
Abstract
Bone marrow contains resident cellular components that are not only involved in bone maintenance but also regulate hematopoiesis and immune responses. The immune system and bone interact with each other, coined osteoimmunology. Hashimoto’s thyroiditis (HT) is one of the most common chronic autoimmune diseases which is accompanied by lymphocytic infiltration. It shows elevating thyroid autoantibody levels at an early stage and progresses to thyroid dysfunction ultimately. Different effects exert on bone metabolism during different phases of HT. In this review, we summarized the mechanisms of the long-term effects of HT on bone and the relationship between thyroid autoimmunity and osteoimmunology. For patients with HT, the bone is affected not only by thyroid function and the value of TSH, but also by the setting of the autoimmune background. The autoimmune background implies a breakdown of the mechanisms that control self-reactive system, featuring abnormal immune activation and presence of autoantibodies. The etiology of thyroid autoimmunity and osteoimmunology is complex and involves a number of immune cells, cytokines and chemokines, which regulate the pathogenesis of HT and osteoporosis at the same time, and have potential to affect each other. In addition, vitamin D works as a potent immunomodulator to influence both thyroid immunity and osteoimmunology. We conclude that HT affects bone metabolism at least through endocrine and immune pathways.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In the bone homeostasis, osteoclasts act in concert with osteoblasts and osteocytes. Osteoclasts mediate bone resorption and osteoblasts mediate bone formation, playing predominant role in the maintenance of bone homeostasis[1]. Osteocytes, deriving from mature osteoblasts, regulate both osteoblast and osteoclast function. Bone marrow functions as a maturation site for mesenchymal stem cells that give rise to osteoblasts and monocyte/macrophage lineage that give rise to osteoclasts. It also contains resident cellular components that are not only involved in bone maintenance but also regulate hematopoiesis and immune responses[2]. B cells and T cells regulate the coupling activity of osteoblasts and osteoclasts, both of them express strong inhibitors or agonist to influence osteoclastogenesis[3]. With the discovery of receptor activator of nuclear factor-kappaB (RANK) ligand (RANKL), a molecule expressed on immune cells (particularly T cells) and a major differentiation factor for osteoclasts[4], people have gradually realized that osteoporosis is not only a metabolic disease but a disease closely related to immune system. In a sense, the difference between a tolerogenic and an immunogenic encounter with antigen relies on the differential upregulation of co-stimulatory pathways[5]. As a kind of co-stimulatory molecule[5], RANKL-mediated effects on osteoclasts are regulated by a series of immune receptors that are also important in host defenses and aggravation of inflammation[4]. This link helps to clarify why many autoimmune diseases characterized by excessive immunoglobulin production with enhanced bone loss and fracture risk[6]. Because the production of autoimmune immunoglobulin is a manifestation of the aberrant activation of co-stimulatory pathways. Science that studies the influence of the immune system on bone is known as “osteoimmunology”, a term coined by Arron and Choi[7].
Chronic inflammation, such as rheumatoid arthritis and Crohn’s disease, has been widely reported to accelerate bone loss and increase fracture risks, suggesting that chronic immune activation is detrimental to bone[6]. And glucocorticoids, widely used as anti-inflammatory treatments, will aggravate the situation. Chronic lymphocytic thyroiditis, also named Hashimoto’s thyroiditis (HT), is the classic type of autoimmune thyroiditis (AIT) and is one of the most common autoimmune diseases. Euthyroidism is critical for skeletal development and maintenance of bone homeostasis. The role of different thyroid statuses of HT and thyroid autoimmunity itself in the process of bone metabolism have been underestimated.
In this review, we summarized the mechanisms of action of thyroid hormones (THs), the long-term effects of HT on bone, and the relationship between thyroid autoimmunity and osteoimmunology. We speculate that it works in at least two ways: as an endocrine disease, HT influences bone remodeling through thyroid function or thyroid stimulating hormone (TSH) levels; as an immune dysregulated disease, HT affects bone metabolism by affecting osteoimmunology.
2 Materials and methods
We performed a literature search for articles published until August 2022 in PubMed by using the search terms “bone metabolism”, “osteoporosis”, “bone mineral density”, “osteoimmunology”, “fractures”, “Hashimoto’s thyroiditis”, “thyroid autoimmunity”, “thyroiditis”, “TPOAb” and “TgAb”. A total of 1213 articles were initially retrieved. Articles on various topics of the relationship between bone metabolism, osteoimmunology and thyroid function, thyroid autoimmunity were included. Articles that focus on secondary osteoporosis, thyroid dysfunction unrelated to Hashimoto’s thyroiditis, and articles that do not conform to the topic of this review will be excluded. Titles of interest were further reviewed by abstract. Two-hundred fourteen articles were finally included in this review.
3 Literature Review
3.1 The endocrine function of the thyroid and bone metabolism
It is known that with the stimulation of TSH, the thyroid gland synthesizes and secrets THs. THs are important hormones that play their function in bone metabolism. With the help of TH transporters, THs are effectively transported into cells, and thyroxine (T4) is then deiodinated to the biologically active form of TH, triiodothyronine (T3). T3 interacts with thyroid hormone nuclear receptors (TRs) to exert its biological effects maily through genomic action, thereby activating the downstream signaling pathways. In addition, TSH may have direct effect on bone. Any abnormality in this process may affect bone metabolism (Fig. 1; Table 1). Thyroid dysfunction will also lead to bone metabolism disorders.

The role of thyroid hormone and TSH on bone. By Figdraw
3.1.1 Thyroid hormones and bone metabolism
3.1.1.1 Thyroid hormone transporters
As a kind of fat-soluble molecule, THs can enter cells directly or via TH transporters, proteins facilitating the cellular importation of THs. They include members of the polyspecific organic anion transporting polypeptide (OATP) family, the monocarboxylate transporter (MCT) family and the solute carriers(SLC) family[8]. The important and efficient thyroid hormone transporters are: MCT8, MCT10, OATP1C1, and SLC17A4[9]. However, the roles of MCT8 and MCT10 with regards to bone biology have been investigated more deeply. TH transporters affect bone by regulating local TH concentrations and may also have unique effects on bone. In a mouse model, MCT8 mRNA was expressed in chondrocytes, osteoblasts and osteoclasts at all stages of cell differentiation[10]. Global Mct8-knockout (Mct8-KO) mice were reported to demonstrate lower trabecular bone loss[11]. In vivo, conditional Mct8-KO in osteoblast and osteoclast progenitors resulted in trabecular bone gain in 12-week mice[11]. In addition, conditional Mct8-KO mice of different ages exhibited different skeletal phenotypes. For example, Mct8 deficiency in osteoclast precursors led to spine trabecular bone gain in 6-week mice but led to femur and vertebra trabecular bone loss in 12-week mice[8]. However, in vitro studies of osteoblasts showed no significant changes in osteogenic marker gene expression and mineralization capacity. Therefore, the role of MCT8 in the process of bone metabolism needs more researches. Besides, there might be compensatory factors that could help to import THs. MCT10, another protein that functions as a TH transporter[12], was shown to be another site- and age-dependent regulator of bone mass and turnover[13]. The exact effects of TH transporters on bone need to be investigated in greater detail in future studies.
3.1.1.2 Deiodinases
THs mainly refer to T3 and T4. Genomic actions of TH are mediated in the nucleus by T3, and non-genomic actions are mediated by T4 or T3 at the cell membrane or cytoplasm[14]. Deiodinases, kinds of selenoenzymes, regulate TH concentrations in different tissues. Type 1 iodothyronine deiodinase (DIO1) and type 2 iodothyronine deiodinase (DIO2) both function by converting T4 to T3 and removing iodine from the 5′ position or outer ring, while type 3 iodothyronine deiodinase (DIO3) removes iodine from the inner ring of T4 and T3, thus resulting in T3 inactivation[15]. Studies found that Dio1 mRNA and its enzyme activity were both undetectable in primary chondrocytes, osteoblasts and osteoclasts[10], as well as in cell lines[16], indicating that DIO1 has an insignificant physiological role in bone. A study reported that DIO1 deficiency contributes to excess TH-induced differences in peak bone mass accrual between DIO1-deficient mice and normal C57BL/6J mice, indicating that DIO1 assumes an indirect but relevant role in bone mass when systemic homeostasis is challenged by excess THs[17]. DIO2 activity was only present in mature osteoblasts[10]. Bones from adult Dio2-KO mice are brittle and exhibit a 50% reduction in bone formation and a generalized increase in mineralization, uncovering an essential role for DIO2 in osteoblasts in the optimization of bone strength and mineralization[18]. In addition, DIO2 also played a role in the perichondrium during early skeletal development, and the degradation of DIO2 promoted growth plate chondrocyte differentiation[19]. Adult mice lacking the Dio2 have increased subchondral bone but normal articular cartilage[20]. A cohort study enrolling 6,022 participants found that the DIO2 Thr92Ala polymorphism was associated with decreased bone mineral density in tibial in female subjects[21]. However, a genome-wide association (GWA) meta-analysis including 27,061 subjects found no association between DIO2 and bone mineral density or fractures[22]. One study sequenced the critical exons of DIO2 in subjects with relevant high BMD, indicating that DIO2 mutations are unlikely to be a common cause of increased BMD in euthyroid postmenopausal women[23]. DIO3 activity was detectable throughout chondrocyte, osteoblast and osteoclast differentiation in primary cell cultures[10], suggesting that DIO3-mediated regulation of TH activity plays an important role during skeletal development. In fact, the effect of deiodinase on bone still acquires further study.
3.1.1.3 Thyroid hormone nuclear receptors
It is known that the hypothalamic-pituitary-thyroid (HPT) axis is involved in bone remodeling[24]. T3 action is mediated by TRs, which activate T3 target gene transcription. TRs are composed of two subunits, TRα and TRβ, encoded by Thra and Thrb respectively, and each subunit has subtypes. TRα and TRβ are expressed to varying degrees in almost all tissues. TRα1, TRα2, and TRβ1 are expressed in osteoblasts, osteoclasts, and chondrocytes [25, 26]. TRs are also expressed in skeletal mast cells, and their localizations are not affected by changes in thyroid status, suggesting that disrupted endochondral ossification in hypothyroidism may be mediated in part by skeletal mast cells[27] for their ability to synthesize and store matrix-degrading enzymes. Previous studies have suggested that TRβ is mainly expressed in the pituitary and hypothalamus to negatively regulate the HPT axis. In the skeleton, TRα is expressed at higher levels than TRβ and mediates T3 action on bone and cartilage[28]. Analysis of T3 target gene expression revealed skeletal hypothyroidism in TRα(0/0) mice but skeletal thyrotoxicosis in TRβ-/- mice[29], demonstrating that bone loss in thyrotoxicosis was mediated mainly by TRα. Further research confirmed that TRβ mediated the effects of transient TH concentration changes on bone remodeling, whereas TRα mediated the long-term effects of chronic changes in TH metabolism[30]. When a point mutation of the Thra allele was introduced to weaken its function, severely delayed skeletal development and high adult bone mass were shown, whereas point mutations in the Thrb gene showed accelerated bone formation and mineralization followed by rapid bone loss[31]. The PV mutation is an insertion mutation of the Thrb gene resulting in TRβ dysfunction. Homozygous ThrbPV/PV mutant mice had elevated circulating TH levels and accelerated skeletal development[32]. A microarray study found elevated activity of the canonical Wnt/β-catenin pathway in the skeleton of ThrbPV/PV mice in vivo and in ThrbPV/PV osteoblasts in vitro, while T3 inhibited the Wnt pathway by facilitating proteasomal degradation of β-catenin and preventing its accumulation in the nucleus[33]. This finding demonstrated that Wnt signaling pathways were involved in the T3-mediated regulation of bone formation. In addition, the TRβ-specific agonist GC-1 not only helped to reduce the number of bone marrow-derived adipocytes in Tshr-/- mice but also increased the expression of several brown/beige fat markers in bone marrow mesenchymal stromal or mouse marrow stromal ST2 cells, indicating that TH regulation of marrow adiposity was mediated at least in part via activation of TRβ signaling[34].
3.1.1.4 Thyroid hormone
The skeleton is an important TH-targeted tissue. THs play important roles in linear growth and skeletal maturation and enhance the response to growth hormone and insulin-like growth factor-1 (IGF-1)[35]. In addition, T3 induces FGFR1 expression and FGF-induced MAPK signaling in osteoblasts[36]. For children, severe untreated hypothyroidism leads to delayed skeletal development, defective endochondral ossification, and short stature[1]. Timely diagnosis and TH replacement therapy helped to reverse the situation. For adults, THs are key regulators of bone health; the excess caused high bone turnover-mediated bone loss with a shortened remodeling time[26], and the deficiency caused low bone turnover-mediated bone loss for reduced osteoclastic bone resorption together with decreased osteoblastic activity[1]. Meanwhile, T3 promotes the differentiation and activation of osteoclasts directly[37] or indirectly by acting on osteoblasts[38]. Some researchers hold that the action of T3 on osteoclastic bone resorption is mediated predominantly by osteoblast-derived osteotrophic cytokines[39]. Thyroid homeostasis is critical for the maintenance of bone homeostasis.
3.1.2 TSH and bone metabolism
Currently, the role of TSH itself in bone homeostasis has been debated. There is growing evidence showing that TSH has physiologically relevant effects on bone or bone cells and may exert a direct protective effect against osteoporosis (Fig. 1; Table 1). In euthyroid postmenopausal Greek women, higher TSH levels were associated with a lower risk of vertebral fracture[40]. TSH in the low-normal range correlated with a high incidence of vertebral[41], hip[42], spine, forearm, and humerous fractures[43]. Similarly, the results of the OPENTHYRO cohort showed a relevance between the risk of fractures and decreasing levels of TSH in euthyroid people[43]. By using genetic biobank data, genetically raised serum TSH concentrations were noticed to causally associated with decreased bone fracture risk in men [44]. Another study found that TSH levels were positively correlated with BMD in men with normal thyroid function[45]. In addition, TSH was positively correlated with osteoprotegerin (OPG), indicating that the level of OPG was directly affected by TSH[46]. However, a study including 993 euthyroid postmenopausal females and 968 euthyroid males found no association between TSH and BMD[47].
Therefore, further researches are needed to determine the role of TSH in bone. Compared with ovariectomized controls, intermittent injection of TSH significantly improved BMD in ovariectomized rats or mice[48]. Baliram et al. found greater bone loss and resorption in Tshr-KO hyperthyroid mice than in wild-type hyperthyroid mice, demonstrating that the absence of TSH signaling contributed to bone loss[49]. In ex vivo cultures of bone marrow-derived osteoclast precursors, the inhibitory action of TSH on osteoclasts lasted more than 4 weeks after the cessation of TSH exposure, manifesting as reduced expression of key genes for osteoclastogenesis[48]. TSH inhibits osteoclast formation and survival by attenuating JNK/c-jun and NFκB signaling triggered in response to RANK-L and tumor necrosis factor-alpha (TNF-α)[50]. TSH also directly inhibited TNF-α production and reduced the number of TNF-α-producing osteoclast precursors[51]. In the process of osteoblast differentiation, TSH stimulates osteoblast differentiation[45], primarily through the activation of protein kinase Cδ and the upregulation of the noncanonical Wnt components frizzled and Wnt5a[52]. Further investigations showed that the presence of a macrophage-derived TSH-β splice variant (TSH-βv) exerted an osteoprotective effect by inducing osteoblastogenesis[53]. However, some researchers have suggested that TSH also inhibits osteoblast differentiation and type 1 collagen expression in a runt-related transcription factor 2 (RUNX2) and osterix independent manner by downregulating Wnt (LRP-5) and VEGF (Flk) signaling[50]. The exact mechanisms of action need further investigation. In addition, small molecules have received increasing attention as therapeutic options for modulating TSHR signaling. It has been reported that signal activation mediated by the small molecule β-arrestin-1 is involved in the TSH-induced enhancement of osteoblast differentiation[54].
3.1.3 Thyroid function status and bone metabolism
Some patients with HT have euthyroid function. Others present as subclinical or overt hypothyroidism, with a low progression of subclinical to overt hypothyroidism occurring at an average rate of 5% per year. In addition, few HT patients have concurrent hyperthyroidism at the same time, named “Hashitoxicosis”. Hashitoxicosis is the hyperthyroid phase of HT, caused by the destruction of thyroid follicles for thyroid inflammation[55], usually followed by a euthyroid or hypothyroid state[56], and it resolves in a few weeks to months[57]. Therefore, HT patients may have an effect on bone for various thyroid function statuses.
HT was the most common cause of hypothyroidism in the USA[58]. In addition to the autoimmune condition of HT, hypothyroidism also affects the bone. For patients with overt hypothyroidism, the duration of bone resorption was twofold longer, and the duration of bone formation and subsequent bone mineralization was fourfold longer[59]. From this perspective, hypothyroidism has the ability to boost bone mass and bone stiffness. However, with the increase in bone stiffness, the risk of fractures also increases[35]. A nationwide follow-up study in 16,249 patients demonstrated that fracture risk increased in hypothyroidism patients[60]. In addition, a study using mice carrying mutations showed that THs lacking rather than excess thyrotropin may cause abnormal skeletal development in hypothyroidism[61]. Whether hypothyroidism leads to osteoporosis remains controversial. Bone microarchitecture in women with hypothyroid HT did not differ considerably from healthy control subjects[62]. Hypothyroidism in premenopausal females was report to have no influence on bone density[63]. For subclinical hypothyroidism, there is no consensus on its role in osteoporosis. A meta-analysis found no association between subclinical hypothyroidism and fracture risk[64]. However, some researchers noticed that subclinical hypothyroidism appeard to decrease serum calcium and low bone density[65]. Postmenopausal women with subclinical hypothyroidism tended to have a higher risk for low-trauma hip fracture[66] and a lower heel quantitative ultrasound[67].
At present, there is a consensus that patients with thyrotoxicosis tend to have a higher risk of osteoporosis and fracture, with high bone turnover and shortened bone remodeling. The uncoupling of osteoclast and osteoblast activities results in a 10% loss of bone per remodeling cycle[68]. Subclinical hyperthyroidism is commonly seen in patients undergoing TSH suppression therapy with levothyroxine (LT4) after thyroid tumor surgery. Previous studies have suggested that long-term LT4 therapy and decreased TSH levels usually have negative effects on bone metabolism and bone mass[69, 70]. However, a cross-sectional study suggests that long-standing suppressive therapy with LT4 may have no significant adverse effects on bone mineral density or microarchitecture[71]. A meta-analysis suggested that for patients with differentiated thyroid cancer, TSH suppression therapy was associated with lower BMD of the spine and total hip in postmenopausal women but not in premenopausal women and men[72]. Similarly, evidence from observational studies suggests that postmenopausal women treated with TSH suppression therapy are at risk for lower BMD[73]. Therefore, more researches are required to figure out the relationship between thyroid status and bone, and whether the status of HT will aggravate the influence.
3.2 HT patients’ thyroid autoimmunity and bone metabolism
Current studies indicate that for patients with HT, the bone is affected not only by thyroid function but also by the setting of the autoimmune background. HT patients could develop thyroid dysfunctions, such as hyperthyroidism and hypothyroidism, that will lead to the disturbance of bone homeostasis. Meanwhile, abnormal immune activation in HT patients will also affect bone metabolism. Autoimmune diseases are a heterogeneous group of diseases characterized by inflammatory responses directed against self-tissues by the activation of abnormal immune cells[74]. Based on the main mechanisms and cell types involved in the pathogenesis, they can generally be characterized as antibody-mediated humoral immunity and T lymphocyte-mediated cellular immunity[75]. Immune cells are capable of producing various cytokines and interacting with other cells, further regulating and aggravating immune disorders. The destruction of thyroid cells in autoimmune thyroid disease (AITD) involves different mechanisms, including autoreactive T-lymphocytes, natural killer (NK) cells, and cytokines[75]. HT is a typical T-cell-mediated disease with severe parenchymal inflammatory infiltrates[76]. Antibodies against thyroglobulin (TgAb) and antibodies against thyroid peroxidase (TPOAb) are produced. In HT, the cytokines IL-1β, IL-17, and IL-23 are the key players[77]. Thyroid autoimmunity interacts with bone through these molecules (Fig. 2; Table 1). The pathophysiological mechanisms between thyroid autoimmunity and bone metabolism have not been well ascertained, and they might involve multiple factors.

Thyroid autoimmunity could interact with bone through these molecules. By Figdraw
3.2.1 Thyroid autoantibodies
The presence of thyroid autoantibodies is one of the main characteristics of HT. A positive thyroid autoantibody test with a normal thyroid function often indicates an early-stage of HT. At this stage, abnormal autoimmune status is sufficient to affect bone homeostasis. TPOAb induces cell death through complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC) to attack thyroid tissues, accompanied by a changed immune microenvironment. Thyroid autoantibodies may have direct effects on bone function with unclear mechanisms. For euthyroid patients with HT, the BMD values of the lumbar spine and femoral neck were lower than those without HT, and the longer the course of HT, the higher the prevalence of osteoporosis[78]. The presence of TPOAb was associated with decreased BMD at both the spine and hip in euthyroid postmenopausal women[79]. Lambrinoudaki et al. reported that positivity for TgAb and TPOAb was associated with the risk of vertebral fractures in postmenopausal women. TSH and TgAb both independently help to predict the presence of vertebral fractures[40]. Polovina et al. found that the presence of TPOAb was associated with a higher Fracture Risk Assessment Tool (FRAX) score in postmenopausal women[66]. It is konwn that FRAX score is used to quantify a patient’s 10-year probability of a hip or major osteoporotic fracture, which is applicable to both postmenopausal women and men aged 40 to 90 years[80]. As HT is also one of the most common disease affecting middle-aged and elderly population. It is unknown whether the positivity of TPOAb should be added and what proportion they should occupy in the FRAX score. In addition, For patients with subclinical hypothyroidism, TSH was a better indicator for future fragility fractures than TPOAb[79]. The effect of thyroid hormone on bone homeostasis is more prominent than that of immune activation, when HT patients developed thyroid dysfunction.
3.2.2 Vitamin D and HT
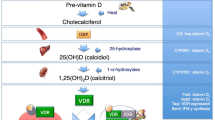
The classical role of vitamin D is in the regulation of calcium and phosphate metabolism and bone mineralization[81]. It was reported that 1,25-dihydroxyvitamin D (1,25(OH)2D) induces the transcriptional activity of the vitamin D receptor (VDR) to increase calcium absorption in the intestine and calcium reabsorption in the kidney. VDR is expressed in osteoblasts, and 1,25(OH)2D modifies the gene expression of various osteoblast differentiation- and mineralization-related genes[82]. 1,25(OH)2D also plays important roles in the formation of osteoclasts through its actions on osteoblastic cells[83]. In recent years, the “noncalcemic” effect of vitamin D has drawn attention. It has been regarded as a potent immunomodulator. Some immune cells, such as B cells, T cells, and antigen-presenting cells, express 1-hydroxylase (CYP27B1) in cells and have the ability to generate the active compound vitamin D, exerting immunomodulatory effects[84]. According to the study of Hossein-nezhad et al., supplementation with vitamin D was closely related to the expression of genes involved in the regulation of more than 160 pathways linked to autoimmune disorders, cancers and cardiovascular diseases[85]. Chronic inflammatory diseases may lead to the deactivation of the VDR[86].
There is growing evidence showing that vitamin D may participate in the pathogenesis of HT. Vitamin D supplementation can be helpful for alleviating disease activity in HT patients[87, 88]. A recent study indicated that vitamin D deficiency or insufficiency can increase the rate of autoimmune diseases such as HT[89]. After vitamin D treatment, TPOAb titers significantly reduced without a change in thyroid function[90]. It could also change the balance of CD4 + T-cell subsets to improve disease control[91]. Therefore, some researchers held that vitamin D supplementation was advisable because it could not only correct vitamin deficiencies but also potentially improve autoimmune status[92].
However, the relationship between vitamin D and HT is still a controversial issue. Some studies found that lymphocytic thyroiditis was associated with neither vitamin D deficiency nor vitamin D levels[93, 94]. A study based on a Caucasian Polish population suggested that vitamin D receptor gene polymorphisms were not a major susceptibility factor for AIT development[95]. However, a study based in India found that vitamin D levels were low in AITD patients and vitamin D supplementation in HT patients increased thyroid autoantibody titers, reduced serum TSH levels, and increased free T4 levels[96]. In fact , the effect of vitamin D on thyroid function is also controversial. Some studies reported that Vitamin D treatment may not influence thyroid functions[90, 97]. But other studies reported it may be beneficial to thyroid function, especially during pregnancy and postpartum[98, 99]. Therefore, there is a need for large double-blind clinical trials to investigate the effects of vitamin D supplementation on HT.
3.2.3 RANK/RANKL/OPG axis and HT
It is well known that RANKL and OPG are produced by a variety of tissues, including epithelial and mesenchymal cells, and play important roles in bone metabolism and immune responses. RANKL secreted by osteoblast lineage cells (osteoblasts and osteocytes) binds to its receptor RANK on the cell membrane of osteoclast progenitors, promoting subsequent osteoclast activation[100]. Osteoblasts secrete OPG, blocking RANKL/RANK signaling by acting as a decoy receptor.
Existing studies have shown that, not only in the bone, the RANK/RANKL/OPG axis also functions in the thyroid gland. Sutdies found that RANKL and OPG were produced by thyroid follicular cells, regulated by cytokines and TSH, and capable of modulating dendritic cell functions, indicating that immunoregulatory factors were involved in the pathogenesis of autoimmune thyroid diseases[101]. At the cellular level, pronounced OPG mRNA levels in normal human thyroid were noticed, while RANKL mRNA levels were low[101]. OPG mRNA expression was upregulated and RANKL mRNA was inhibited by TSH stimulation[101]. Patients with hypothyroidism had higher serum levels of OPG, and the correction of thyroid status led to a decrease in OPG[102], which might be related to endothelial dysfunction[103]. A study found that RANKL levels were lower and OPG levels were higher in premenopausal women with AIT than in nonthyroiditis controls[46], indicating that thyroid autoimmunity may regulate the level of RANKL/OPG. However, a study reported no correlation between OPG and RANKL in patients with a history of thyroid diseases[104], and another study did not observe a significant increase in OPG or RANKL levels after recombinant human TSH administration[105]. Therefore, further studies are needed to clarify the actions and mechanisms behind thyroid autoimmunity in the RANK/RANKL/OPG axis and to determine whether the change results in alterations of osteoimmunology.
3.2.4 Cytokines
Osteoporosis has been considered a chronic immune-mediated inflammatory disease with the discovery of the RANK/RANKL/OPG axis engaging in bone homeostasis[106]. The etiology of thyroid autoimmunity and osteoimmunology is complex and involves a number of immune cells, cytokines, and chemokines. T lymphocytes and their secreted cytokines play vital roles in modulating the immune response. The same cytokines may affect and connect different tissues and organs. Proinflammatory cytokines might be primary mediators of accelerated bone loss in postmenopausal osteoporosis[107].
IL-6: Interleukin (IL)-6 is involved in the pathogenesis of osteoporosis, which is characterized by a negative balance between bone resorption and formation[108]. In ovariectomized mice, IL-6 appears to be crucial in bone turnover acceleration[109]. It increases RANKL mRNA expression and inhibits bone formation[101]. IL-6 may exert its inhibitory effect on bone formation directly through gp130-STAT 1/3 signaling[110] or indirectly by influencing the balance between the RANKL/RANK/OPG pathway[111]. In addition, IL-6 suppressed the differentiation of osteoclast precursors into osteoclasts by inducing chondrocytic PGE(2) production[112]. A study found a positive correlation between IL-6 and RANKL and a negative correlation between IL-6 and TSH[46]. When the status of thyroid function changes or thyroid autoimmunity occurs, cytokine levels altered. For example, hyperthyroidism leads to increased levels of IL-1 and IL-6, which are vital for bone resorption. It also plays a role in mediating the bone loss that results from excess THs[113].
IL-17: Recent studies on thyroiditis have focused on the imbalance between Th17 and Treg cells. Treg cells suppress immune responses. Th17 cells are a newly discovered lineage of effector CD4 + T cells that are different from Th1 and Th2 cells[114]. Th17 cells are necessary for the removal of extracellular bacteria and play an essential role in the pathogenesis of AITD[115]. The combination of IL-17 and IL-17 A motivates MAPK signaling, recruits macrophages and neutrophils, and results in proliferation of T cells and inflammation of the thyroid[116]. For patients with HT, significantly elevated levels of TH17 cells and IL-17 A were noticed in both the peripheral blood and thyroid tissues[114, 116,117,118]. The reciprocal role of Th17 cells and Treg cells is involved in the pathogenesis of osteoporosis in ovariectomized mouse models[119]. Th17 cells promote osteoclastogenesis, while Treg cells exhibit anti-osteoclastogenic activity[119]. In experimental periodontitis, Th17 cells and the level of IL-17 were pathogenic drivers of inflammatory bone loss[120]. Increased Th17 cells and IL-17 levels are associated with low BMD in postmenopausal women[119].However, the function of IL-17 on osteoblasts is controversial. Some studies have reported that IL-17 inhibited osteoblast differentiation and bone regeneration in rats[[121, 122]. Another study reported that IL-17 stimulated osteoblast differentiation and osteoblast-dependent osteoclastogenesis in vitro[123]. Since AIT accompanies elevated serum IL-17, whether this change is involved in the pathogenesis of osteoporosis has not been explored. Similar to chronic skin inflammation leading to bone loss by IL-17-mediated inhibition of Wnt signaling in osteoblasts[124], there could also be a connection between AIT and osteoporosis.
IL-23: IL-23 is known to stimulate the development of Th17 cells and the production of IL-17, to obtain a pathogenic and sustained phenotype[125]. The IL-23/IL-17 axis plays a role in the pathogenesis of HT[77, 114, 126]. The data of Gerenova et al. showed that the significance of IL-23 was more pronounced than that of IL-17 in HT development and severity[114]. Furthermore, the IL-23/IL-17 axis also plays a role in the pathogenesis of inflammation and bone destruction[127]. In mice, systemic overexpression of IL-23 induced chronic arthritis and increased osteoclast differentiation and systemic bone loss[128]. According to Priyanka Shukla et al., the efficacy of anti-IL-23 therapy supported bone protective effects[127]. IL-23 directly mediates bone loss by augmenting osteoclast activity and RANKL expression in T cells[129], and promotes osteoclastogenesis in an osteoblast-osteoclast coculture system[130]. However, Razawy et al. demonstrated that IL-23R-deficient mice had a temporary defect in their bone formation, which resulted in temporal effects on trabecular bone and long-term effects on cortical bone[131]. Kamiya et al. reported that IL-23 and IL-27, both partly and indirectly through activated T cells, inhibited osteoclastogenesis[132]. IL-23 may have different effects on bone in different situations. So what is the role of IL-23 in the contest of HT needs more exploration.
IL-33: Recently, the IL-33/suppression of tumorigenicity 2 protein (IL-33/ST2) pathway revealed its participation in the process of several autoimmune diseases. ST2 is the receptor of IL-33. Wang et al. found that the elevated mRNA expressions of plasma IL-33 and sST2 perpetuated chronic inflammation in HT[133]. However, the results of elevated IL-33 and sST2 in bone are still debatable. Mun el at. reported that IL-33 stimulated the formation of functional osteoclasts from human CD14(+) monocytes[134]. Malcolm et al. found that IL-33 exacerbated periodontal disease through the induction of RANKL, leading to bone loss in a RANKL-dependent manner[135]. However, Zaiss held that IL-33 shifted the balance from osteoclast to alternatively activated macrophage differentiation and protects against TNF-α-mediated bone loss[136]. Therefore, how abnormal levels of IL-33 caused by thyroid autoimmunity affect bone is inconclusive thus far.
IL-1β: The IL-1 family is known to play a unique role in innate immunity. IL-1α and IL-1β are two individual forms of IL-1 which are isolated from two distinct cDNAs but are indistinguishable in terms of biological functions[137]. Previous studies have shown that IL-1 stimulates thyroid cell growth[138], inhibits the synthesis and release of THs[139], promotes the exposure of hidden autoantigens to the immune system[140], and inhibits the expression of thyroglobulin[141] and thyroid peroxidase[142]. IL-1β is a potent proinflammatory cytokine involved in a variety of cellular activities, including cell apoptosis and the promotion of autoimmune diseases[143]. Upregulated tissue expression of IL-1β was noticed in the thyroid, and it may be an active etiologic factor in the pathogenesis of HT[144]. IL-1β contributes to autoimmune arthritis by inducing osteoclastogenic capacity in Tregs[145]. In the early stages of fracture healing, IL-1β leads to impaired recruitment of osteoblasts[146]. In human periodontal ligament fibroblasts, IL-1β inhibits BMP-9-induced osteoblastic differentiation[147]. However, the effect of IL-1β on bone marrow mesenchymal stem cells is unclear[148, 149]. IL-1β is involved in multiple processes of HT autoimmunity and bone loss caused by various chronic inflammatory diseases. In patients with HT, whether the abnormal expression of IL-1β is involved in the process of osteoporosis needs further study.
TNF-α: TNF-α is known to be produced by thyroid-infiltrating CD4 + T cells and B cells[150]. The secretion of TNF-α by infiltrating T cells in the thyroid is higher than that in normal human lymph nodes, indicating that TNF-α could directly damage human thyroid follicular epithelial cells[151]. The ratio of TNF-α/IL-6 in HT patients was higher than that in healthy patients or those with Graves’ disease[152]. TNF-α and transforming growth factor-β promoted hyperplasia and the proliferation of thyrocytes in IFN-γ -/-Non-Obese Diabetic.H-2h4, resulting in a chronic state of thyroid dysfunction and fibrosis[153]. Local and systemic production of cytokines, including TNF-α, IL-1, IL-6 and IL-17, functions to recruit osteoclast precursors into the bone microenvironment, which is suitable for differentiation into mature osteoclasts[154]. TNF-α-mediated bone destruction has been confirmed, especially in rheumatoid arthritis, and its therapeutic monoclonal antibodies have been widely used in the clinic. In addition, TNF-α directly inhibits osteoblast differentiation and bone nodule formation by suppressing the expression of positive regulators of bone development and growth[45]. TNF-α has a promoting effect on thyroid immunity and bone loss, so whether the increase in TNF-α caused by one disease will lead to the aggravation of another disease requires more research.
IFN-γ: Interferon-gamma (IFN-γ), another immunogenic cytokine, is involved in both innate and adaptive immune responses. It plays a role by activating macrophages, inducing antigen presentation, upregulating adhesion molecules, and recruiting Th1 cells to sites of inflammation, etc.[155]. A previous study showed that IFN-γ played a dual role in the development of lymphocytic-spontaneous autoimmune thyroiditis (L-SAT). It is required for the development of L-SAT, and it also functions to inhibit thyroid epithelial cell hyperplasia and proliferation[156, 157]In addition, IFN-γ mediates thyroid follicular cell apoptosis by upregulating the expression of Fas in HT[158]. Coincidentally, it also has dual functions in bone. IFN-γ not only blunts osteoclast formation by directly influencing osteoclast precursors but also indirectly promotes osteoclast differentiation and bone resorption by stimulating antigen-dependent T-cell activation and T-cell secretion of the osteoclastogenic factors RANKL and TNF-α[159]. IFN-γ promotes the degradation of tumor necrosis factor receptor associated factor 6 (TRAF6) via the proteasome, leading to blocked activation of the downstream transcription factors NF-κB and JNK of the RANK-RANKL signaling axis and ultimately leading to reduced osteoclastogenesis[160]. Interestingly, this inhibition of osteoclastogenesis could be rescued by overexpression of TRAF6 in precursor cells. In addition, IFN-γ increases the expression of osteoblast-related genes, including RUNX2, osterix, alkaline phosphatase, and osteocalcin[161]. In the condition of estrogen deficiency and chronic inflammation, the net effect of IFN-γ is that of stimulating bone resorption and bone loss[159]. IFN-γ has dual effects on the regulation of both thyroid immunity and bone metabolism. How IFN-γ regulates bone in the setting of chronic thyroid immune dysfunction is unclear.
In addition to the cytokines discussed above, there are many cytokines or small molecules involved in the pathogenesis of HT and osteoporosis at the same time, and potentially affect both diseases by interacting with each other, such as TNF-β, IL-10, IL-18, IL-34, IL-38, et al. However, it remains unclear whether changes in circulating immune complexes caused by HT have an impact on the development of osteoporosis.
3.3 Reduce the effects of HT on osteoporosis
HT is a slowly progressive chronic disease that affects bone differently at different disease stages. At the early stage, patients often maintain euthyroid status but have a disordered immune system, manifesting as abnormal levels of relevant cytokines or positivity for thyroid-related immunoglobulins. At this time, disturbance of the immune system caused by HT interacts with osteoimmunology, thereby affecting bone remodeling. As the disease progresses, when the aggravation of thyroid immune inflammation leads to thyroid dysfunction, patients gradually develop permanent hypothyroidism, while a few patients show a short stage of hyperthyroidism. At this time, HT affects patients through TSH and/or THs in addition to the immune system, which significantly exacerbates the impact on bone.
Therefore, different treatment regimens at different stages should be applied to reduce the impact of HT on bone. However, no effective treatment for immune abnormalities in the early stage of HT has been developed. Some studies have proven that selenium (Se) has an immunomodulatory effect. For HT patients, Se supplementation was able to decrease TPOAb, TgAb, and TSH levels, increase glutathione peroxidase 3, increase activated Tregs[162], decrease INF-γ and increase IL-1β[163], exerting a beneficial effect on thyroid autoantibodies and thyroid function by increasing antioxidant activity and upregulating activated Treg cells[162]. In vitro studies showed that Se nanoparticles (SeNPs) promoted osteoblast differentiation by modulating alkaline phosphatase (ALP) activity and promoting calcium nodule formation and collagen content[164]. In vivo studies have shown that SeNPs stimulate osteoblast differentiation via the BMP-2/MAPKs/β-catenin pathway in diabetic osteoporosis[164]. In addition, inverse associations of Se levels with osteoporosis risk were observed[165]. Within the reference range, Se levels were positively correlated with BMD in healthy elderly men[166]. In euthyroid postmenopausal women, Se levels were inversely related to bone turnovers and positively correlated with BMD[167]. A study, based on population in the US, reported that higher Se status was correlated with lower FRAX scores and a lower incidence of previous bone fractures[168]. Therefore, Se supplement to patients in the early stage of HT may not only delay its progression but also may reduce the possibility of the occurrence and progression of osteoporosis. In addition, when HT patients develop abnormal thyroid function, it is particularly important to correct thyroid status.
4 Conclusion
Osteoporosis is not only a disease of bone-metabolic abnormalities, but also a disease featuring immune disorders. HT is the most common thyroid autoimmune disease and is also a common autoimmune disease. Immune changes caused by HT may be one of the factors that exacerbate osteoporosis. In this review, we summarized the mechanisms of the long-term effects of thyroid dysfunction on bone and the relationship between thyroid autoimmunity and osteoimmunology. We speculate that HT affects the occurrence and development of osteoporosis at least through endocrine and immune pathways. However, there are currently no drugs that can effectively modulate immunity in HT. Long-term thyroid function monitoring should be performed in patients with HT to avoid thyroid dysfunction promoting or exacerbating osteoporosis.
Abbreviations
- HT:
-
Hashimoto’s thyroiditis
- RANK:
-
receptor activator of nuclear factor-kappaB
- RANKL:
-
receptor activator of nuclear factor-kappaB ligand
- AIT:
-
autoimmune thyroiditis
- TH:
-
thyroid hormones
- TSH:
-
thyroid stimulating hormone
- T4:
-
thyroxine
- T3:
-
triiodothyronine
- OATP:
-
organic anion transporting polypeptide
- MCT:
-
monocarboxylate transporter
- SLC:
-
solute carriers
- DIO1:
-
type 1 iodothyronine deiodinase
- DIO2:
-
type 2 iodothyronine deiodinase
- DIO3:
-
type 3 iodothyronine deiodinase
- GWA:
-
genome-wide association
- HPT:
-
hypothalamic-pituitary-thyroid
- TR:
-
thyroid hormone nuclear receptor
- IGF-1:
-
insulin-like growth factor-1
- OPG:
-
osteoprotegerin
- LT4:
-
levothyroxine
- NK:
-
natural killer
- TgAb:
-
antibodies against thyroglobulin
- TPOAb:
-
antibodies against thyroid peroxidase
- CDC:
-
complement-dependent cytotoxicity
- ADCC:
-
antibody-dependent cell-mediated cytotoxicity
- FRAX:
-
Fracture Risk Assessment Tool
- 1,25(OH)2D:
-
1,25-dihydroxyvitamin D
- VDR:
-
vitamin D receptor
- IL:
-
interleukin
- ST2:
-
suppression of tumorigenicity 2 protein
- TNF-α:
-
tumor necrosis factor- alpha
- IFN-γ:
-
interferon-gamma
- L-SAT:
-
lymphocytic- spontaneous autoimmune thyroiditis
- TRAF6:
-
tumor necrosis factor receptor associated factor 6
- RUNX2:
-
runt-related transcription factor 2
- Se:
-
selenium
- SeNPs:
-
Se nanoparticles
- ALP:
-
alkaline phosphatase
References
Delitala AP, Scuteri A, Doria C. Thyroid Hormone Diseases and Osteoporosis. J Clin Med, 2020. 9(4).
Engblom C, et al., Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science, 2017. 358(6367).
Schett G, David JP. The multiple faces of autoimmune-mediated bone loss. Nat Rev Endocrinol. 2010;6(12):698–706.
Takegahara N, Kim H, Choi Y. RANKL biology Bone. 2022;159:116353.
Edner NM, et al. Targeting co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discov. 2020;19(12):860–83.
Schett G, Takayanagi H. Editorial overview: Osteoimmunology Bone. 2022;162:116466.
Arron JR, Choi Y. Bone versus immune system. Nature. 2000;408(6812):535–6.
Lademann F, et al. Bone cell-specific deletion of thyroid hormone transporter Mct8 distinctly regulates bone volume in young versus adult male mice. Bone. 2022;159:116375.
Groeneweg S, et al., Thyroid Hormone Transporters. Endocr Rev, 2020. 41(2).
Williams AJ, et al. Iodothyronine deiodinase enzyme activities in bone. Bone. 2008;43(1):126–34.
Lademann F, et al. Lack of the thyroid hormone transporter Mct8 in osteoblast and osteoclast progenitors increases trabecular bone in male mice. Thyroid. 2020;30(2):329–42.
Abe S, et al. Monocarboxylate transporter 10 functions as a thyroid hormone transporter in chondrocytes. Endocrinology. 2012;153(8):4049–58.
Lademann F, et al., The thyroid hormone transporter MCT10 is a Novel Regulator of Trabecular Bone Mass and bone turnover in male mice. Endocrinology, 2022. 163(1).
Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31(2):139–70.
Lavado-Autric R, et al. Deiodinase activities in thyroids and tissues of iodine-deficient female rats. Endocrinology. 2013;154(1):529–36.
Gouveia CH, et al. Type 2 iodothyronine selenodeiodinase is expressed throughout the mouse skeleton and in the MC3T3-E1 mouse osteoblastic cell line during differentiation. Endocrinology. 2005;146(1):195–200.
Zaitune CR, et al. Abnormal thyroid hormone status differentially affects bone Mass Accrual and Bone Strength in C3H/HeJ mice: a mouse model of type I deiodinase Deficiency. Front Endocrinol (Lausanne). 2019;10:300.
Bassett JH, et al. Optimal bone strength and mineralization requires the type 2 iodothyronine deiodinase in osteoblasts. Proc Natl Acad Sci U S A. 2010;107(16):7604–9.
Dentice M, et al. The hedgehog-inducible ubiquitin ligase subunit WSB-1 modulates thyroid hormone activation and PTHrP secretion in the developing growth plate. Nat Cell Biol. 2005;7(7):698–705.
Waung JA, Bassett JH, Williams GR. Adult mice lacking the type 2 iodothyronine deiodinase have increased subchondral bone but normal articular cartilage. Thyroid. 2015;25(3):269–77.
Kang YE, et al. Type 2 deiodinase Thr92Ala polymorphism is associated with a reduction in bone mineral density: a community-based korean genome and epidemiology study. Clin Endocrinol (Oxf). 2020;93(3):238–47.
Zhang L, et al. Multistage genome-wide association meta-analyses identified two new loci for bone mineral density. Hum Mol Genet. 2014;23(7):1923–33.
Gogakos A, et al. THRA and DIO2 mutations are unlikely to be a common cause of increased bone mineral density in euthyroid post-menopausal women. Eur J Endocrinol. 2014;170(4):637–44.
Bassett JH, Williams GR, Critical role of the hypothalamic-pituitary-thyroid axis in bone Bone, 2008. 43(3): p. 418 – 26.
Abu EO, et al. The expression of thyroid hormone receptors in human bone. Bone. 1997;21(2):137–42.
Lademann F, et al. Disruption of BMP Signaling prevents Hyperthyroidism-Induced Bone loss in male mice. J Bone Miner Res. 2020;35(10):2058–69.
Siebler T, et al. Thyroid status affects number and localization of thyroid hormone receptor expressing mast cells in bone marrow. Bone. 2002;30(1):259–66.
Nicholls JJ, et al. The skeletal consequences of thyrotoxicosis. J Endocrinol. 2012;213(3):209–21.
Bassett JH, et al. Thyroid hormone excess rather than thyrotropin deficiency induces osteoporosis in hyperthyroidism. Mol Endocrinol. 2007;21(5):1095–107.
Monfoulet LE, et al. Thyroid hormone receptor β mediates thyroid hormone effects on bone remodeling and bone mass. J Bone Miner Res. 2011;26(9):2036–44.
Bassett JH, et al. Thyroid status during skeletal development determines adult bone structure and mineralization. Mol Endocrinol. 2007;21(8):1893–904.
Kaneshige M, et al. Mice with a targeted mutation in the thyroid hormone beta receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci U S A. 2000;97(24):13209–14.
O’Shea PJ, et al. Advanced bone formation in mice with a dominant-negative mutation in the thyroid hormone receptor β gene due to activation of Wnt/β-catenin protein signaling. J Biol Chem. 2012;287(21):17812–22.
Lindsey RC, Mohan S. Thyroid hormone acting via TRβ induces expression of browning genes in mouse bone marrow adipose tissue. Endocrine. 2017;56(1):109–20.
Bassett JH, Williams GR. Role of thyroid hormones in skeletal development and bone maintenance. Endocr Rev. 2016;37(2):135–87.
Stevens DA, et al. Thyroid hormone activates fibroblast growth factor receptor-1 in bone. Mol Endocrinol. 2003;17(9):1751–66.
Mundy GR, et al. Direct stimulation of bone resorption by thyroid hormones. J Clin Invest. 1976;58(3):529–34.
Allain TJ, et al. Tri-iodothyronine stimulates rat osteoclastic bone resorption by an indirect effect. J Endocrinol. 1992;133(3):327–31.
Siddiqi A, et al. Serum cytokines in thyrotoxicosis. J Clin Endocrinol Metab. 1999;84(2):435–9.
Lambrinoudaki I, et al. Thyroid function and autoimmunity are associated with the risk of vertebral fractures in postmenopausal women. J Bone Miner Metab. 2017;35(2):227–33.
Mazziotti G, et al. Serum TSH values and risk of vertebral fractures in euthyroid post-menopausal women with low bone mineral density. Bone. 2010;46(3):747–51.
Leader A, et al. Thyrotropin levels within the lower normal range are associated with an increased risk of hip fractures in euthyroid women, but not men, over the age of 65 years. J Clin Endocrinol Metab. 2014;99(8):2665–73.
Abrahamsen B, et al. Low serum thyrotropin level and duration of suppression as a predictor of major osteoporotic fractures-the OPENTHYRO register cohort. J Bone Miner Res. 2014;29(9):2040–50.
Soto-Pedre E, et al. Evidence of a causal relationship between serum thyroid-stimulating hormone and osteoporotic bone fractures. Eur Thyroid J. 2021;10(6):439–46.
Deng T, et al. Thyroid-stimulating hormone decreases the risk of osteoporosis by regulating osteoblast proliferation and differentiation. BMC Endocr Disord. 2021;21(1):49.
Konca Degertekin C, et al. RANKL/Osteoprotegerin system and bone turnover in Hashimoto Thyroiditis. Calcif Tissue Int. 2016;99(4):365–72.
Grimnes G, et al. The relationship between serum TSH and bone mineral density in men and postmenopausal women: the Tromsø study. Thyroid. 2008;18(11):1147–55.
Sun L, et al. Intermittent recombinant TSH injections prevent ovariectomy-induced bone loss. Proc Natl Acad Sci U S A. 2008;105(11):4289–94.
Baliram R, et al. Hyperthyroid-associated osteoporosis is exacerbated by the loss of TSH signaling. J Clin Invest. 2012;122(10):3737–41.
Abe E, et al. TSH is a negative regulator of skeletal remodeling. Cell. 2003;115(2):151–62.
Hase H, et al. TNFalpha mediates the skeletal effects of thyroid-stimulating hormone. Proc Natl Acad Sci U S A. 2006;103(34):12849–54.
Baliram R, et al. Thyroid-stimulating hormone induces a Wnt-dependent, feed-forward loop for osteoblastogenesis in embryonic stem cell cultures. Proc Natl Acad Sci U S A. 2011;108(39):16277–82.
Baliram R, et al. Thyroid and bone: macrophage-derived TSH-β splice variant increases murine osteoblastogenesis. Endocrinology. 2013;154(12):4919–26.
Boutin A, et al. β-Arrestin-1 mediates thyrotropin-enhanced osteoblast differentiation. Faseb j. 2014;28(8):3446–55.
Iddah MA, Macharia BN. Autoimmune thyroid disorders. ISRN Endocrinol. 2013;2013:509764.
Dunne C, De Luca F. Long-term Follow-Up of a child with autoimmune thyroiditis and recurrent hyperthyroidism in the absence of TSH receptor antibodies. Case Rep Endocrinol. 2014;2014:749576.
Shahbaz A, et al. Prolonged duration of hashitoxicosis in a patient with Hashimoto’s Thyroiditis: a Case Report and Review of Literature. Cureus. 2018;10(6):e2804.
Hennessey JV, et al. The Association between switching from Synthroid(®) and clinical outcomes: US evidence from a retrospective database analysis. Adv Ther. 2021;38(1):337–49.
Eriksen EF, Mosekilde L, Melsen F. Kinetics of trabecular bone resorption and formation in hypothyroidism: evidence for a positive balance per remodeling cycle. Bone. 1986;7(2):101–8.
Vestergaard P, Mosekilde L. Fractures in patients with hyperthyroidism and hypothyroidism: a nationwide follow-up study in 16,249 patients. Thyroid. 2002;12(5):411–9.
Bassett JH, et al. A lack of thyroid hormones rather than excess thyrotropin causes abnormal skeletal development in hypothyroidism. Mol Endocrinol. 2008;22(2):501–12.
Obling ML, et al. Restoration of euthyroidism in women with Hashimoto’s thyroiditis changes bone microarchitecture but not estimated bone strength. Endocrine. 2021;71(2):397–406.
Tuchendler D, Bolanowski M. Assessment of bone metabolism in premenopausal females with hyperthyroidism and hypothyroidism. Endokrynol Pol. 2013;64(1):40–4.
Blum MR, et al. Subclinical thyroid dysfunction and fracture risk: a meta-analysis. JAMA. 2015;313(20):2055–65.
Liang LB, et al. Changes of bone mineral density and bone metabolic marker in patients with subclinical hypothyroidism. Sichuan Da Xue Xue Bao Yi Xue Ban. 2014;45(1):83. 66 – 9,, , ( : p.
Polovina S, et al. Frax score calculations in postmenopausal women with subclinical hypothyroidism. Horm (Athens). 2013;12(3):439–48.
Nagata M, et al. Subclinical hypothyroidism is related to lower heel QUS in postmenopausal women. Endocr J. 2007;54(4):625–30.
Mosekilde L, Eriksen EF, Charles P. Effects of thyroid hormones on bone and mineral metabolism. Endocrinol Metab Clin North Am. 1990;19(1):35–63.
Reverter JL, et al. Lack of deleterious effect on bone mineral density of long-term thyroxine suppressive therapy for differentiated thyroid carcinoma. Endocr Relat Cancer. 2005;12(4):973–81.
Appetecchia M. Effects on bone mineral density by treatment of benign nodular goiter with mildly suppressive doses of L-thyroxine in a cohort women study. Horm Res. 2005;64(6):293–8.
Mendonça M, de Barros G, et al. Bone mineral density and bone microarchitecture after long-term suppressive levothyroxine treatment of differentiated thyroid carcinoma in young adult patients. J Bone Miner Metab. 2016;34(4):417–21.
Yoon BH, et al. Influence of thyroid-stimulating hormone suppression therapy on bone Mineral density in patients with differentiated thyroid Cancer: a Meta-analysis. J Bone Metab. 2019;26(1):51–60.
Ku EJ, et al. Effect of TSH suppression therapy on bone Mineral density in differentiated thyroid Cancer: a systematic review and Meta-analysis. J Clin Endocrinol Metab. 2021;106(12):3655–67.
Davidson A, Diamond B. Autoimmune diseases. N Engl J Med. 2001;345(5):340–50.
Ganesh BB, et al. Role of cytokines in the pathogenesis and suppression of thyroid autoimmunity. J Interferon Cytokine Res. 2011;31(10):721–31.
Crane IJ, Forrester JV. Th1 and Th2 lymphocytes in autoimmune disease. Crit Rev Immunol. 2005;25(2):75–102.
Zake T, et al. Upregulated tissue expression of T helper (th) 17 pathogenic interleukin (IL)-23 and IL-1β in Hashimoto’s thyroiditis but not in Graves’ disease. Endocr J. 2019;66(5):423–30.
Huifang S. Study on the correlation between Hashimoto’s thyroiditis and bone mineral density in postmenopausal women. Qingdao University; 2021.
Polovina SP, et al. The impact of thyroid autoimmunity (TPOAb) on bone density and fracture risk in postmenopausal women. Horm (Athens). 2017;16(1):54–61.
Siris ES, Baim S, Nattiv A. Primary care use of FRAX: absolute fracture risk assessment in postmenopausal women and older men. Postgrad Med. 2010;122(1):82–90.
Koehler VF, Filmann N, Mann WA. Vitamin D status and thyroid autoantibodies in Autoimmune Thyroiditis. Horm Metab Res. 2019;51(12):792–7.
van de Peppel J, van Leeuwen JP. Vitamin D and gene networks in human osteoblasts. Front Physiol. 2014;5:137.
Takeda S, et al. Stimulation of osteoclast formation by 1,25-dihydroxyvitamin D requires its binding to vitamin D receptor (VDR) in osteoblastic cells: studies using VDR knockout mice. Endocrinology. 1999;140(2):1005–8.
Bikle D. Nonclassic actions of vitamin D. J Clin Endocrinol Metab. 2009;94(1):26–34.
Hossein-nezhad A, Spira A, Holick MF. Influence of vitamin D status and vitamin D3 supplementation on genome wide expression of white blood cells: a randomized double-blind clinical trial. PLoS ONE. 2013;8(3):e58725.
Lechner J, Aschoff J, Rudi T. The vitamin D receptor and the etiology of RANTES/CCL-expressive fatty-degenerative osteolysis of the jawbone: an interface between osteoimmunology and bone metabolism. Int J Gen Med. 2018;11:155–66.
Chahardoli R, et al., Can Supplementation with Vitamin D Modify Thyroid Autoantibodies (Anti-TPO Ab, Anti-Tg Ab) and Thyroid Profile (T3, T4, TSH) in Hashimoto’s Thyroiditis? A Double Blind, Randomized Clinical Trial Horm Metab Res, 2019. 51(5): p. 296–301.
Fang F, et al. Vitamin D deficiency is associated with thyroid autoimmunity: results from an epidemiological survey in Tianjin, China. Endocrine. 2021;73(2):447–54.
Khozam SA, et al. Association between vitamin D Deficiency and Autoimmune thyroid disorder: a systematic review. Cureus. 2022;14(6):e25869.
Jiang H, et al. Effects of vitamin D treatment on thyroid function and autoimmunity markers in patients with Hashimoto’s thyroiditis-A meta-analysis of randomized controlled trials. J Clin Pharm Ther. 2022;47(6):767–75.
Nodehi M, et al. Effects of vitamin D supplements on frequency of CD4(+) T-cell subsets in women with Hashimoto’s thyroiditis: a double-blind placebo-controlled study. Eur J Clin Nutr. 2019;73(9):1236–43.
Altieri B, et al. Does vitamin D play a role in autoimmune endocrine disorders? A proof of concept. Rev Endocr Metab Disord. 2017;18(3):335–46.
Sarmiento-Ramón MP, et al. Characterization of serum vitamin D levels in pediatric patients with chronic lymphocytic thyroiditis. Bol Med Hosp Infant Mex. 2022;79(3):161–9.
Kaan Demircioglu M, et al. Is vitamin D Deficiency Associated with chronic lymphocytic thyroiditis? Sisli Etfal Hastan Tip Bul. 2021;55(4):510–5.
Maciejewski A, et al. Vitamin D receptor gene polymorphisms and autoimmune thyroiditis: are they Associated with Disease occurrence and its features? Biomed Res Int. 2019;2019:8197580.
Behera KK, et al. Effect of vitamin D supplementation on thyroid autoimmunity among subjects of autoimmune thyroid disease in a Coastal Province of India: a Randomized Open-label trial. Niger Med J. 2020;61(5):237–40.
Azam, Amini, et al. The effect of vitamin D replacement on Musculoskeletal Pain in hypothyroid patients with vitamin D Deficiency. Int J Sci Eng Res. 2017;8(5):640–2.
Wang H, et al., Associations between dynamic vitamin D level and thyroid function during pregnancy. Nutrients, 2022. 14(18).
Chen Y, et al. Vitamin D categories and postpartum thyroid function in women with hypothyroidism. Front Nutr. 2022;9:953745.
Xu Z, et al. SMURF2 regulates bone homeostasis by disrupting SMAD3 interaction with vitamin D receptor in osteoblasts. Nat Commun. 2017;8:14570.
Hofbauer LC, et al. Detection and characterization of RANK ligand and osteoprotegerin in the thyroid gland. J Cell Biochem. 2002;86(4):642–50.
Nagasaki T, et al. Increased levels of serum osteoprotegerin in hypothyroid patients and its normalization with restoration of normal thyroid function. Eur J Endocrinol. 2005;152(3):347–53.
Guang-da X, et al. Changes in plasma concentrations of osteoprotegerin before and after levothyroxine replacement therapy in hypothyroid patients. J Clin Endocrinol Metab. 2005;90(10):5765–8.
Giusti M, et al. Serum osteoprotegerin and soluble receptor activator of nuclear factor kappaB ligand levels in patients with a history of differentiated thyroid carcinoma: a case-controlled cohort study. Metabolism. 2007;56(5):699–707.
Giusti M, et al. Recombinant human thyroid stimulating hormone does not acutely change serum osteoprotegerin and soluble receptor activator of nuclear factor-kappabeta ligand in patients under evaluation for differentiated thyroid carcinoma. Horm (Athens). 2007;6(4):304–13.
Ginaldi L, De Martinis M. Osteoimmunology and Beyond. Curr Med Chem. 2016;23(33):3754–74.
Brincat SD, et al. The role of cytokines in postmenopausal osteoporosis. Minerva Ginecol. 2014;66(4):391–407.
Chen B, Li HZ. Association of IL-6 174G/C (rs1800795) and 572 C/G (rs1800796) polymorphisms with risk of osteoporosis: a meta-analysis. BMC Musculoskelet Disord. 2020;21(1):330.
Poli V, et al. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. Embo j. 1994;13(5):1189–96.
Sims NA, et al. Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. J Clin Invest. 2004;113(3):379–89.
Liu XH, et al. Interactive effect of interleukin-6 and prostaglandin E2 on osteoclastogenesis via the OPG/RANKL/RANK system. Ann N Y Acad Sci. 2006;1068:225–33.
Honda K. Interleukin-6 and soluble interleukin-6 receptor suppress osteoclastic differentiation by inducing PGE(2) production in chondrocytes. J Oral Sci. 2011;53(1):87–96.
Lakatos P, et al. Serum interleukin-6 and bone metabolism in patients with thyroid function disorders. J Clin Endocrinol Metab. 1997;82(1):78–81.
Gerenova J, Manolova I, Stanilova S. SERUM LEVELS OF INTERLEUKIN – 23 AND INTERLEUKIN – 17 IN HASHIMOTO’S THYROIDITIS. Acta Endocrinol (Buchar). 2019;-5(1):74–9.
Li Q, et al. The pathogenesis of thyroid autoimmune diseases: new T lymphocytes - cytokines circuits beyond the Th1-Th2 paradigm. J Cell Physiol. 2019;234(3):2204–16.
Pan Y, et al. A chinese patent Medicine JiaYanKangTai alleviates inflammatory lesions of experimental autoimmune thyroiditis by regulating Interleukin-17 signaling. Front Endocrinol (Lausanne). 2021;12:794568.
Liu Y, et al. Elevated MicroRNA-326 levels regulate the IL-23/IL-23R/Th17 cell Axis in Hashimoto’s Thyroiditis by targeting a disintegrin and metalloprotease 17. Thyroid. 2020;30(9):1327–37.
Figueroa-Vega N, et al. Increased circulating pro-inflammatory cytokines and Th17 lymphocytes in Hashimoto’s thyroiditis. J Clin Endocrinol Metab. 2010;95(2):953–62.
Bhadricha H, et al. Increased frequency of Th17 cells and IL-17 levels are associated with low bone mineral density in postmenopausal women. Sci Rep. 2021;11(1):16155.
Ikeuchi T, Moutsopoulos NM. Osteoimmunology in periodontitis; a paradigm for Th17/IL-17 inflammatory bone loss. Bone, 2022: p. 116500.
Kim YG, et al. IL-17 inhibits osteoblast differentiation and bone regeneration in rat. Arch Oral Biol. 2014;59(9):897–905.
Zhang JR, et al. Different Modulatory Effects of IL-17, IL-22, and IL-23 on osteoblast differentiation. Mediators Inflamm. 2017;2017:5950395.
Kim HJ, et al. IL-17 promotes osteoblast differentiation, bone regeneration, and remodeling in mice. Biochem Biophys Res Commun. 2020;524(4):1044–50.
Uluçkan Ö, et al. Chronic skin inflammation leads to bone loss by IL-17-mediated inhibition of wnt signaling in osteoblasts. Sci Transl Med. 2016;8(330):330ra37.
Gaffen SL, et al. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14(9):585–600.
Ruggeri RM, et al. Serum interleukin-23 (IL-23) is increased in Hashimoto’s thyroiditis. Endocr J. 2014;61(4):359–63.
Shukla P, Mansoori MN, Singh D. Efficacy of anti-IL-23 monotherapy versus combination therapy with anti-IL-17 in estrogen deficiency induced bone loss conditions. Bone. 2018;110:84–95.
Adamopoulos IE, et al. IL-23 is critical for induction of arthritis, osteoclast formation, and maintenance of bone mass. J Immunol. 2011;187(2):951–9.
Ju JH, et al. IL-23 induces receptor activator of NF-kappaB ligand expression on CD4 + T cells and promotes osteoclastogenesis in an autoimmune arthritis model. J Immunol. 2008;181(2):1507–18.
Kang YK, Zhang MC. IL-23 promotes osteoclastogenesis in osteoblast-osteoclast co-culture system. Genet Mol Res. 2014;13(2):4673–9.
Razawy W, et al. IL-23 receptor deficiency results in lower bone mass via indirect regulation of bone formation. Sci Rep. 2021;11(1):10244.
Kamiya S, et al. Effects of IL-23 and IL-27 on osteoblasts and osteoclasts: inhibitory effects on osteoclast differentiation. J Bone Miner Metab. 2007;25(5):277–85.
Wang X, et al. Dysregulated interleukin – 33/ST2 pathway perpetuates chronic inflammation in Hashimoto’s Thyroiditis. Endocr Metab Immune Disord Drug Targets. 2019;19(7):1012–21.
Mun SH, et al. Interleukin-33 stimulates formation of functional osteoclasts from human CD14(+) monocytes. Cell Mol Life Sci. 2010;67(22):3883–92.
Malcolm J, et al. IL-33 exacerbates Periodontal Disease through induction of RANKL. J Dent Res. 2015;94(7):968–75.
Zaiss MM, et al. IL-33 shifts the balance from osteoclast to alternatively activated macrophage differentiation and protects from TNF-alpha-mediated bone loss. J Immunol. 2011;186(11):6097–105.
Kaneko N, et al. The role of interleukin-1 in general pathology. Inflamm Regen. 2019;39:12.
Mine M, et al. Interleukin-1 stimulates thyroid cell growth and increases the concentration of the c-myc proto-oncogene mRNA in thyroid follicular cells in culture. Endocrinology. 1987;120(3):1212–4.
Enomoto T, et al. Prolonged effects of recombinant human interleukin-1 alpha on mouse thyroid function. Endocrinology. 1990;127(5):2322–7.
Nilsson M, et al. Cytokines and thyroid epithelial integrity: interleukin-1alpha induces dissociation of the junctional complex and paracellular leakage in filter-cultured human thyrocytes. J Clin Endocrinol Metab. 1998;83(3):945–52.
Yamashita S, et al. Interleukin-1 inhibits thyrotrophin-induced human thyroglobulin gene expression. J Endocrinol. 1989;122(1):177–83.
Ashizawa K, et al. Inhibition of human thyroid peroxidase gene expression by interleukin 1. Acta Endocrinol (Copenh). 1989;121(4):465–9.
Zhao R, Zhou H, Su SB. A critical role for interleukin-1β in the progression of autoimmune diseases. Int Immunopharmacol. 2013;17(3):658–69.
Sun L, et al. Elevated interleukin-1β in peripheral blood mononuclear cells contributes to the pathogenesis of autoimmune thyroid diseases, especially of Hashimoto thyroiditis. Endocr Res. 2016;41(3):185–92.
Levescot A, et al., IL-1β-driven osteoclastogenic Tregs accelerate bone erosion in arthritis. J Clin Invest, 2021. 131(18).
Hengartner NE, et al. IL-1β inhibits human osteoblast migration. Mol Med. 2013;19(1):36–42.
Ebe Y, et al. Effect of interleukin-1β on bone morphogenetic protein-9-induced osteoblastic differentiation of human periodontal ligament fibroblasts. Eur J Oral Sci. 2021;129(4):e12792.
Huang J, Chen L. IL-1β inhibits osteogenesis of human bone marrow-derived mesenchymal stem cells by activating FoxD3/microRNA-496 to repress wnt signaling. Genesis, 2017. 55(7).
Ye W, et al. IL-1β-Treated bone marrow mesenchymal stem cells enhances osteogenetic potential via NF-κB pathway. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2017;25(3):890–5.
Hong SH, Braley-Mullen H. Follicular B cells in thyroids of mice with spontaneous autoimmune thyroiditis contribute to disease pathogenesis and are targets of anti-CD20 antibody therapy. J Immunol. 2014;192(3):897–905.
Mariotti S, et al. Recent advances in the understanding of humoral and cellular mechanisms implicated in thyroid autoimmune disorders. Clin Immunol Immunopathol. 1989;50(1 Pt 2):S73–84.
Yongshuang Xie WQ. Researching on the relation between autoimmune thyroid disesae and tumor necrosis factors-alpha/interleukin-6 ratio. J Practical Med Techniques. 2009;16:425–6.
Braley-Mullen H, Yu S. NOD.H-2h4 mice: an important and underutilized animal model of autoimmune thyroiditis and Sjogren’s syndrome. Adv Immunol. 2015;126:1–43.
Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118(11):3537–45.
Boehm U, et al. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–95.
Yu S, Sharp GC, Braley-Mullen H. Dual roles for IFN-gamma, but not for IL-4, in spontaneous autoimmune thyroiditis in NOD.H-2h4 mice. J Immunol. 2002;169(7):3999–4007.
Yu S, Sharp GC, Braley-Mullen H. Thyrocytes responding to IFN-gamma are essential for development of lymphocytic spontaneous autoimmune thyroiditis and inhibition of thyrocyte hyperplasia. J Immunol. 2006;176(2):1259–65.
Zhang X, Zhu L, Sun L, IFN-y mediates thyroid injury by upregulating Fas expression in Hashimoto thyroiditis Acta Universitatis Medicinalis Anhui, 2017. 52(06): p. 806–809.
Gao Y, et al. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. 2007;117(1):122–32.
Takayanagi H, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408(6812):600–5.
Maruhashi T, et al. DCIR maintains bone homeostasis by regulating IFN-γ production in T cells. J Immunol. 2015;194(12):5681–91.
Hu Y, et al. Effect of selenium on thyroid autoimmunity and regulatory T cells in patients with Hashimoto’s thyroiditis: a prospective randomized-controlled trial. Clin Transl Sci. 2021;14(4):1390–402.
Kryczyk-Kozioł J, et al., Assessment of the Effect of Selenium supplementation on production of selected cytokines in women with Hashimoto’s Thyroiditis. Nutrients, 2022. 14(14).
Poleboina S, et al. Selenium nanoparticles stimulate osteoblast differentiation via BMP-2/MAPKs/β-catenin pathway in diabetic osteoporosis. Nanomed (Lond). 2022;17(9):607–25.
Wei M, et al. Manganese, iron, copper, and selenium co-exposure and osteoporosis risk in chinese adults. J Trace Elem Med Biol. 2022;72:126989.
Beukhof CM, et al. Selenium Status is positively Associated with Bone Mineral density in healthy aging european men. PLoS ONE. 2016;11(4):e0152748.
Hoeg A, et al. Bone turnover and bone mineral density are independently related to selenium status in healthy euthyroid postmenopausal women. J Clin Endocrinol Metab. 2012;97(11):4061–70.
Wu CC, et al. Selenium status is independently related to bone mineral density, FRAX score, and bone fracture history: NHANES, 2013 to 2014. Bone. 2021;143:115631.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Statements and declarations
Funding.
This work was supported by grants from the National Natural Science Foundation of China [No. 82273294]; the Science and Technology Department of Sichuan Province (2022YFS0136); the Chengdu Bureau of Science and Technology (2022-YF05-01316-SN); the Sichuan University [No. 2018SCUH0093]; and the 1.3.5 project for discipline of excellence, West China Hospital, Sichuan University [No. 2020HXFH008, No. ZYJC18003].
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, J., Huang, H. & Yu, X. How does Hashimoto’s thyroiditis affect bone metabolism?. Rev Endocr Metab Disord 24, 191–205 (2023). https://doi.org/10.1007/s11154-022-09778-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-022-09778-x