on ammoniation
The addition of NH3 to the endo–exo conformer of R–B(OH)2 and the cis conformer of R–BH(OH) (R = H; NH2, OH, F) to generate a N → B dative bond induces the expected tetrahedral environment surrounding the boron atom (see Fig. 2); optimizations using the trans conformer in place of the cis conformer of R–BH(OH) yield similar results. (In Table 4S of the Supplementary Materials, binding energies of the dative-bonded structures discussed in this article are compared to analogous hydrogen-bonded structures.) However, the distortion from planarity of the R–B(OH)2 and R–BH(OH) moieties, as measured by the deviation from 360° of the sum of the three bond angles surrounding the boron atom, is only ~ 15°. (The sums of the three bond angles at the MP2(FC)/aug-cc-pVTZ[PBE1PBE/aug-cc-pVTZ]{PBE1PBE-GD3BJ/aug-cc-pVTZ} <PBE1PBE/aug-cc-pVTZ SCRF(PBM,Solvent = Water> level for the ammoniated forms of H–B(OH)2, H2N–B(OH)2, HO–B(OH)2, and F–B(OH)2 are 345.4°[345.1°]{345.1°} <339.7>, 346.9°[346.9°]{346.9°} <340.6>, 345.4°[345.2°]{345.4°}, and 344.8°[344.6°]{344.6°}, respectively; the sums for the corresponding borinic acids are 344.4°[343.7°]{343.7°}, 346.6°[346.2°]{346.2°}, 345.4°[345.1°]{345.1°}, and 344.4°[344.1°]{344.1°}. There is a slight increase in the non-planarity of the R–B(OH)2 and R–BH(OH) moieties as the electronegativity of the atom bonded to the boron atom (N → O → F) increases. Furthermore, the two N → B–O angles and the N → B–X angle (X = H, N, O, and F) in H3N → BR(OH)2 as well as the N → B–O, N → B–X, and N → B–H angles in H3N → BRH(OH) are all less than 109.5°, suggesting these structures may be described as distorted trigonal pyramids.
There are significant distortions involving the hydroxyl hydrogen atoms of the boronic acids upon ammoniation. For example, at the MP2(FC)/aug-cc-pVTZ [PBE1PBE/aug-cc-pVTZ] level, the values of the two torsional angles τHOBO are H − 8.2°, 131.8° [− 7.2°, 133.9°]; NH2 + 11.8°, 85.7° [+ 10.5°, 89.2°]; OH − 8.1°, 129.5° [− 6.5°, 131.17°]; F − 28.2°, 141.8° [− 23.9°, 141.2°]. The most extreme case involves R = NH2, presumably a result of steric hindrance involving the hydrogen atoms. This finding persists even when empirical dispersion effects are included, e.g., at the PBE1PBE-GD3BJ/aug-cc-pVTZ level, the values of τHOBO are 10.6° and 89.0°. This distortion in H3N → B(NH2)(OH)2 leads to the longest B–O–H…O–H intermolecular hydrogen bond we found in this investigation, 2.462 Å, and, by far, the largest change in this length from the corresponding isolated boronic acid, 0.071 Å. There is also significant rotation about the B–O bond in B(NH2)H(OH) upon ammoniation; see Fig. 2.
A.3 Changes in boron–oxygen bonding on ammoniation
With the addition of the dative-bonded ammonia molecule to H–B(OH)2, both boron–oxygen bond lengths increase, 0.075 Å (5.5%) and 0.070 Å (5.1%) to 1.439 Å and 1.444 Å; see Table 1S. This increase is accompanied by a 5.2° (4.4%) decrease in the O–B–O angle. These structural changes give rise to a small decrease in the intramolecular H…O distance from 2.403 to 2.373 Å (1.2%). As would be expected, NBO analysis using the HF and QCI densities classify both boron–oxygen bonds in H3N → BH(OH)2 as single bonds. The largest second-order stabilization energies involve delocalization from oxygen lone pair orbitals into a boron–nitrogen antibonding orbital with E [2] values of 19.0 and 17.5 kcal/mol using the HF density. As can be seen from the data in Table 3S, the p-character of the natural atomic boron hybrid orbitals associated with each of the boron–oxygen bonds increases significantly (~ 25.9%) in going from H–B(OH)2 (sp2.24) to H3N → BH(OH)2 (sp2.82), in qualitative agreement with the expected progression toward boron sp3 bonding in the ammoniated structure. For the other boronic acids with lone pairs on the substitutents, R–B(OH)2 (R = NH2, OH, and F), the addition of ammonia results in the calculated lengths of the boron–oxygen bonds increasing by ca. 0.060–0.067 Å (4.4–4.7%), from the 1.36–1.39 Å range to the 1.42–1.45 Å range; see Table 1S; these bond lengths decrease as the electronegativity of the substitutent increases similar to what we observed for the isolated boronic acids. The intramolecular B–O–H…O distance increases upon the addition of ammonia from 2.391 Å in the isolated acid to 2.462 Å for R = NH2 and from 2.446 Å to 2.451 Å for R = F, whereas this distance decreases from 2.432 to 2.401 Å for R = OH. The p-character of the natural atomic boron hybrid orbitals associated with the B–O and B–X (X = N, O, and F) bonds in H3N → BR(OH)2 are all significantly larger compared to those in R–B(OH)2 (see Table 1S), consistent with the expected transition toward sp3 bonding. The p-character of the boron hybrid orbitals associated with the B–O bonds decreases as the electronegativity of X increases while the p-character of B–X orbitals increases [38].
With the addition of the dative-bonded ammonia molecule to the parent borinic acid H2B(OH), the boron–oxygen distance increases from 1.360 to 1.449 Å (6.5%). This boron–oxygen bond is only 0.005 Å longer than the corresponding bond in H3N → BH(OH)2, and NBO analyses using the HF and QCI densities classify it as a single bond. The largest E [2] value using the HF density is relatively small compared to the boronic acid analog and involves delocalization from an oxygen lone pair orbital into a boron–nitrogen antibonding orbital. For the borinic acids R–BH(OH) (R = NH2, OH, and F), ammoniation results in the calculated lengths of the boron–oxygen bonds increasing by 0.082 Å (5.9%), 0.075 Å (5.5%), and 0.072 Å (5.3%), respectively (see Table 1SB); these bond lengths decrease as the electronegativity of the substitutent increases, similar to what we found for the isolated borinic acids. NBO analysis using the HF and QCI densities classify the boron–oxygen bonds in H3N → BRH(OH) for R = NH2, OH, and F as single bonds. The p-character boron hybrid orbitals associated with the B–O and B–X bonds in H3N → BRH(OH) are all significantly larger compared to those in the isolated borinic acid (see Table 3S), consistent with the expected transition toward sp3 bonding. In addition, this p-character is larger in the ammoniated borinic acids than in the corresponding ammoniated boronic acids.
A.4 Ammoniation reaction thermochemistry
In Table 2, we list values of ∆E, ∆\( {H}_{298}^0 \), and ∆ \( {G}_{298}^0 \) for the reactions
and
(R = H; NH2, OH, and F), calculated at the MP2(FC)/aug-cc-pVTZ, MP2(FC)/aug-cc-pVQZ, MP2(FC)/aug-cc-pV5Z, CCSD(FC)/aug-cc-pVTZ, and CCSDT(FC)/aug-cc-pVTZ computational levels, consistently using the MP2(FC)/aug-cc-pVTZ optimized geometries. With the exception of R = NH2, these reactions are exothermic. As can be seen from the data in Table 2, the borinic acid reactions are more exothermic than the corresponding boronic acid reactions; for R = NH2, OH, and F, the values of ∆\( {H}_{298}^0 \) become more negative as the electronegativity of the substitutent increases. The ammoniation reactions for H2N–B(OH)2 and H2N–BH(OH) are endothermic, presumably a result of strong boron–nitrogen bonding in the isolated acids. This finding is consistent with relatively large nN → n*BE [2] delocalization energies and large increases in the boron–nitrogen bond lengths with the addition of ammonia (see Table 1). Indeed, these are the largest bond length increases upon ammonia substitution for any of the B–X bonds we observed in this study. Transition states were located for the formation of H3N → B(NH2)(OH)2 and H3N → B(NH2)H(OH). The structures are shown in Fig. 1S, and thermochemical parameters are given in Table 5S of the Supplementary Materials.
Concluding remarks
In this investigation, we compared computationally the structure and bonding surrounding the trivalent boron atoms in R–B(OH)2 and R–BH(OH), the tetravalent boron atoms in H3N → BR(OH)2 and H3N → BRH(OH) (R = H; NH2, OH, and F), and the thermodynamics of the ammoniation process. The following points may be noted: (1) To a large extent, the NBO classification (single/double) of the bonds surrounding the boron atoms in R–B(OH)2, H3N → BR(OH)2, and the borinic acid analogs (R = H; NH2, OH, and F) is the same regardless of whether the HF/aug-cc-pVTZ or QCI/aug-cc-pVTZ densities are employed; one exception involves F–BH(OH), where the classification of the boron–oxygen bond depends on the density employed. (2) Although boron–oxygen double bonds are seemingly rather scare in nature, NBO analysis of H2B(OH) classifies the boron–oxygen bond as a double bond, suggesting a structure of the form H2B=O+H−. Admittedly, the calculated length of this boron–oxygen bond, 1.360 Å, is much longer than that in the borocycle (–HN=CH–CH=CH–NH–B–)O, 1.270 Å. (3) The addition of an ammonia molecule dative-bonded to the boron atom in R–B(OH)2 or R–BH(OH) (R = H; NH2, OH, and F) leads to the elongation of every bond to the boron atom; see Table 1S and Fig. 1. Even in an acid where there are no lone pairs of electrons on the substitutents, such as H3C–B(OH)2, the boron–carbon bond length increases significantly from 1.574 to 1.606 Å (0.032 Å) with the addition of ammonia. (4) The boron–oxygen bond lengths for R–B(OH)2 and R–BH(OH) (R = H; NH2, OH, and F) increase from ~ 4.5 to ~ 6.5% with the addition of the dative-bonded ammonia molecule. In conjunction with large nO → n*B delocalization energies, it is reasonable to assign some degree of double bond character to these boron–oxygen bonds in both the boronic and borinic acids; the degree of double bond character is greater in the borinic acids than in the boronic acids. (5) The p-character of boron hybrid orbitals associated with the B-X (X = N, O, F) bonds is larger for the isolated borinic acid than for the corresponding isolated boronic acid as well as for the ammoniated species. (6) With the exception of R = NH2, the ammoniation reactions are exothermic and the borinic ammoniations are more exothermic than the corresponding boronic ammoniations.
References
Icli B, Sheepwash E, Riis-Johannessen T, Schenk K, Filinchuk Y, Scopelliti R, Severin K (2011) Dative boron-nitrogen bonds in structural supramolecular chemistry: multicomponent assembly of prismatic organic cages. Chem Sci 2(9):1719–1721
Dhara A, Beuerle F (2015) Reversible assembly of a supramolecular cage linked by boron-nitrogen dative bonds. Chem Eur J 21(48):17391–17396
Nunes M, Gois P, Rosa M, Martins S, Fernandes P, Ribeiro M (2016) Boronic acids as efficient cross linkers for PVA: synthesis and application of tunable hollow microspheres in biocatalysis. Tetrahedron 72(46):7293–7305
Reddy R, Srivastava A, Kumar A (2013) Monosaccharide-responsive phenylboronate-polyol cell scaffolds for cell sheet and tissue engineering applications. PLoS One. 8(10):1–10
James TD, Sandanayake KRA, Shinkai S (1994) A Glucose-Selective Molecular Fluorescence Sensor. Angewandtie Chemie 33(21):2207–2209
Larkin J, Fossey J, James T, Brooks B, Bock C (2010) A computational investigation of the nitrogen-boron interaction in o-(N,N-Dialkylaminomethyl)arylboronate systems. J Phys Chem A 114(47):12531–12539
Chapin B, Metola P, Vankayala S, Woodcock H, Mooibroek T, Lynch V, Larkin J, Anslyn E (2017) Disaggregation is a mechanism for emission turn-on of ortho-aminomethylphenylboronic acid-based saccharide sensors. J Am Chem Soc 139(15):5568–5578
Moller C, Plesset MS (1934) Note on an approximation treatment for manyelectron systems. Phys Rev 46:618–622
Dunning Jr TH (1989) Gaussian basis sets for use in correlated molecular calculations. I. the atoms boron through neon and hydrogen. J Chem Phys 90:1007–1023
Kendal RA, Dunning Jr TH, Harrison RJ (1992) Electron affinities of the first row atoms revisited. J Chem Phys 96:6796–6806
Woon DE, Dunning Jr TH (1993) Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J Chem Phys 98:1358–1371
Peterson KA, Woon DE, Dunning Jr TH (1994) Benchmark calculations with correlated molecular wave functions. IV. The classical barrier height of the H+H2--> H2 + H reaction. J Chem Phys 100:7410–7415
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts RE, Stratmann O, Yazyev AJ, Austin R, Cammi C, Pomelli JW, Ochterski R, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2003) G03; R B.02 ed. Gaussian Inc., Wallingford, CT
Frisch JM, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman RJ, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts RE, Stratmann O, Yazyev AJ, Austin R, Cammi C, Pomelli JW, Ochterski R, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) G09. Gaussian Inc., Wallingford, CT
Bartlett RJ, Purvis GD (1978) Many body perturbation theory, coupled pair many electron theory, and the importance of quadruple excitations for the correlation problem. Int J Quantum Chem 14:561–581
Grimme, S.; Ehrlich, S.; Goerigk, L., (2011)Effect of damping function in dispersion corrected density functional theory. J. Comput. Chem. 32(7):1456–1465
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate abinitio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132(15):154104
Becke AD (1993) A new mixing of Hartree-Fock and local density functional theories. J Chem Phys 98:1372–1377
Yanai T, Tew D, Handy N (2004) A new hybrid exchange-correlation functional using the coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett 393:51–57
Perdew JP, Burke K, Ernzerhoff M (1997) Errata: generalized gradient approximation made simple. Phys Rev Lett 78(7):1396
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868
Grimme S (2006) Semi-empirical hybrid density functional with perturbative second-order correlation. J Chem Phys 124(3):034108
Goerigk L, Grimme S (2011) Efficient and accurate double-hybrid-meta-GGA density functionals—evaluation with the extended GMTKN30 database for general main group thermochemistry, kinetics, and noncovalent interactions. J Chem Theory Comput 7:291–309
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Accounts 120:215–241
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735–746
Foster J, Weinhold F (1980) Natural hybrid orbitals. J Am Chem Soc 102:7211–7218
Carpenter J, Weinhold F (1988) Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure. J Mol Struct THEOCHEM 169:41–62
Weinhold F, Glendening ED (2001) NBO 5.0 program manual: natural bond orbital analysis programs. Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin, Madison, WI, p 53706
Rao NZ, Larkin JD, Bock CW (2016) A comparison of the structure and bonding in the aliphatic boronic R-B(OH)2 and borinic R-BH(OH) acids (R=H, NH2, OH, and F): a computational investigation. Struct Chem 27:1081–1091
Demaison J, Lievin J, Csaszar AG, Gutle C (2008) Equilibrium structure and torsional barrier of BH3NH3. J Phys Chem A 112(19):4477–4482
Staubitz A, Robertson APM, Manners I (2010). Chem Rev 110:4079–4124
Klooster WT, Koetzle TF, Siegbhan PEM, Richards TB, Crabtree RH (1999) Study of the N-H---H-B dihydrogen bond including the crystal structure of BH3NH3 by neutron diffraction. J Am Chem Soc 121:6337–6343
Boese R, Niederprum N, Blaser D, Maksic ZB, Eckert-Masic M, Horwood E (1992) In molecules in natural science and medicine. Chichester
Thorne LR, Suenrum RD, Lovas FJ (1983) Microwave spectrum, torsional barrier, and structure of BH3NH3. J Chem Phys 78(1):167
Dixon DA, Gutkowski M (2005). J Phys Chem A 109:5129–5135
Horvath V, Kovacs A, Hargittai I (2003). J Phys Chem A 107:1197–1202
Bent HA (1961) An appraisal of valence-bond structures and hybridization in compounds of the first-row elements. Chem Rev 61:275–311
Acknowledgements
This research was supported in part by the National Science Foundation through XSEDE resources provided by the XSEDE Science Gateways program. The PQS Cluster Facility at Jefferson University was also used for the calculations described in this manuscript. J.D.L would like to thank the National Heart, Lung, and Blood Institute of the National Institutes of Health for generous support under Award Number K22HL113045. J.D.L. would also like to thank the National Science Foundation for support through grant CHE-1531590.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 170 kb)
Rights and permissions
About this article

Cite this article
Larkin, J.D., Bock, C.W. A comparison of the structure and bonding in the donor–acceptor complexes H3N → BR(OH)2 and H3N → BRH(OH) (R = H; NH2, OH, and F): a computational investigation. Struct Chem 30, 361–368 (2019). https://doi.org/10.1007/s11224-018-1205-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-018-1205-2