
Abstract
Key Message
Present study revealed a complex relationship among histone H3 methylation (examined using H3K4/K27me3 marks), cytosine DNA methylation and differential gene expression during Lr28 mediated leaf rust resistance in wheat.
Abstract
During the present study, genome-wide histone modifications were examined in a pair of near isogenic lines (NILs) (with and without Lr28 in the background of cv. HD2329). The two histone marks used included H3K4me3 (an activation mark) and H3K27me3 (a repression mark). The results were compared with levels of expression (using RNA-seq) and DNA methylation (MeDIP) data obtained using the same pair of NILs. Some of the salient features of the present study include the following: (i) large scale differential binding sites (DBS) were available for only H3K4me3 in the susceptible cultivar, but for both H3K4me3 and H3K27me3 in its resistant NIL; (ii) DBSs for H3K27me3 mark were more abundant (> 80%) in intergenic regions, whereas DBSs for H3K4me3 were distributed in all genomic regions including exons, introns, intergenic, TTS (transcription termination sites) and promoters; (iii) fourteen (14) genes associated with DBSs showed co-localization for both the marks; (iv) only a small fraction (7% for H3K4me3 and 12% for H3K27me3) of genes associated with DBSs matched with the levels of gene expression inferred from RNA-seq data; (v) validation studies using qRT-PCR were conducted on 26 selected representative genes; results for only 11 genes could be validated. The proteins encoded by important genes involved in promoting infection included domains generally carried by R gene proteins such as Mlo like protein, protein kinases and purple acid phosphatase. Similarly, proteins encoded by genes involved in resistance included those carrying domains for lectin kinase, R gene, aspartyl protease, etc. Overall, the results suggest a very complex network of downstream genes that are expressed during compatible and incompatible interactions; some of the genes identified during the present study may be used in future validation studies involving RNAi/overexpression approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Molecular mechanism underlying disease resistance in plants has been an active area of research. Although, the mechanism for resistance against biotrophic and necrotrophic pathogens may differ in detail, the basic outline of the mechanism is the same. It is now widely accepted that the R gene-mediated race-specific resistance is managed through two layers of immunity, namely pathogen associated molecular patterns (PAMP) triggered immunity (PTI) and effector triggered immunity (ETI). The gene R provides resistance, only when there is a corresponding avirulence (Avr) gene in the prevailing race of the pathogen. This has been described as gene-for-gene relationship (Flor 1942, 1971). In both PTI and ETI, the plant cells generally respond to the pathogen attack through nucleotide binding site-leucine rich repeats (NBS-LRR) (also called nod-like receptors = NLR), followed by downstream signaling pathways. These pathways form a network that is not fully understood, although some pathways including salicylic acid (SA), jasmonic acid (JA) and ethylene (Et) pathways are known to operate. More recently, formation of a resistosome with R gene encoded NBS-LRR protein has also been suggested (Wang et al. 2019a, b, c). Another recent study also demonstrated that on pathogen attack, the cell death is really caused due to NADase activity of the Toll/Interleukin receptor 1 (TIR) domain at the N-terminus of NBS-LRR receptor (Horsefield et al. 2019; Wan et al. 2019); NADase causes cleavage of the oxidized form of nicotinamide adenine dinucleotide (NAD+) leading to cell death and hypersensitive reaction (HR), thus providing protection against biotrophs.
In wheat, leaf rust is an important disease and causes major losses in yield (Draz et al. 2015). At least ~ 80 Lr genes for leaf rust resistance in wheat are already known (see Gill et al. 2019) and more genes are being regularly discovered. These genes include both the race-specific seedling resistance (Sr; also described as all stage resistance) genes and also the race-non-specific adult plant resistance (APR) genes. An important leaf rust resistance gene, which has been deployed for development of a number of resistant cultivars in India is Lr28. In order to understand the molecular mechanism underlying leaf rust resistance due to Lr28, a pair of NILs in the background of wheat cultivar HD2329 was developed at ICAR-Indian Agricultural Research Institute (ICAR-IARI), India. This pair of NILs has already been utilized in our laboratory for studies involving transcriptome, DNA methylation, histone modification and non-coding RNAs (miRNAs and lncRNAs). Using transcriptomics approach, we discovered a large number of downstream genes, which are either induced or repressed in the susceptible and resistant NILs following the attack by the pathogen (Dhariwal et al. 2011; Chandra et al. 2016; Singh et al. 2017; Sharma et al. 2018a).
During our earlier transcriptome studies involving the above pair of NILs, some evidence of epigenetic regulation of Lr28-mediated leaf rust resistance was available. This evidence included Lr28-mediated differential expression of genes encoding methyltransferases, non-coding RNAs and histone acetylases/methyltransferases. This prompted us to undertake epigenetic studies using the same experimental material. For instance, the role of histone acetylation was examined by us in controlling the expression of six genes involved in interaction between wheat and leaf rust pathogen (Sharma et al. 2018b). Studies are also available in rice, Arabidopsis and cowpea where the role of histone methylation was examined during abiotic/biotic stresses (Kim et al. 2008; Zong et al. 2013; Ayyappan et al. 2015). However, the role of histone methylation in regulation of leaf rust resistance in wheat has never been examined.
Histone acetylation is more generally correlated with open chromatin and therefore more active transcription (reviewed by Zentner and Henikoff 2013). By contrast, histone methylation can result in different transcriptional outcomes, depending on the specific individual amino acids that are modified and the degree of modification (reviewed by Li et al. 2007). Among histone methylation marks, two histone trimethylation marks (H3K4me3 and H3K27me3) have been widely used in understanding the epigenetic regulation of development, vernalization and abiotic stress tolerance, nitrogen use efficiency (NUE) in Arabidopsis, rice, wheat, Brachypodium distachyon, etc. (Kim et al. 2008; Diallo et al. 2012; Zong et al. 2013; Zhang et al. 2009; You et al. 2017; Huan et al. 2018; Wu et al. 2020). These two histone trimethylation marks have also been used in animal systems including mouse (Yang and Wilson 2018). H3K4me3 is widely known to be associated with promoter sequences and gene activation, while H3K27me3 is known to be associated with silenced genes or non-genic regions causing heterochromatinization and repression of gene expression through trans-regulation (Heintzman et al. 2007 and Zhou and Hu 2010). However, there are only few reports where the role of these two histone marks in the regulation of specific genes during biotic stress (disease resistance) has been examined. For example, the removal of repression mark H3K27me3 through Jumonji C domain protein JMJ705, which encodes H3K27me2/3 demethylase resulted into activation of defense related genes during infection by the bacterial pathogen Xanthomonas oryzae in rice (Li et al. 2013). However, no studies involving genome-wide histone profiling in relation to biotic stress have been conducted in cereals although a solitary genome-wide study for H3K9me2 and H4K12ac is available for rust (Uromyces appendiculatus) infected Phaseolus vulgaris (Ayyappan et al. 2015).
The present study was undertaken to examine the genome-wide occurrence of H3K4/K27 trimethylations and their effect on expression of downstream genes following inoculation by the most virulent race 77-5. A pair of NILs involving susceptible wheat cv. HD2329 and its resistant NIL HD2329 + Lr28 was used to study the effect of the presence/absence of the gene Lr28 on the occurrence of H3K4me3/K27me3 and resulting regulation of genome-wide expression of genes. A large number of genes were found to be differentially expressed due to binding of these two H3 trimethylation marks.
Materials and methods
Plant material and sample preparation
The details of the plant material used in the present study are available in an earlier report (Sharma et al. 2018a). In brief, the seedlings of susceptible cv. HD2329 and its resistant NIL (HD2329 + Lr28) were raised in a growth-chamber under controlled conditions of 16 h light (240 mmol m−2 s−1) with 25 °C and 8 h dark with 18 °C at the National Phytotron Facility, ICAR-IARI, New Delhi. The seedlings (14 days old) were inoculated by the most prevalent and virulent pathotype 77-5 (syn. 121R63-1) of Puccinia triticina Erikss. & E. Henn. (for details, see Sharma et al. 2018a), and leaf samples were collected as detailed below.
Eight leaf samples from seedlings included two biological replicates each for the following four treatments: (i) S0 (susceptible cv. HD2329 at 0 hbi), (ii) S96 (susceptible cv. HD2329 at 96 hai), (iii) R0 (resistant NIL HD2329 + Lr28 at 0 hbi) and (iv) R96 (resistant NIL HD2329 + Lr28 at 96 hai). Leaf samples were collected and fixed in formaldehyde buffer (1% formaldehyde, 0.4 M sucrose, 10 mM Tris–HCl, 1 mM EDTA, 1 mM PMSF) followed by vacuum infiltration.
Chromatin immunoprecipitation (ChIP) and preparation of ChIP-seq libraries
Antibodies specific for H3K4Me3 and H3K27Me3 marks were procured from Abcam, USA. The workflow for ChIP and preparation of ChIP-seq libraries included the following steps: (i) chromatin was isolated (using Digenode Plant ChIP-seq kit) from all the eight samples (as above); (ii) isolated chromatin was sheared and immuno-precipitated using each of the above two antibodies, thus making 16 samples; input DNA (total genomic DNA used as control) was also isolated from the above four samples (without biological replicates) following precipitation of chromatin using Digenode Plant ChIP-seq Kit; in this manner 20 samples were available. (iii) DNA was isolated from all the 20 ChIPed samples and was used for constructing ChIP-Seq libraries using Microplex Library Prep Kit V2; (iv) Quantity and quality check (QC) of each amplified library was done by Bioanalyzer2100 (Agilent Technology) using high sensitivity (HS) DNA chip based on Qubit concentration and mean peak size; (v) sequencing of 20 DNA libraries was outsourced to Xcelris Genomics Pvt. Ltd, Ahmedabad (India), who used Illumina platform for cluster generation and sequencing; paired-end (PE) sequencing (2 × 75 bp) was undertaken, which allowed template fragments to be sequenced in both forward and reverse directions. Sequencing data was generated with ~ 11–90 million reads/sample. Data in FASTQ files for all the 20 libraries was subjected to detailed bioinformatics analysis (for details of 20 libraries, see Table S1).
ChIP-seq data analysis
The filtered reads from each library were mapped to wheat reference genome-Ref-Seq v1.0 (IWGSC 2018; https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Assemblies/v1.0/) using Bowtie2.0 (Langmead and Salzberg 2012). Pearson’s correlation coefficients were calculated for all replicates/treatments to check reproducibility in the aligned reads between the replicates by deepTools (Ramírez et al. 2014). Scores for fraction of reads in peak (FRiP) were also calculated for each replicate for each treatment; > 1% FRiP was used as a cutoff. Peaks marked with H3K4me3 and H3K27me3 were identified using MACS2.0 (Model-based Analysis of ChIP-Seq by Zhang et al. 2008). The narrow/sharp peaks represented H3K4me3 and broad peaks represented H3K27me3 (Rintinch et al. 2014; Nakato and Shirahige 2017; Laczik et al. 2016). The peaks were localized into exon, intron, promoter (− 1 kb to + 100 bp), TTS—Transcription Termination Sites (− 100 bp to + 1 kb) and intergenic regions using HOMER (Hypergeometric Optimization of Motif EnRichment; Heinz et al. 2010). Differential binding sites (DBSs) for the two histone marks were extracted by using DiffBind package of Bioconductor (Stark and Brown 2011) at P value < 0.01. Perl-script was used to find known high confidence genes using IWGSC RefSeq v1.0 data; some genes were found overlapping with DBSs for both histone marks. In order to study their role in wheat-leaf rust interaction, the high confidence genes were categorized into the following 10 categories based on their functions in biotic stress response: (i) resistance, (ii) transcription factor (TF), (iii) oxidative stress response, (iv) transport, (v) photosynthesis, (vi) epigenetic regulation, (vii) metabolism, (viii) defense, (ix) plant hormone response, and (x) miscellaneous class.
The ChIP-Seq raw reads of susceptible genotype (HD2329) and resistant NIL (HD2329 + Lr28) at 0 hbi and 96 hai were deposited in Sequence Read Archive (SRA), National Center for Biotechnology Information (NCBI) and can be accessed through Bioproject Database under the accession number PRJNA588134.
Gene ontology analysis and identification of transposable elements (TEs)
Blast2GO (Conesa and Götz 2008) was used for functional annotation of genes associated with DBSs. Based on annotation, the genes were placed into the three well-known classes: cellular component, molecular function and biological process. The DBSs were also searched for TEs utilizing IWGSC RefSeq using Perl script.
Co-localized DBSs for H3K4me3 and H3K27me3
A DBS was considered co-localized for both the marks only if peak coordinates for both H3K4me3 and H3K27me3 had an overlap of at least one base. Genes were considered as co-localized, only if both marks occur within the gene, even if the marks do not overlap.
RNA-seq analysis and relationship of histone methylation with gene expression
In order to examine the relationship of histone methylation with gene expression, RNA-seq analysis was also conducted for the same four treatments (S0, S96, R0 and R96). For this purpose, RNA samples from two biological replicates of each treatment were pooled and the sequencing was outsourced to Xcelris Genomic Pvt. Ltd (pooling was done to reduce sequencing costs). The libraries were prepared with ~ 1 μg input total RNA using Illumina TruSeq Standard Total RNA Library Preparation Kit as per the manufacturer’s protocol. Briefly, the protocol involved the following steps (i) total RNA was Ribo depleted using plant rRNA removal mix and rRNA removal beads; (ii) RNA was subjected to purification, fragmentation and priming for cDNA synthesis; (iii) Ribo-depleted and fragmented RNA was converted into first-strand cDNA, followed by second-strand cDNA synthesis, A-tailing, adapter-index ligation; (iv) ligated RNA was amplified using recommended number of PCR cycles; (v) library quality and quantity was checked using Agilent DNA High Sensitivity Assay Kit. The pipeline used for bioinformatics analysis and identification of differentially expressed genes is presented in Fig. S1. For identification of differentially expressed genes (DEGs), log FC > 0 criterion was followed. The RNA-seq raw reads of susceptible genotype (HD2329) and resistant NIL (HD2329 + Lr28) at 0 hbi and 96 hai were deposited in Sequence Read Archive (SRA), National Center for Biotechnology Information (NCBI) and can be accessed through Bioproject Database under the accession number PRJNA588134.
In order to examine the relationship of histone methylation and gene expression, the genes associated with DBSs were compared with differentially expressed genes (DEGs) identified from RNA-seq data (as above). A DEG was considered overlapped with DBS only if it overlaps with peak coordinates at least by one base. This allowed us to identify genes, which were associated with modified histone marks and exhibited differential expression, assuming activation due to H3K4me3 and repression due to H3K27me3.
Relationship of histone methylation with DNA methylation
For examining the relationship of histone methylation with DNA methylation, differentially methylated genes (DMGs) identified using mathylated DNA immuno precipitation (MeDIP) analysis from our earlier study (Saripalli et al. 2020) were compared with the differentially histone modified (DH3M) genes. The MeDIP raw reads of susceptible (HD2329) and resistant (HD2329 + Lr28) NILs at 0 hbi and 96 hai were deposited in Sequence Read Archive (SRA), NCBI and can be accessed through Bioproject Database under the accession number PRJNA517568.
Chromatin states and genome annotation analysis
In order to study the chromatin states using epigenomic information (DNA methylation, H3K4me3, H3K27me3), ChromHMM (Ernst and Kellis 2012) was utilized. This exercise involved the following steps; (i) file for chromosome size was prepared and COORDS and ANCHORFILES for wheat were prepared utilizing IWGSC v1.1 annotation file; Perl scripts were used for this purpose; (ii) genome wise Cellmarkfiletable was prepared utilizing controls for histone marks; (iii) binarization was performed on BAM files by BinarizeBam separately for DNA methylation and histone modifications; (iv) binary files for DNA methylation and histone modifications were merged chromosome-wise; (v) model was learned using LearnModel for 15 chromatin states, which are biologically interpretable.
Validation by quantitative RT-PCR (qRT-PCR)
Identification of genes for qRT-PCR
Out of the genes associated with DBSs, 26 genes showing FDR < 0.05 or P < 0.01 with a FC > 2.5 or < − 2.5 were selected for validation using qRT-PCR. These 26 genes included 17 genes for H3K4me3 and 9 genes for H3K27me3. Primers for the above genes were designed using Primer Express (ver. 3.0) software (Applied Biosystems, USA). Details of primers are available in Table S2.
RNA isolation, cDNA synthesis and qRT-PCR
For qRT-PCR, total RNA was isolated using TRIzol reagent (Ambion, USA) following manufacturer’s guidelines and treated with RNAse-free DNAse I (Invitrogen, USA) for 15 min to degrade any residual genomic DNA. First-strand cDNA was synthesized from this RNA using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, USA) according to manufacturer’s instructions. qRT-PCR was performed in 96 well plates with Step OnePlus Real Time PCR system using SYBR Green (both from Applied Biosystem, USA). All reactions were performed using three biological and three technical replicates. Target gene expression (fold-change between two treatments) was calculated using 2−∆∆CT method using actin gene for normalization (Livak and Schmittgen 2001). Heat maps were generated using online tool clustvis, and were used to depict the fold changes in differential binding/expression of genes following qRT-PCR/ChIP-Seq.
Results
ChIP sequencing and alignment with wheat genome
ChIP sequencing data from 20 libraries (including 16 IpDNA and 4 input DNA) had ~ 950 million paired end reads with an average of ~ 48 million reads/sample. An average of 89.88% reads from each library were mapped with wheat reference genome. The correlations between aligned reads of two replicates ranged from 0.54 to 0.85 (Table S1); the correlation between different treatments was also high (> 0.5); this was not unexpected, since the susceptible (S) and resistant (R) lines differed only for Lr28 gene and had almost similar genetic background (Fig. S2).
Differential binding sites (DBSs) with modified histone marks
Chromosomal distribution of DBSs
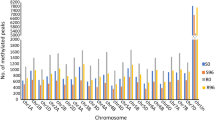
Differential binding sites (DBSs) for the two histone marks were distributed on all the 21 chromosomes (Fig. 1). Some DBSs (1061) could not be assigned to specific individual chromosomes and were labelled as ‘chromosome Un’. For H3K4me3, the number of DBSs ranged from 251 on the unidentified chromosome (Un) to 455 on chromosome 2A; for H3K27me3, this number ranged from 14 on chromosome 4D to 559 on the unidentified chromosome (Un). The number of DBSs for H3K4me3 in individual treatment pairs ranged from 296 (S96 vs R96) to 4428 (S0 vs. S96); for H3K27me3, it ranged from 295 (S96 vs. R96) to 733 (S0 vs. R0).

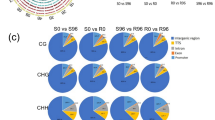
Distribution of DBSs in different treatment pairs on all the 21 chromosomes (1A to 7D) and the DBSs unassigned to any of the 21 chromosomes are indicated as Un. The DBSs indicated in each of the two histone marks, starting from outermost circle, (i) first circle indicates the treatment pair S0 vs S96, (ii) the second circle: S0 vs R0; (iii) the third circle S96 vs R96 and (iv) fourth circle R0 vs R96. Bars facing outwards indicate increased affinity and bars facing inwards indicate decreased affinity to H3K4me3 in outer four circles in red, and to H3K27me3 marks in inner four circles in blue. S0: HD2329 at 0 hbi; S96: HD2329 at 96 hai; R0: HD2329 + Lr28; R96: HD2329 + Lr28 at 96 hai
DBSs in S (HD2329) and R (HD2329 + Lr28) lines
For H3K4me3, DBSs showing increased affinity at 96 hai (relative to 0 hbi) in each of the two NILs (S0 vs. S96 and R0 vs. R96) were abundant, relative to those between two NILs (S0 vs. R0 and S96 vs. R96). In the former two comparisons involving the same NILs at two time points, when comparisons were made between S and R lines, binding sites decreased at 96 hai in R NIL relative to S line with the passage of time (R0 vs. R96 relative to S0 vs. S96). For the H3K27me3, in S96 and R0, the binding sites showing decreased affinity were more abundant relative to S0, whereas in R96 the binding sites with increased affinity were more abundant relative to S96 and R0 (Figs. 1 and 2).

Binding sites showing increased and decreased affinity of histone marks: a H3K4me3 and b H3K27me3 in each of the four treatment comparison; in each comparison, second treatment is compared to first
Distribution of DBSs in genomic regions
Relative frequencies of DBSs in five genomic regions (promoter, exon, intron, TTS and intergenic regions) for H3K4/K27me3 are depicted in Fig. 3. The DBSs of H3K27me3 were more frequently found in the intergenic region (84–88%) than in genic and promoter regions in comparison to H3K4me3 (18–23%). Binding of the H3K4me3 mark occurred with higher frequency in the exons relative to introns. In contrast, the binding of H3K27me3 in exons and the introns did not differ. Details of DBSs in exons, intron, promoter and TTS for both the marks are available in Tables S3–S10; whereas details of DBSs in intergenic regions for both the marks are available in Tables S11–S18. DBSs in the intergenic regions (associated with H3K27me3) were found to be involved in trans-regulation of genes that were located at > 50 kb away (upstream as well as downstream).

Proportions of different genomic regions, occupied by DBSs for two histone marks: a H3K4me3 and b H3K27me3 in each of the four treatment comparisons
Differentially modified genes due to H3 histone marks (DH3M genes)
Differential binding sites (DBSs) other than those falling in intergenic regions were used for identification of differentially H3 modified (DH3M) genes; IWGSC reference sequence was used for this purpose. Many more DH3M genes (3869) were associated with H3K4me3 relative to those associated with H3K27me3 (186). Majority of DH3M genes in individual treatment pairs were unique; only a few genes occurred in more than one treatment pairs (Fig. 4; prepared based on protein domain information). For instance, if we consider the two important comparisons, namely S0 vs S96 and R0 vs R96, there were 2259 unique DH3M genes associated with H3K4me3 and 53 unique DH3M genes associated with H3K27me3 in S96 (relative to S0) whereas in R96 (relative to R0), 800 unique DH3M genes associated with H3K4me3 and 9 unique DH3M genes associated with H3K27me3. However, some of these unique DH3M genes shared common domains indicating their similar functions. Based on their domains, the DH3M genes were classified into 10 categories (Fig. 5). Some important categories of DH3M genes that may be used for further study of wheat-leaf rust interaction are listed in Table S3. Maximum DH3M genes associated with H3K4me3 belonged to the miscellaneous category in all the four treatment pairs; minimum number of DH3M genes belonged to growth hormone response category. However, in case of H3K27me3, many more genes belonged to defense response and metabolism categories in all the four treatment pairs.

Venn diagrams showing number of DH3M genes marked with a H3K4me3 and b H3K27me3. These venn diagrams have been prepared using the protein domains encoded by the genes. Therefore, the genes with the common domains in the same treatment pair have been omitted. The detailed list of gene IDs has been presented in Tables S3–S10

Ten and nine different categories of genes identified in DBSs associated to a H3K4me3 and b H3K7me3 in four different treatment pairs, respectively
Gene ontology analysis of DH3M genes
GO terms of DH3M genes for H3K4/K27me3 are shown in Fig. 6a and b. The DH3M genes can be broadly classified as follows: (i) DH3M genes for H3K4me3: in this category, the genes involved in binding activity were most abundant followed by catalytic activity, metabolic process and cellular process when compared to others. (ii) DH3M genes for H3K27me3: in this category, genes involved in binding activity, catalytic activity, metabolic processes, cellular processes and those belonging to different cellular components were predominant (for details, see Tables S19–S26).

GO analysis of DH3M genes associated with a H3K4me3 and b H3K27me3. The GO categories are distributed in three categories, namely, (i) cellular component (ii) molecular function and (iii) biological process
Transposable elements (TE) in DBSs
TEs in DBSs were identified and the results are summarized in Table 1; as can be seen, TEs in each case could be placed in two categories, TEs with nested repeats (NR) and those with simple repeats (SR). It can also be seen that for H3K4me3, maximum number of TEs (775) were available in DBSs from the treatment S0 vs. S96 and minimum TEs (65) were available in DBSs of S96 vs. R96. The corresponding figures for H3K27m3 were 158 in S0 vs. R0 and 90 in S96 vs R96.
Chromatin states associated with H3K4/K27me3 marks and DNA methylation
The results of analysis of chromatin states involving three epigenetic marks (DNA methylation, H3K4me3, H3K27me3) and four treatments (S0, S96, R0, R96) are depicted in Fig. 7. As many as 15 chromatin states were identified, which provided sufficient resolution to understand biologically meaningful patterns, across four different treatments. We used this model to produce five genome-wise chromatin state annotations, which are summarized in Table 2 and depicted as heat maps in Fig. 7. From this figure, following notable features of the analysis involving chromatin states were observed: (i) chromatin state 2, which represents repressed enhancers located 2 kb upstream of the TSS carrying H3K27me3, is predominant in the treatment R96; (ii) chromatin state 8, which represents transcribed genic region enriched with DNA methylation in TTS seems to be common in all the four treatments (S0, S96, R0 and R96), although the probability seems to be relatively low in R0; (iii) chromatin state 10, which represents repressed genic region with all the three epigenetic marks is common in all the four treatments; (iv) chromatin state 12 involving strong promoter enriched with H3K4me3 in TSS, seems to be common in S0, S96 and R0, but its probability is rather high in S0; (v) chromatin state 13 having the same attributes as state 12 is particularly common in R96; (vi) chromatin state 14 is particularly common in S0 suggesting the presence of weak promoters in many genes marked with H3K4me3; (vii) chromatin state 15 involving repetitive region enriched for DNA methylation and H3K4me3 seems to have high probability in R0.

a A heatmap of emission probabilities, where each row represents a different chromatin state, and each column represents a different epigenetic mark in individual treatments; the darker regions correspond to a greater probability of observing the mark in the state; b heat map of emission probablities displaying enrichment for six different external genomic annotations based on RefSeq and 15 chromatin states; c a heat map of transition parameters, where each row and column represents a chromatin state
Differentially expressed genes identified using RNA-seq
RNA-seq analysis produced > 105 million reads for all the four treatments with maximum number of reads obtained for S96 (2,989,116). The mapping efficiency of these reads ranged from 89.19% (R96) to 93.49% (S0). The DEGs in the treatment pairs S0 vs R0 and R0 vs R96 were nearly similar but were relatively higher when compared with the remaining two treatment pairs, namely S0 vs S96 and S96 vs R96 (Table 3). A large number of genes were also expressed in only one of the two treatments of a pair in all the four treatment comparisons. The DEGs largely belonged to the following different classes of genes: (i) genes encoding receptor like kinases, TFs, and important enzymes involved in metabolic pathways, (ii) growth hormone responsive genes, defense response R genes, etc.
Relationship between histone trimethylation and gene expression
As many as 346 DH3M genes associated with H3K4me3 marks and 34 genes associated with H3K27me3 marks were also found to be differentially expressed during RNA-seq analysis. Summary of a comparison of number of DH3M genes during ChIP-seq analysis which were also found to be differentially expressed using RNA-seq is given in Table 4 (for details see Tables S27 and S28). It can be seen that among DH3M genes for H3K4me3, 232 DH3M genes with increased affinity showed high expression and 9 DH3M genes with reduced affinity showed lower expression. Similarly, for H3K27me3, two DH3M genes with increased affinity showed lower expression and 14 DH3M genes with reduced affinity showed higher expression. An opposite relationship was observed for the remaining 123 DH3M genes (105 associated with H3K4me3 and 18 associated with H3K27me3). Overall, only ~ 0.6% of the DEGs (identified using RNA-Seq) were found to be influenced due to histone modifications (both H3K4/K27me3).
Co-localized or bivalent DBSs for H3K4me3 and H3K27me3
As many as 14 DH3M genes associated with 125 DBSs exhibited association with both, H3K4me3 and H3K27me3; these were described as colocalized/bivalent DBSs (Table 5); the distribution of these 14 genes in treatment pairs was as follows: (i) 4 genes (having decreased affinity with both the marks) belonged to S0 vs R0; (ii) 6 genes (showing opposite affinities with the two marks) belonged to S0 vs S96; (iii) 3 genes (showing increased affinity to both the marks) belonged to R0 vs R96 and (iv) 2 genes (showing decreased affinity to both the marks) belonged to S96 vs R96.
Validation of identified genes by qRT-PCR
As mentioned earlier, a representative sample of 26 genes was used for qRT-PCR. Among 17 genes marked with H3K4me3 and used for qRT-PCR, expression of only 6 genes could be validated (Fig. 8a). Similarly, among 9 genes marked with H3K27me3 and used for qRT-PCR, expression of only 5 genes (Fig. 8b) could be validated. For H3K4me3, among six validated genes, two genes with increased affinity showed enhanced expression, while remaining four genes with reduced affinity showed reduced expression. Similarly, for the H3K27me3 three genes with increased affinity showed reduced expression while remaining two genes with reduced affinity showed enhanced expression. The functions of these genes validated using qRT-PCR are summarized in Table 6.

Heatmap showing relationship of histone modifications due to a H3K4me3 mark and b H3K27me3 (using ChIP-Seq) with gene expression (using qRT-PCR). Genes highlighted in yellow were validated using qRT-PCR analysis
Discussion
Molecular mechanism for disease resistance in wheat-leaf rust pathosystem is not fully understood; this is particularly true of the signal transduction pathways that follow the initial interaction between the protein encoded by an individual Lr gene and the corresponding effector produced by the pathogen. It is, however, known that an attack by the leaf rust fungus (P. triticina) is perceived by the host cells through cell-surface and intracellular receptors; the cell surface receptors have recently been shown to form a resistosome, which provides defense through cell death mediated hypersensitive reaction, the so called HR (Wang et al. 2019a, b, c). The perception signal also triggers differential activation/repression of a large number of downstream genes involved in more than one signal transduction pathways. Differential expression of these downstream genes has been a subject of intensive research. It has also been shown that the differential expression of genes is partly regulated by epigenetic modifications. The present study is a part of such studies, where role of two H3 histone modification marks (H3K4me3 and H3K27me3) in regulation of expression of these downstream genes of the host has been examined. For this purpose, we used a susceptible cv. HD2329 and its resistant NIL, the latter carrying the leaf rust resistance gene Lr28 that has been widely used for imparting resistance against leaf rust in many Indian cultivars. This gene, Lr28 which is located on chromosome arm 4AL was transferred from Aegilops speltoides and could not be cloned and characterized so far. However, in one of our own studies involving RNA-seq analysis using the same experimental material (Sharma et al. 2018a) and based on highly expressed genes identified on chromosome 4AL in resistant NIL, this gene was speculated to encode an ATP binding serine threonine protein kinase (receptor like kinase).
It is widely known that the epigenetic control of gene expression is broadly mediated by DNA methylation, ncRNAs and histone modifications. The present study is the first study, which involved identification of downstream genes, whose expression and crosstalk is regulated by the association of H3K4/27me3, primarily due to presence/absence of the gene Lr28. The study involved genome-wide ChIP sequencing to identify genomic regions, the so-called differential binding sites (DBS), associated with each of the two histone marks.
While studying association of modified histones, generally we do not know whether the histone marks are the cause or the result of activation or repression of relevant genes. Available information indicates the prevalence of both the possibilities. Alternatively, the histone marks may simply cause uncoiling or coiling of DNA in the form of nucleosomes, thus influencing expression of genes rather indirectly. For instance, available evidence indicates that H3K4me3 may not be the cause, but only the result of active transcription, since it has been shown that loss of H3K4me3 from genes involved in active transcription does not lead to reduction in transcription of differentially expressed genes (reviewed in Howe et al. 2017). In rice, it has also been shown that H3K4me3 is strongly associated with active transcription and removal of H3K4me3 by histone demethylase leads to gene repression (Chen et al. 2013).
In view of the above, we feel that caution should be exercised in interpreting the results of ChIP-seq and in comparing these results with the results of gene expression obtained from RNA-seq. During the present study also, the expected results of gene expression available from RNA-seq data do not always match with the results of ChIP-seq, if we assume that H3K4me3 is an activation mark and H3K27me3 is a repression mark. Such unexpected results were also reported earlier in common bean (Ayyappan et al. 2015), rice (Zong et al. 2013) and Arabidopsis (Brusslan et al. 2012).
DBSs for the two histone marks (H3K4/K27me3)
In the present study, a large number of DBSs in the genome were available for H3K4me3; the frequency of DBSs for H3K27me3 were fewer suggesting that relatively more genes are activated than repressed for infection to be successful. Also, there seem to be many more DBSs (4379) in S line relative to R line (2184), that are associated with H3K4me3 relative to those which are associated with H3K27me3, suggesting that in the S line, many more genes are activated relative to those which are repressed (Fig. 2). The results of H3K27me3 are largely in agreement with this conclusion, except that with the passage of time, there is many fold decrease in the number of DBSs for both the marks, suggesting that different sets of almost equal number of genes perhaps get downregulated and upregulated in the R line also. This also suggests complexity of the interacting genes following inoculation in both NILs.
Genomic distribution of DBSs
Distribution of DBSs in the different genomic regions (promoters, exons, introns, TTS and intergenic regions) also had some distinctive features including the following (Fig. 3; for details see Tables S3–S18).
-
(i).
DBSs in promoter regions The frequency of DBSs for H3K4me3 associated with promoter regions was much higher than those with H3K27me3. This is partly in agreement with the available results for yeast, fruit fly, Arabidopsis and mouse (Yang and Wilson 2018). This observation was further supported by chromatin state analysis (Fig. 7a). For instance, the chromatin state 12 mainly involving promoter regions associated with H3K4me3 in the TSS was common in S0, S96 and R96; probability being particularly high in S96. This suggested activation of a number of genes in susceptible cultivar due to binding of H3K4me3 in promoter region. High expression of such genes (with high affinity DBSs in promoter region) was also obtained in RNA-seq analysis; some of the proteins encoded by these genes include the following: ABC transporter family, polyubiquitin, sodium hydrogen exchanger, F-box protein, alpha/beta hydrolase, etc. While comparing these results with those of chromatin state analysis, it was observed that chromatin state 13 was exclusively enriched for strong promoter with H3K4me3 in R96 indicating the activation of some important genes perhaps involved in providing resistance. In contrast, the chromatin state 2 involving repressed enhancers located 2 kb upstream of TSS carrying H3K27me3 was predominant in S0, although it was present in other treatments also. This indicated repression of genes, which need to have low expression in susceptible genotype in the absence of the pathogen; these genes mainly encoded proteins for photosystem II, Ankyrin repeats, pentatricopeptide repeats, etc.
-
(ii)
DBSs in intergenic regions The frequency of DBSs located in the intergenic regions was much higher for H3K27me3 (84–88%) relative to those for H3K4me3 (28–46%); these results are not very different from those recently reported for mouse (Yang and Wilson 2018). Earlier in wheat also, similar situation was observed with these histone marks indicating that these marks may also be involved in trans-regulation of gene activity (Li et al. 2019). The abundance of the DBSs for H3K27me3 in the intergenic region may also explain the possible repression of transcription of the transposable elements (TEs), thus leading to genome stability (Walter et al. 2016). For some of DBSs in the intergenic regions, the frequencies were similar for both the epigenetic markers; these results are also in partial agreement with the results of chromatin state analysis, since chromatin state 4 showed high enrichment of both the marks in the intergenic regions. Later, we discuss the role of intergenic regions in trans-regulation of gene expression.
-
(iii)
DBSs in exons, introns and TTS The proportions of DBSs located in exons, introns and TTS for the two histone marks also differed with relative abundance of H3K4me3. Thus, overall, H3K4me3 mark is more spread out in the genome whereas H3K27me3 is mostly restricted to intergenic region. The distribution of the binding sites for activation (H3K4me3) and repression (H3K27me3) marks in the present study in wheat is in agreement with the distribution of the other activation (H4K12ac) and repression (H3K9me2) marks in the genome examined during rust infection in cowpea (Ayyappan et al. 2015). On individual chromosomes also, the DBSs for H3K4me3 are distributed all along the length of individual chromosomes whereas the DBSs for H3K27me3 had more clustered distribution (see Fig. 1).
Mechanism of action of H3K4me3 and H3K27me3
In view of the above, we believe that the mechanism of action of the two histone marks may differ. In fact, H3K27me3 and the associated intergenic sequences (> 50 kb from the DBS) may be involved in trans-regulators (for list of genes see Tables S11–S18). In contrast, H3K4me3 may facilitate the associated gene sequences in performing their function as cis-regulatory elements. The interactions between specific intergenic regions associated with H3K27me3 and distantly located genes, may actually be facilitated through at least following two different known mechanisms: (i) The folding of chromatin in space through an unknown mechanism may bring the DBSs in the intergenic region in close proximity to the genes to be trans-regulated; this may be further investigated through a study of 3D structure of chromatin conformations using the available techniques like 3C (Dekker et al., 2002; Miele and Dekker 2009), Hi-C (van Berkum et al. 2010) and Dip-C (Tan 2019). (ii) Trans-regulation through DBSs in the intergenic region carrying H3K27me3 may also involve lncRNAs, which are known to be derived from non-coding regions of the genome and act in trans by recruiting polycomb repressor complex 2 (PRC2), which mediates methylation of H3K27me3; similarly, PRC1 seems to limit the access of chromatin to transcription factors by binding with other nucleosome, thus affecting the expression of a distant gene (Lehmann et al. 2012; Brockdorfi 2013; Roberts et al. 2013). LncRNA mediated regulation of leaf rust disease resistance genes is also being investigated by us in a separate study.
Expression of DH3M genes due to H3K4/K27me3 in S and R lines
During the present study, as many as 2259 unique DH3M genes (out of a total of 2565 DH3M genes; based on the protein domains) associated with H3K24me3 were identified in the susceptible cultivar. In contrast, only 800 unique DH3M genes (out of total 1200 DH3M genes) were associated with H3K4me3 in resistant NIL. These DH3M genes must have been active, although the exact role of H3K4me3 following an attack by the pathogen cannot be inferred (Fig. 4). Further analysis also revealed that the number of DH3M genes that were associated with H3K4me3 differed in susceptible (S) and resistant (R) NILs. We like to discuss these results under four different categories of genes. (i) First, in S lines, the number of genes activated are almost three-fold the number of genes that are activated in the R line. That susceptibility accompanies activation of relatively large number of genes was observed in our earlier studies also, which involved either the transcriptome studies involving APR gene Lr48 or DNA methylation (MeDIP) studies involving Lr28; this activation may partly be attributed to association with H3K4me3, although loss of DNA methylation could be another factor, as reported by us in another study (Saripalli et al. 2020). In fact, a complex network of genes may be involved, which needs to be elucidated. (ii) Second, in R line at 96 hai, activation of some genes and repression of some other genes seem to play important roles in providing resistance. This was apparent from the results of differential binding of H3K4me3 and H3K27me3 in R96 line relative to those in either R0 or S96. This conclusion is based on abundance of sites showing either increased affinity or decreased affinity for H3K4me3 relative to those for H3K27me3. (iii) Third, S line and R line had 245 common DH3M genes associated with H3K4me3 (no such common genes were available for H3K27me3). These genes must be involved in important functions but may not be causal in determining resistance or susceptibility, unless trans-regulatory elements were involved, which could not be examined in the present study. The role of these genes shared by both NILs need to be subjected to further study. (iv) Fourth, in both S and R lines, there were many more unique DH3M genes associated with H3K4me3 relative to those associated with H3K27me3; these included 53 in S96 (relative to S0) and mere 9 in R96 (relative to R0)]. The implications are that relatively many more genes are activated than repressed in both the reactions (S0 vs S96 and R0 vs R96). The factors involved in activation of these genes will have to be elucidated, which at least in some cases may be due to histone H3 acetylation, as shown by us in our earlier study (Sharma et al. 2018b). However, some unique DH3M genes in the two opposite interactions perform different functions in the compatible and incompatible interactions, although these unique genes may contain one or more common domains.
The genes regulated by two histone modifications in S and R genotypes belonged to 10 different categories that are depicted in Fig. 5. The same 10 categories (but not necessarily the same genes) were also noticed for genes that were found to be differentially expressed in our earlier study on transcriptome analysis (Sharma et al. 2018a) and differentially methylated in our earlier study on MeDIP analysis (Saripalli et al. 2020). A more detailed study of the functions of DH3M genes (inferred through ChIP-Seq) and their assignment to three well known categories of functions (cellular, molecular and biological) suggested that majority of these genes were involved in catalytic or binding functions and metabolic (molecular functions) or cellular/biological processes (Fig. 6).
Relationship of histone modifications with gene expression (RNA-seq)
It is apparent from the data presented in Table 4 that a very small fraction of genes associated with the two histone marks (7% and 12%) exhibited the expected expression patterns in the transcriptome (RNA-seq) data. This is not surprising, keeping in view that a complex network of regulatory systems controls the expression of genes, and that the two histone marks used in the present study make only a tiny fraction of the overall regulatory system involving DNA methylation, siRNA and other histone marks (Takahashi et al. 2018; Sharma et al. 2018b). Similar comparisons were earlier made in rice and Arabidopsis; this match accounted for 24–26% genes in rice, under drought stress involving H3K4me3 mark (Zong et al. 2013) and 10–15% genes in Arabidopsis for leaf senescence involving H3K4/K27me3 marks (Brusslan et al. 2012). A number of studies in mammals also did not find a match between H3K4me3 binding and RNA-Seq data (for a review see Howe et al. 2017). According to some studies, H3K4me3 binding and transcription are two independent events; H3K4me3 seems to be involved in post-transcriptional events like splicing, transcription, termination, etc. (Howe et al. 2017). H3K4me3 has also been shown to be involved in other functions like DNA repair and targeting of double stranded breaks during meiotic recombination (Howe et al. 2017). In the present study also, a number of H3K4me3 associated genes were found to contain important domains involved in DNA repair (e.g. flap endonuclease) and targeting double stranded breaks. Thus, exclusive role of H3K4me3 in gene activation seems to be doubtful and needs detailed further investigation.
Co-localization of H3K4/27me3 marks (bivalent chromatin)
In a very small proportion of genes (14 out of 5328/305 genes; for complete list of DH3M genes and their gene IDs, refer to supplementary Tables S3 to S10); co-localization of the two histone marks (i.e. bivalent domains) was observed in coding/promoter regions (Table 5). Co-localization of histone H3 marks has also been reported in FLC gene for flowering, camalexin biosynthesis genes and a large number of other genes in Arabidopsis (Jiang et al. 2008; Zhang et al. 2009; Zhao and Rhee 2018); many more reports of this co-localization of the two histone H3 marks are available in mammalian genomes (Sachs et al. 2013; Dattani et al. 2018; Vestenhouw and Schier 2012). It has been also suggested that in these bivalent domains, the association of H3K4me3 with an hitherto inactive gene may help in subsequent activation. Some reports involving plant systems also suggest that transcription can take place even in the presence of repressive marks like H3K27me3, which is not sufficient for silencing of genes (Schubert et al. 2006; Adrian et al. 2010). In other cases, a hitherto active gene due to H3K4me3 may also be repressed due to subsequent association of H3K27me3 (Akkers et al. 2009). Together, the expression of genes in the presence of bivalent histone marks seems to be a complex process.
Even though the genes with co-localized histone marks in this study constitute only a tiny fraction of the total genes associated with DBSs, some of these genes also have a role in binding of colocalized histone marks. Such genes included genes like those encoding PHD domains (Qian et al. 2018); these genes showed increased affinity with H3K4me3 in the present study. Therefore, efforts have been made to study the mechanism involved in binding of two histone marks on the same site. For instance, it is known that certain proteins with domains described as ‘writer’, ‘eraser’ and ‘reader’ may be involved respectively in adding, removing and recognizing modified histones. In a recent study, Qian et al. (2018) demonstrated that a plant-specific histone ‘reader’ called SHORT LIFE (SHL) recognizes both H3K4me3 and H3K27me3, the former with the help of its domain PHD (plant homeodomain) and the latter with the help of its domain BAH (bromo-adjacent homology).
In the resistant NIL, colocalization of the two chromatin marks was observed for four genes encoding the following proteins: U-box protein (E3 ligase), ubiquitin carboxyl-terminal hydrolase-2 (UCH2), protein containing coiled-coil domain (e.g. NLR) involved in a variety of functions including plant immunity and poly[ADP-ribose] polymerase (PARP) involved in plant immunity (Table 5). Interestingly U-box protein is involved in ubiquitination, whereas UCH2 belonging to the deubiquitinase (DUB) superfamily has a role in deubiquitination (see Zhou and Zeng 2017). Genes encoding these two proteins were also found to exhibit high expression in RNA-seq data during expression of resistance to leaf rust. In rice, ubiquitination (involving some of the U-box proteins) has been shown to promote host immunity by degradation of avirulence effector AvrPiz-t of Magnaporthe oryzae, although ubiquitination is known to often attenuate PTI signalling through proteasome dependent degradation of pattern recognition receptor (for a review, see Zhou and Zeng 2017). Similarly, UCHs (causing deubiquitintion) have been shown to have a role in both the biotic and abiotic tolerance. For example, UCH60 was reported in seeds harvested from field grown disease infested wheat (Kamal et al. 2010). Similarly, the UCHs also seem to be involved in providing tolerance to heat stress in an important vegetable chieh-qua (Benincasa hispidsa) and drought stress in maize (Hu et al. 2015; Wang et al. 2019a, b, c). Further, the deubiquitinating enzymes such as AtUBP12 and NtUBP12 through their deubiquitinating activity have been implicated in suppression of plant cell death in Arabidopsis and tobacco (Ewans et al. 2011). Together these observations suggest a possible role of ubiquitination and deubiquitination in leaf rust resistance. The remaining two genes, namely, gene with coiled-coil domain and PARP gene are widely known to be involved in plant immunity (Hu et al. 2017; De Block et al. 2005; Amor et al. 1998). Therefore, we assume that these genes must also be involved in resistance, directly or indirectly.
In the susceptible cultivar, the bivalent chromatin associated genes encoding three different disease resistance-like proteins (R genes) and S-acyltransferase were identified. Two of these R-like genes containing TIR-NBS-LRR and disease resistance protein with RPM1 domains showed high expression during RNA-seq using the same treatments as used for ChIP-seq. The H3K4me3 mark also showed increased affinity with these DH3M genes, suggesting the possible role of the epigenetic regulation of these genes conferring susceptibility (for more details, see later).
Relationship between H3 lysine methylation and cytosine DNA methylation
A comparison of results of the present study with the results of our earlier study on the role of cytosine DNA methylation using MeDIP (Saripalli et al. 2020) allowed identification of 43 genes in S and R reactions which were perhaps regulated by differential histone methylation as well as DNA methylation (Table 7). Information on the expression of these genes in our RNA-seq data was not available and therefore, we could not correlate the patterns of histone/DNA methylation with the pattern of expression of these genes. However, in our present study, nearly equal number of genes showed antagonistic and synergistic relationship with differential DNA methylation. Results of H3K4me3 and H3K27me3 will be separately compared with DNA methylation results. (i) H3K4me3 mark was earlier shown to have a positive relationship with DNA hypomethylation in Arabidopsis (Zhang et al. 2009; Mathieu et al. 2005). This hypomethylation may be due to inhibition of MET1 (methyltransferase 1) activity by H3K4me3, since MET1 is involved in CG methylation in plants (Finnegan et al. 1996; Saze et al. 2003). (ii) The relationship of H3K27me3 mark with DNA methylation is still poorly understood and both positive and negative relationship are reported. The DNA methylation is a negative modulator of PRC2-chromatin interaction, so that hypomethylation occurs in the presence of H3K27me3 (Mendenhall et al. 2010; Meissner et al. 2008). However, a positive relationship between DNA methylation and histone modification has actually been reported in humans, where genes that are de novo methylated in cancer cells were found to be enriched with H3K27me3 mark (Schlesinger et al. 2007). The chromatin-state analysis also showed a distinct pattern in chromatin state 10, where repressed genic regions were enriched with all the three epigenetic marks (H3K4me3, H3K27me3 and DNA methylation) in TTS region, although the genes associated with the two different marks differed in the present study (Fig. 7).
Important genes involved in wheat-leaf rust interaction
The present study also allowed us to identify some important genes that are differentially expressed due to association with H3K4/K27s; these genes encode proteins with domains involved in biotic stress response and can be grouped into the following five classes. (i) Genes with increased association with H3K4me3 and showing high expression in both susceptible and resistant lines following inoculation. (ii) Genes showing decline in association with H3K27me3 and showing high gene expression in susceptible cultivar or resistant NIL. (iii) Genes showing decline in association with H3K4me3 and showing low expression in resistant NIL. (iv) Genes showing increased association with H3K4me3 and hypomethylated in susceptible cultivar. (v) Genes associate with two co-localized histone marks including genes showing increased association with H3K4me3 and decline in association with H3K27me3 in susceptible genotype or increased association with both the marks in resistant NIL (genes in this category discussed above). The role of some of these important genes will be discussed in relation to susceptibility and resistance to leaf rust.
Genes involved in susceptibility
Some of the genes belonging to four of the above five classes are known to be involved in sensitivity/susceptibility to biotic stresses in general (Lorang et al. 2007; Faris et al. 2010; Gupta et al. 2018; Pessina et al. 2016; Ravichandran et al. 2015) (See Supplementary Tables S27 and S28; Tables 5 and 7; Fig. 9). The most important of these genes that are identified during the present study and may be involved in susceptibility to leaf rust include three genes that encode proteins containing three different known domains, namely CC-NBS LRR, TIR-NBS-LRR and RPM1; these genes show an increase in affinity with H3K4me3 and a decline in affinity with H3K27me3. Two of these three genes (CC-NBS-LRR and RPM1 containing genes) also showed high expression in susceptible cultivar. This indicated that during the compatible interaction, these genes are perhaps induced due to pathogen attack only (due to their increased affinity with activation mark H3K4me3); otherwise these genes remain repressed. Thus, these genes might have an important role in susceptibility and hence may actually be susceptibility genes (S genes) with NBS-LRR domains (Gupta et al. 2018; Faris et al. 2010; Pessina et al. 2016). Such S genes have also been shown to confer susceptibility to Cochliobolus victoriae in Arabidopsis (Lorang et al. 2007), and to necrotrophic pathogens in wheat (Faris et al. 2010; Gupta et al. 2018). However, no such S-genes conferring susceptibility against leaf rust pathogen, have so far been reported in wheat, although several sensitivity gene including Tsn1 conferring susceptibility to spot blotch and Tsc1 against tan spot have been reported (McDonald et al. 2018). When Tsn1 interacts with ToxA gene in the pathogen, it leads to susceptibility of wheat to spot blotch. This ToxA-Tsn1 system represent an inverse-gene-to-gene relationship (Nawathe et al. 2020). We speculate that such a sensitivity gene for leaf rust in wheat may be discovered in future. Such genes encoding purple acid phosphatases, receptor like kinases, cytochrome P450, F-box, Mlo like protein, etc. were up-regulated and also exhibited increased affinity with H3K4me3 in susceptible cultivar (S96), indicating their possible role in susceptibility. Over-expression of purple acid phosphatase in Arabidopsis was earlier shown to inhibit expression of PR1, accumulation of salicylic acid and increased accumulation of H2O2 enhancing susceptibility to Pseudomonas syringeae (Ravichandran et al. 2015). The gene encoding (Mlo)-like protein is another important gene whose dominant allele acts as a susceptibility gene for powdery mildew infection in barley (Buschges et al. 1997) and in grapevine, the RNAi mediated loss of function mutations in MLO6 and MLO7 genes contributed to powdery mildew resistance (Pessina et al. 2016). Another gene encoding for phloem protein A10 was found to exhibit high expression in susceptible cultivar as revealed through qRT-PCR analysis. Phloem proteins are actually the lipid binding proteins involved in lipid mediated signalling in plants (Barbaglia et al. 2016). However, their role in increased susceptibility towards leaf rust infection or other biotic stresses is not known.

Modified from our earlier study, Sharma et al. (2018a)
A network of genes based on the common genes with DBSs that are differentially expressed or differentially methylated during compatible (S0 vs S96) and/or incompatible interactions (S96 vs. R96 or R0 vs. R96). Genes that are differentially expressed/DNA methylated/histone methylated in R lines (green boxes), S lines (yellow boxes), and in both the NILs (blue boxes). Genes highlighted in red font were associated with H3K4me3 mark, genes highlighted in purple were associated with H3K27me3 mark and genes highlighted in black font showed colocalization of both the H3K4/K27me3 marks.
In addition to the above genes, there were also genes (carrying domains for bHLH TF, auxin response factor, F-box, etc.) which had high affinity with H3K4me3 in susceptible genotype and thus are perhaps involved in promoting leaf rust infection. Earlier, overexpression of bHLH in wheat and Arabidopsis was shown to exhibit enhanced susceptibility to P. striiformis (in wheat) and Pseudomonas syringae (in Arabidopsis) (Wang et al. 2015).
Genes involved in resistance
Out of a number of DH3M genes associated with resistance or defence responses (R0 vs R96) one of the genes encodes a protein for alpha–beta hydrolase (for details of genes refer to Supplementary Tables S27 and S28 and Fig. 9). These proteins provide core structure for phytohormone/ligand receptors for two signalling pathways, namely gibberellin and strigolactone signalling pathways that are known to play an important role in defense (Mindrebo et al. 2016). For instance, gibberellin is known to act as both positive and negative regulator of disease resistance. Transgenic rice over-expressing GA deactivating enzyme displayed enhanced resistance to Mo and Xoo pathogens suggesting the role of GA as negative regulator of disease resistance; in contrast, GA was shown to act as a positive regulator of disease resistance during infection with necrotrophic root pathogen like P. graminicola (for further details, refer to review by Bruyne et al. 2014). The role of strigolactones in defense response was examined in tomato, where the mutants deficient in strigolactone were found to be more susceptible to foliar fugal pathogens like Botrytis cinerea and Alternaria alternate. These tomato mutants also exhibited reduction in the content of defense hormones like jasmonic acid, salicylic acid and abscisic acid (Torres-Vera et al. 2014).
Another class of genes that were identified during the present study and may be involved in resistance encode proteins carrying domains for GDSL esterase/lipases that are known to be involved in providing resistance against Alternaria brassisicola in Arabidopsis (Oh et al. 2005). Resistance through GDSL esterase/lipases are mainly provided through signalling involving phytohormone like ethylene (Kwon et al. 2009); negative regulation through auxin signalling has also been reported (Lee et al. 2009).
Genes which showed low affinity with H3K4me3 and associated with reduced expression were also found to be associated with disease resistance (S96 vs R96). For instance, the genes encoding proteins carrying domains like TIR-NBS-LRR domain, C-type lectin domain, aspartyl protease domain perhaps serve as negative regulators of disease resistance. Similarly, WAK (wall associated kinase) and exocyst subunit 70 showing high affinity with H3K4me3 were repressed (perhaps due to their hypermethylation) in incompatible interaction (Table 7), suggesting that their repression is involved in providing leaf rust resistance. Some of these genes have actually been reported to provide resistance through their negative regulation. For instance, there are genes which encode proteins containing lectin domain (e.g., C-type lectin domain containing proteins) and function as receptor like kinases involved in perception and transduction of environmental stimuli (for a review see Singh and Zimmerli 2013). Such genes include Lec-RK V.5 (receptor kinase V.5) that were earlier reported to act as a negative regulator of Pseudomonas syringeae infection through stomatal closure in Arabidopsis, since plants lacking this gene were found to be resistant (Desclos-Theveniau et al. 2012). WAK is another receptor kinase, exemplified by OsWAK112d in rice, which has been shown to act as a negative regulator of blast disease, so that its down regulation enhances disease resistance (Delteil et al. 2016). Some other genes which could be validated using qRT-PCR seem to be involved in resistance, since, these were induced in resistant NIL (Table 6).
Another group of genes carrying domains for bHLH TF, auxin response factor, F-box, etc. (for details of genes see Table S26) which were associated with high affinity DBSs of H3K27me3 in resistant NIL perhaps act as negative regulators of leaf rust disease resistance assuming that H3K27me3 leads to repression of gene expression. Interestingly, the same set of genes were shown to promote leaf rust infection when associated with H3K4me3 (see above). This suggested that activation and repression of the same set of genes may be modulated by different histone marks during susceptible and resistant reaction. In addition, the genes carrying domains for ethylene response TF, F-box, transmembrane protein, etc. were activated in resistant NIL indicating their role in defense response. Earlier also, the overexpression of ethylene response TF was shown to provide enhanced tolerance against infection by Rhizoctonia cerealis (Zhu et al. 2014).
Genes involved in binding of H3K4/K27me3 to the chromatin
A number of DH3M genes associated with H3K4me3 are known to encode proteins that regulate the H3 histone lysine trimethylation. These DH3M genes mainly encode histone lysine methyltransferases/SET domain containing proteins, PHD (plant homeodomain) finger like proteins, S-adenosyl-l-methionine-dependent methyltransferase (SAM), etc. Histone lysine methyltransferase/SET domain containing proteins are preferentially recruited to the 5′ coding regions of transcriptionally active genes, where they catalyze trimethylation of H3K4 (Ng et al. 2003). Similarly, proteins carrying PHD (plant homeodomain) help in recognizing modified histones like H3K4me3 and trap K4me3 in an aromatic cage like binding pocket (Musselman and Kutateladze, 2018). This subsequently helps in normal gene expression with the help of other proteins (Sanchez and Zhou 2011). The SAM proteins mainly help in the transfer of methyl group from methionine of ATP to H3 lysine causing histone trimethylation.
Conclusions and summary
The present study on wheat leaf-rust pathosystem is the first of its kind, where we studied genome-wide gene regulation mediated by histone H3 methylation coupled with DNA methylation and chromatin state analysis. Our results showed that susceptibility to leaf rust infection seems to be controlled mainly through activation of a large number of genes, whereas leaf rust resistance/defense response seems to be controlled by activation and repression of relatively fewer genes as revealed by the association of H3K4/K27me3 marks. Our earlier findings also showed the involvement of histone acetylation and DNA methylation in regulating the expression of genes involved in wheat-leaf rust pathosystem. The results of differential histone modification, chromatin state analysis and DNA methylation also suggested that more than one epigenetic marks may be involved in wheat-leaf rust pathosystem. We also identified 43 DH3M genes, which also carried DNA methylation marks; there were also genes that are regulated uniquely by one of the two histone modifications. Although, we do not expect complete correspondence between the genes available in ChIP-seq and RNA-seq libraries, but the presence of merely 7% (H3K4me3) and 12% (H3K27me3) genes that were common to the RNA-seq data do indicate the possible role of other epigenetic mechanisms in the regulation of gene expression, which need to be examined. This small fraction of genes (and the encoded proteins) enriched by two histone methylation marks included the following: (i) RLKs and cell wall modifying enzymes, (ii) protein kinases (S/TPKs) and phosphatase (purple phosphatase), (iii) ENTH/VAS family protein involved in phospholipid signaling, (iv) TFs (scarecrow, wuschell, NAC, MYB), (v) genes involved in ROS homeostasis (cytB5, GST, cytP450), (vi) growth hormone responsive genes (alpha beta hydrolase, GDSL esterase/lipase), and (vii) R genes (NBS-LRR containing). These genes may be involved in PTI and ETI signalling in a sequential manner. Some of these genes (e.g. acid purple phosphatase, ROS homeostasis genes, etc.) are perhaps involved in promoting infection leading to susceptibility mostly by suppressing the activity of other defense response genes like PR1 or due to oxidative burst. Some other genes are involved in defense response either through negative regulation (e.g. lectin domain containing genes, RLKs like WAK) where the low expression leads to resistance or through positive regulation where high expression leads to resistance. Evidence of trans-regulation of genes due to intergenic DBSs of both these marks was also noticed. With a view to examine the role of the remaining epigenetic mechanisms, we are currently examining the role of ncRNA (miRNAs and lncRNAs) and DNA methylation (at the level of a single base) using whole genome bi-sulfite sequencing. Together, the genes identified during the present study and those to be identified by the future studies involved in wheat-leaf rust interaction will hopefully be used in future studies thus expanding the knowledge-base for plant immunity in general and wheat-leaf rust pathosystem in particular.
Data availability
The ChIP-Seq and RNA-Seq raw reads of susceptible genotype (HD2329) and resistant NIL (HD2329 + Lr28) at 0hbi and 96hai were deposited in Sequence Read Archive (SRA), National Center for Biotechnology Information (NCBI) and can be accessed through Bioproject Database under the accession number PRJNA588134. MeDIP raw sequencing reads of susceptible (HD2329) and resistant (HD2329 + Lr28) NILs at 0 hbi and 96 hai were deposited in Sequence Read Archive (SRA), NCBI and can be accessed through Bioproject Database under the accession number PRJNA517568. Some data related to this study is also available in Supplementary Tables S1 to S28.
References
Adrian J, Farrona S, Reimer JJ, Albani MC, Coupland G, Turck F (2010) Cis-regulatory elements and chromatin state co-ordinately control temporal and spatial expression of FLOWERING LOCUS T in Arabidopsis. Plant Cell 22:1425–1440
Agarwal PK, Agarwal P, Reddy MK, Sopory SK (2006) Role of DREB transcription factors in abiotic and biotic stress tolerance in plants. Plant Cell Rep 25:1263–1274
Akkers RC, van Heeringen SJ, Jacobi UG, Janssen-Megens EM, Francoij KJ, Stunnenberg HG, Veenstra GJ (2009) A hierarchy of H3K4me3 and H3K27me3 acquisition in spatial gene regulation in Xenopus embryos. Dev Cell 17:234–425
Amor Y, Babiychuk E, Inze D, Levine A (1998) Involvement of poly (ADP-ribose) polymerase in oxidative stress response in plants. FEBS Lett 440:1–7
Ayyappan A, Kalavacharla V, Thimmapuram J, Bhide KP, Sripathi VR, Smolinksi TG, Manoharan M, Thurston Y, Todd A, Kingham B (2015) Genome-wide profiling of histone modifications (H3K9me2 and H4K12ac) and gene expression in rust (Uromyces appendiculatus) inoculated common bean (Phaseolus vulgaris L). PLoS ONE 10:e0132176
Barbaglia AM, Tamot B, Greve V, Benning SH (2016) Phloem proteomics reveals new lipid binding proteins with putative role in lipid mediated signalling. Front Plant Sci 7:563
Bozkurt O, Unver T, Akkaya MS (2007) Genes associated with resistance to wheat yellow rust disease identified by differential display analysis. Physiol Mol Plant Pathol 71:254–259
Brockdorfi N (2013) Non coding RNA and polycomb recruitment. RNA 19:429–442
Brown EJ, Batchtrog D (2014) The chromosome landscape of Drosophila: comparison between species, sexes and chromosomes. Genome Res 24:1125–1137
Brusslan JA, Rus Alvarez-Canterbury AM, Nair NU, Rice JC, Hitchler MJ, Pellegrini M (2012) Genome-wide evaluation of histone methylation changes associated with leaf senescence in Arabidopsis. PLoS ONE 7:e33151
Bruyne LD, Hofte M, Vleesschauwer D (2014) Connecting growth and defence: the emerging roles of brassinosteroids and gibberellins in plant innate immunity. Mol Plant 7:943–959
Buschges R, Hollricher K, Panstruga R, Simons G, Wolter M et al (1997) The barley Mlo gene: a novel control element of plant pathogen resistance. Cell 88:695–705
Chandra S, Singh D, Pathak J, Kumari S, Kumar M, Poddar R, Balyan HS, Gupta PK, Prabhu KV, Mukhopadhyay K (2016) De novo assembled wheat transcriptomes delineate differentially expressed host genes in response to leaf rust infection. PLoS ONE 11:e0148453
Chen Q, Chen X, Wang Q, Zhang F, Lou Z et al (2013) Structural basis of a histone h3 lysine 4 demethylase required for stem elongation in rice. PLoS Genet 9:e1003239
Cheng Q, Dong L, Gao T, Liu T, Li N, Wang L, Chang X, Wu J, Xu P, Zhang S (2018) The bHLH transcription factor GmPIB1 facilitates resistance to Phytophthora sojae in Glycine max. J Exp Bot 69:2527–2541
Conesa A, Götz S (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genom. https://doi.org/10.1155/2008/619832
Dattani A, Kao D, Mihaylova Y, Abnave P, Huges S, Lai A, Sahu S, Aboobaker AA (2018) Epigenetic analysis of planarian stem cells demonstrate conservation of bivalent histone modifications in animal stem cells. Genome Res 28:1543–1554
De Block M, Verduyn C, De Brouwer D, Cornelissen M (2005) Poly (ADP-ribose) polymerase in plants affects energy homeostasis, cell death and stress tolerance. Plant J 41:95–106
Dekker J, Rippe K, Dekker M, Kleckner N (2002) Capturing chromosome conformation. Science 295:1306–1311
Delteil A, Gobbato E, Cayrol B, Estevan J, Romiti CM, Dievart A, Kroj T, Morel JB (2016) Several wall-associated kinases participate positively and negatively in basal defense against rice blast fungus. BMC Plant Biol 16:17
Desclos-Theveniau M, Arnaud D, Huang TY, Lin GJ, Chen WY, Lin YC, Zimmerli L (2012) The Arabidopsis lectin receptor kinase LecRK-V.5 represses stomatal immunity induced by Pseudomonas syringae pv. tomato DC3000. PLoS Pathog 8:e1002513
Dhariwal R, Vyas S, Govindraj RB, Jha SK, Khurana JP, Tyagi AK, Prabhu KV, Balyan HS, Gupta PK (2011) Analysis of differentially expressed genes in leaf rust infected bread wheat involving seedling resistance gene Lr28. Funct Plant Biol 38:1–14
Diallo AO, Ali-Benali MA, Badawi M, Houde M, Sarhan F (2012) Expression of vernalisation responsive genes in wheat is associated with histone H3 trimethylation. Mol Genet Genom 287:575–590
Draz I, Abou-Elseoud MS, Kamara AEM, Alaa-Eldein OA, El-Bebany A (2015) Screening of wheat genotypes for leaf rust resistance along with grain yield. Ann Agric Sci 60:29–39
Ernst J, Kellis M (2012) ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 28:215–216
Ewans R, Pangestuti R, Thornber S, Craig A, Carr C, O'Donnell L, Zhang C, Sadanandom A (2011) Deubiquitinating enzymes AtUBP12 and AtUBP13 and their tobacco homologue NtUBP12 are negative regulators of plant immunity. New Phytol 191:92–106
Faris JD, Zhang Z, Lu H, Lu S, Reddy L et al (2010) A unique wheat disease-resistance like gene governs effector triggered susceptibility to necrotrophic pathogens. Proc Natl Acad Sci USA 107:13544–13549
Finnegan EJ, Peacock WJ, Dennis ES (1996) Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc Natl Nat Acad Sci USA 93:8449–8454
Flor HH (1942) Inheritance of pathogenicity in Melampsora lini. Phytopathology 32:653–669
Flor HH (1971) Current status of the gene-for-gene concept. Annu Rev Phytopathol 9:275–296
Geddes J, Eudes F, Laroche A, Selinger LB (2008) Differential expression of proteins in response to the interaction between the pathogen Fusarium graminearum and its host, Hordeum vulgare. Proteomics 8:545–554
Gill HS, Li C, Sidhu JS, Liu W, Wilson D, Bai G, Gill BS, Sehgal SK (2019) Fine mapping of the wheat leaf rust resistance gene Lr42. Int J Mol Sci 20:2445
Gupta PK, Chand R, Vasistha NK, Pandey SP, Kumar U, Mishra VK, Joshi AK (2018) Splot blotch disease of wheat: the current state of research on genetics and breeding. Plant Pathol 67:508–531
Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD et al (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in human genome. Nat Genet 39:311–318
Heinz S, Benner C, Spann N, Bertolino E et al (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38:576–589
Horsefield BH, Zhang X, Manik MK, Shi Y, Chen J, Qi T et al (2019) NAD+ cleavage activity by animal and plant TIR domains in cell death pathways. Science 365:793–799
Howe FS, Fischl H, Murray SC, Mellor J (2017) Is H3K4me3 instructive for transcription activation? BioEssays 39:1–12
Huan Q, Mao Z, Chong K, Zhang J (2018) Global analysis of H3K4me3/H3K27me3 in Brachypodium distachyon reveals VRN3 as critical epigenetic regulation point in vernalization and provides insights into epigenetic memory. New Phytol 219:1373–1387
Hu L, Wu Y, Wu D, Rao W et al (2017) The coiled-coil and nucleotide binding domains of BROWN PLANTHOPPER RESISTANCE14 function in signaling and resistance against planthopper in rice. Plant Cell 29:3157–3185
Hu X, Wu L, Zhao F, Zhang D, Li N, Zhu G, Li C, Wang W (2015) Phosphoproteomic analysis of the response of maize leaves to drought, heat and their combination stress. Front Plant Sci 6:298
Jiang D, Wang Y, Wang Y, He Y (2008) Repression of FLOWERING LOCUS C and FLOWERING LOCUS T by the Arabidopsis polycomb repressive complex 2 components. PLoS ONE 3:e3404
Kamal AHM, Kim KH, Shin KH, Kim DE, Oh MW, Choi JS, Hirano H, Heo HY, Woo SH (2010) Proteomics-based dissection of biotic stress responsive proteins in bread wheat (Triticum aestivum L.). Afr J Biotechnol 9:7239–7255
Kawahara Y, Oono Y, Kanamori H, Matsumoto T, Itoh T, Minami E (2012) Simultaneous RNA-seq analysis of a mixed transcriptome of rice and blast fungus interaction. PLoS ONE 7:e49423
Kim JM, To TK, Ishida J et al (2008) Alterations of lysine modifications on the histone h3 n-tail under drought stress conditions in Arabidopsis thaliana. Plant Cell Physiol 49:1580–1588
Kwon SJ, Jin HC, Lee S, Nam MH, Chung JH, Kwon SI, Ryu CM, Park OK (2009) GDSL lipase-like 1 regulates systemic resistance associated with ethylene signaling in Arabidopsis. Plant J 58:235–245
Laczik M, Hnedrickx J, Veillard AC, Tammoh M, Marzi S, Poncelet D (2016) Iterative fragmentation improves the detection of ChIP-seq peaks for inactive histone marks. Bioinform Biol Insights 10:209–224
Langmead B, Salzberg S (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Lee DS, Kim BK, Kwon SJ, Jin HC, Park OK (2009) Arabidopsis GDSL lipase 2 plays a role in pathogen defense via negative regulation of auxin signaling. Biochem Biophys Res Commun 379:1038–1042
Lehmann L, Ferrari R, Vashisht AA, Wohlschlegel JA, Kurdistani SK, Carey M (2012) Polycomb repressive complex 1 (PRC1) disassembles RNA polymerase II preinitiation complexes. J Biol Chem 287:35784–35794
Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128:707–719
Li T, Chen X, Zhong X, Zhao Y, Liu X, Zhou S et al (2013) Jumonji C domain protein JMJ 705-mediated removal of histone H3 lysine 27 trimethylation is involved in defense-related gene activation in rice. Plant Cell 25:4725–4736
Li Z, Wang M, Lin K et al (2019) The bread wheat epigenomic map reveals distinct chromatin architectural and evolutionary features of functional genetic elements. BMC Genom Biol 20:139
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-DDC. Methods 25:402–408
Lorang JM, Sweat TA, Wolpert TJ (2007) Plant disease susceptibility conferred by a “resistance” gene. Proc Natl Acad Sci USA 104:14863
Margueron R, Reinberg D (2011) The polycomb complex PRC2 and its mark in life. Nature 469:343–349
Mathieu O, Probst AV, Paszowski J (2005) Distinct regulation of histone H3 methylation at lysines 27 and 9 by CpG methylation in Arabidopsis. EMBO J 24:2783–2791
McDonald MC, Ahren D, Simpfendorfer S, Milgate A, Solomon PS (2018) The discovery of the virulence gene ToxA in the wheat and barley pathogen Bipolaris sorokiniana. Mol Plant Pathol 19:432–439
Meissner A, Mikkelsen TS, Gu H et al (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454:766–770
Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, Ku M, Bernstein BE (2010) GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet 14:e1001244
Miele A, Dekker J (2009) Mapping cis- and trans- chromatin interaction networks using chromosome conformation capture (3C). Methods Mol Biol 464:105–121
Mindrebo JT, Nartey CM, Seto Y, Burkart MD, Noel JP (2016) Unveiling the functional diversity of the alpha/beta hydrolase superfamily in the plant kingdom. Curr Opin Struct Biol 41:233–246
Musselman CA, Kutateladze TG (2018) A histone reader becomes the readout. J Biol Chem 293:7486–7487
Nakato R, Shirahige K (2017) Recent advances in ChIP-Seq analysis: from quality management to whole genome annotation. Brief Bioinform 18:279–290
Nawathe S, Yadav PS, Chand R, Mishra VK, Vasistha NK, Meher PK, Joshi AK, Gupta PK (2020) ToxA–Tsn1 interaction for spot blotch susceptibility in Indian wheats: an example of inverse gene-for-gene relationship. Plant Dis 104:71–81
Ng HH, Robert F, Young RA, Struhl K (2003) Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell 11:709–719
Oh IS, Park AR, Bae MS, Kwon SJ, Kim YS et al (2005) Secretome analysis reveals an Arabidopsis lipase involved in defense against Alternaria brassicicola. Plant Cell 17:2832–2847
Pessina S, Lenzi L, Perazzolli M, Campa M et al (2016) Knockdown of MLO genes reduces susceptibility to powdery mildew in grapevine. Hortic Res 3:16016
Qian S, Lv X, Scheid RN, Lu L, Yang Z, Chen W et al (2018) Dual recognition of H3K4me3 and H3K27me3 by a plant histone reader SHL. Nature Commun 9:2425
Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T (2014) deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42:W187–W191
Ravichandran S, Stone SL, Benkel B, Zhang J, Berrue F, Prithviraj B (2015) Optimal level of purple acid phosphatase5 is required for maintaining complete resistance to Pseudomonas syringeae. Front Plant Sci 6:658
Rintinch C, Heinig M, Bauerfeind A, Schafer S, Meith C et al (2014) Natural variation of histone modification and its impact on gene expression in the rat genome. Genome Res 24:942–953
Roberts TC, Morris KV, Weinberg MS (2013) Perspectives on the mechanism of transcriptional regulation by long non-coding RNAs. Epigenetics 9:13–20
Sachs M, Onodera C, Blaschke K, Ebata KT et al (2013) Bivalent chromatins mark developmental regulatory genes in the mouse embryonic germline in vivo. Cell Rep 3:1777–1784
Sanchez R, Zhou MM (2011) The PHD finger: a versatile epigenome reader 36:P364–372
Saripalli G, Sharma C, Gautam T, Singh K, Jain N, Prasad P, Roy JK, Sharma JB, Sharma PK, Prabhu KV, Balyan HS, Gupta PK (2020) Complex relationship of DNA methylation with gene expression during wheat-leaf rust interaction in a pair of NILs differing for Lr28. Mol Bio Rep 47:1339–1360
Saze H, Mittelsten SO, Paszkowski J (2003) Maintenance of CPG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet 34:65–69
Schlesinger Y, Straussman R, Keshet I, Farkash S et al (2007) Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 29:232–236
Schubert D, Primavesi L, Bishopp A, Roberts G, Doonan J, Jenuwein T, Goodrich J (2006) Silencing by plant Polycomb-group genes requires dispersed trimethylation of histone H3 at lysine 27. EMBO J 25:4638–4649
Sharma C, Saripalli G, Kumar S, Gautam T, Kumar A, Rani S, Jain N, Prasad P, Raghuvanshi S, Jain M, Sharma JB, Prabhu KV, Sharma PK, Balyan HS, Gupta PK (2018a) A study of transcriptome in leaf rust infected bread wheat involving seedling resistance gene Lr28. Funct Plant Biol 45:1046–1064
Sharma C, Kumar S, Saripalli G, Jain N, Raghuvanshi S, Sharma JB, Prabhu KV, Sharma PK, Balyan HS, Gupta PK (2018b) H3K4/K9 acetylation and Lr28-mediated expression of six leaf rust responsive genes in wheat (Triticum aestivum). Mol Genet Genom 294:227–241
Singh D, Kumar D, Satapathy L, Pathak J, Chandra S, Riaz A, Bhaganangre G, Dhariwal R, Kumar M, Prabhu KV, Balyan HS, Gupta PK, Mukhopadhyay K (2017) Insights of Lr28 mediated wheat leaf rust resistance: transcriptomic approach. Gene 637:72–89
Singh P, Zimmerli L (2013) Ectin receptor kinases in plant immunity. Front Plant Sci 4:124
Stark R, Brown GD (2011) DiffBind: differential binding analysis of ChIP-Seq peak data. Bioconductorhttps://bioconductor.org/packages/release/bioc/html/DiffBind.html
Takahashi S, Osabe K, Fukushima M, Takuno S et al (2018) Genome-wide characterization of DNA methylation, small RNA expression, and histone H3 lysine nine di-methylation in Brassica rapa L. DNA Res 25:511–520
Tan L (2019) Three-dimensional genome structure of a single cell. Science 366(6468):964–965
The International Wheat Genome Sequencing Consortium (IWGSC), Appels R, Eversole K et al (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:6403
Torres-Vera R, Garcia JM, Pozo MJ, Lopez-Raez JA (2014) Do strigolactones contribute to plant defence? Mol Plant Pathol 15:211–216
van Berkum NL, Lieberman-Aiden E, Williams L, Imakaev M, Gnirke A, Mirny LA, Dekker J, Lander ES (2010) Hi-C: a method to study the three-dimensional architecture of genomes. J Vis Exp 39:e1869
Vestenhouw NL, Schier AF (2012) Bivalent histone modifications in early embryogenesis. Curr Opin Cell Biol 24:374–386
Walter M, Teissendier A, Perez-Palacios R, Bourschis D (2016) An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. eLife 5:e11418
Wan L, Essuman K, Anderson RG, Sasaki Y, Monteiro F, Chung EH, Nishimura EO et al (2019) TIR domains of plant immune receptors are NAD+-cleaving enzymes that promote cell death. Science 365:799–803
Wang F, Lin R, Feng J, Qiu D, Chen W, Xu S et al (2015) Wheat bHLH transcription factor gene, TabHLH060, enhances susceptibility of transgenic Arabidopsis thaliana to Pseudomonas syringae. Physiol Mol Plant Pathol 90:123–130
Wang J, Hu M, Wang J, Ki J, Han Z, Wang G, Qi Y, Wang HW, Zhou JM, Chai J (2019a) Reconstitution and structure of a plant NLR resistosome conferring immunity. Science 364:eaav5870
Wang M, Jiang B, Liu W, Lin Y, Liang Z, He X, Peng Q (2019b) Transcriptome analyses provide novel insights into heat stress responses in Chieh-Qua (Benincasa hispida Cogn. var. Chieh-Qua How). Int J Mol Sci 20:883
Wang M, Yang D, Ma F et al (2019c) OsHLH61–OsbHLH96 influences rice defense to brown planthopper through regulating the pathogen-related genes. Rice 12:9
Wang X, Tang C, Zhang G et al (2009) cDNA-AFLP analysis reveals differential gene expression in compatible interaction of wheat challenged with Puccinia striiformis f. sp. tritici. BMC Genom 10:289
Wu F, Sheng P, Tan J, Chen X et al (2015) Plasma membrane receptor-like kinase leaf panicle 2 acts downstream of the drought and salt tolerance transcription factor to regulate drought sensitivity in rice. J Exp Bot 66:271–281
Wu K, Wang S, Song W, Zang J, Wang Y et al (2020) Enhanced sustainable green revolution yield via nitrogen responsive chromatin modulation in rice. Science 367:eaaz2046
Yang Y, Wilson MJ (2018) Genome-wide analysis of H3K4me3 and H3K27me3 modifications throughout the mouse urinogenital ridge at E II.5.biorixv. https://doi.org/10.1101/347880
You Y, Sawikowska A, Neumann M, Pose D, Capovilla G, Langenecker T, Neher RA, Krajewski P, Schmid M (2017) Temporal dynamics of gene expression and histone marks at the Arabidopsis shoot meristem during flowering. Nat Commun 8:15120
Zentner GE, Henikoff S (2013) Regulation of nucleome dynamics by histone modification. Nat Struct Mol Biol 20:259–266
Zhang X, Barnatavichute YV, Cokus S, Pellegrini M, Jacobsen SE (2009) Genome-wide analysis of mono, di and tri-methylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol 10:R62
Zhang X, Fu J, Hiromasa Y, Pan H, Bai G, Yan W (2013) Differentially expressed proteins associated with fusarium head blight resistance in wheat. PLoS ONE 8(12):e82079
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers R, Brown M, Li W, Liu XS (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9:R137
Zhao K, Rhee SY (2018) Epigenomic landscape of Arabidopsis thaliana metabolism reveals bivalent chromatin on specialized metabolic genes. bioRxiv. https://doi.org/10.1101/589036
Zhou B, Zeng L (2017) Conventional and unconventional ubiquitination in plant immunity. Mol Plant Pathol 18:1313–1330
Zhou DX, Hu Y (2010) Regulatory function of histone modifications in controlling rice gene expression and plant growth. Rice 3:103–111
Zhu X, Qi L, Liu X, Cai S, Xu H, Huang R et al (2014) The wheat ethylene response factor transcription factor pathogen-induced erf1 mediates host responses to both the necrotrophic pathogen Rhizoctonia cerealis and freezing stresses. Plant Physiol 164:1499–1514
Zong W, Zong X, You J, Xiong L (2013) Genome-wide profiling of histone H3K4-tri-methylation and gene expression in rice under drought stress. Plant Mol Biol 81:175–188
Acknowledgements
The work was carried out, when GS and TG held SRF position under a research project funded under NASF-ICAR program of Government of India. KS held RA position in a project funded by Department of Biotechnology, Govt. of India, New Delhi. PKG was awarded Hony Scientist position and HSB was awarded Senior Scientist position both from Indian National Science Academy (INSA). Funds received from NASF-ICAR (File No. NASF/ABP-6006/2016-17), New Delhi, India for carrying out this study are greatfully acknowledged. Bioinformatics Infrastructure Facility (BIF) laboratory was also used for carrying out a part of the bioinformatics work. Head, Department of Genetics and Plant Breeding, CCS University, Meerut, provided the necessary infrastructure.
Author information
Authors and Affiliations
Contributions
PKG and HSB conceived the experiment with the help of SR and PKS. GS conducted ChIP experiment and prepared the ChIP-Seq libraries with the help of SK. KS conducted data analysis and wrote the first draft of the MS jointly with GS. TG helped GS in conducting qRT-PCR analysis. PP provided inoculum for leaf rust pathotype 77-5 and NJ raised and provided the seedling tissues.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Gautam Saripalli and Kalpana Singh have contributed equally for this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Saripalli, G., Singh, K., Gautam, T. et al. Genome-wide analysis of H3K4me3 and H3K27me3 modifications due to Lr28 for leaf rust resistance in bread wheat (Triticum aestivum). Plant Mol Biol 104, 113–136 (2020). https://doi.org/10.1007/s11103-020-01029-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-020-01029-4