
Abstract
Differential DNA methylation due to Lr28 was examined in susceptible (S) wheat cv. HD2329 and its resistant (R) near isogenic line (NIL) (HD2329+Lr28) using two approaches: methylation sensitive amplified polymorphism (MSAP) and methylated DNA immunoprecipitation (MeDIP). S/R lines each had a large number of hypomethylated genes and relatively fewer hypermethylated genes at 96 hai (hours after inoculation) relative to 0 hbi (hours before inoculation), suggesting activation of many genes during the passage of time (96 hai), although identity of genes may differ in S and R lines. When R NIL was compared with S cultivar, there were many hypermethylated and fewer hypomethylated genes in R NIL relative to S cultivar, suggesting that many genes that are active in S cultivar are silenced in R NIL, both at 0 hbi and at 96 hai. Level of methylation was generally abundant in intergenic regions followed by that in promoters, transcription termination sites (TTSs) and exons/introns. Hypermethylation in promoter and gene body regions was not always associated with inhibition of gene expression and vice-versa, indicating that more than one regulatory mechanisms may control the expression of genes due to pathogen attack in presence and absence of Lr28. MSAP analysis also showed abundance of mCG methylation in S cultivar and that of mCCG methylation in R NIL (at 96 hai), suggesting differences in methylation context in NILs with and without Lr28. The results of the present study improved our understanding of the epigenetic control of leaf rust resistance in wheat.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Wheat is one of the most important crops world-wide; its production has shown remarkable growth during the last 50 years, both globally and in South-East Asia. However, a sharp decline in average annual growth in production from ~ 3% in the past to ~ 0.7% in recent decades has been witnessed. This is a matter of concern, keeping in view the projected global demand for substantial increase in wheat production by the year 2050 [1]. The decline in the growth of wheat production is attributed to a number of biotic and abiotic stresses [2]. Among biotic stresses, leaf rust of wheat (also described as brown rust) caused by Puccinia triticina is an important disease, which is responsible for major loss in grain yield [3]. As many as ~ 80 Lr genes (Lr1–Lr80) conferring resistance to leaf rust in wheat are known [4]; these genes include those conferring race-specific resistance and also those for non-race specific adult plant resistance (APR).
During the last two decades, significant progress has also been made in understanding the molecular mechanism of disease resistance including leaf rust in wheat. For leaf rust, four Lr genes (Lr1, Lr10, Lr21, Lr34) were cloned and characterized, providing a better understanding of the molecular mechanism of leaf rust resistance in wheat [5,6,7,8]. Recently, avirulence genes (Avr) interacting with two Sr genes in wheat (Sr35 and Sr50) were also identified [9, 10], providing information about specific gene-for-gene interactions between Sr genes and Avr genes of the pathogen causing stem rust. However, no Avr gene against any Lr gene in wheat has so far been identified or cloned, which is therefore an important area for future research. In Arabidopsis, recent evidence suggested formation of a resistosome through oligomerization of nucleotide binding leucine rich repeat containing nod-like receptor (NLR), named Hopz Activated Resistance 1 (ZAR1) with other unique proteins during infection by the microbial pathogen Xanthomonas campestris bacteria [11]. Formation of such a resistosome may turn out to be a common feature in all pathosystems in crop plants including leaf rust in wheat. Another major recent finding is that a TIR (Toll/Interleukin receptor) domain in NLR protein functions as NADase for cleavage of NAD+ and is responsible for cell death leading to hypersensitive reaction during infection by biotrophs like leaf rust pathogen [12, 13].
There is a growing realization that resistance to diseases in plants has an element of epigenetic control in addition to the classical genetic control through Mendelian R genes. One of these important epigenetic marks is cytosine methylation in DNA, which is known to play an important role during plant development by regulating gene expression [14, 15]. In plants, it occurs at CG, CHG and CHH islands (C and G indicate cytosine and guanine, respectively, and H indicates any nucleotide other than guanine) and differs from that in mammals, where it occurs mainly at CG sites and only rarely it occurs at non-CG sites [16, 17]. The regulation of gene expression by DNA methylation mainly occurs by silencing specific genes either at the transcriptional level or at the post-transcriptional level. The former involves inhibition of transcription due to hypermethylation in the promoter region, while the latter involves post-transcriptional degradation of mRNA using non-coding RNAs [18]. DNA methylation occurs both in gene body and intergenic regions, but its relationship with gene expression is not well understood. Generally, gene body methylation is known to induce gene expression rather than its suppression, involving alternate splicing [19]. Similarly, the role of intergenic DNA methylation in prevention of aberrant gene transcription has been reported in Arabidopsis [20]. Thus, the relationship of DNA methylation with gene expression seems to be quite complex as shown in rice during drought stress [21]. Similar complexity was also reported in humans [22].
Differential DNA methylation in any plant system can be studied using several techniques including methylation sensitive amplified polymorphism (MSAP), methylated DNA immunoprecipitation (MeDIP) and bisulphite sequencing (BiS-Seq). MSAP is a modified amplified fragment length polymorphism (AFLP) technique [23], while MeDIP and BiS-Seq require DNA sequencing. MSAP makes use of a pair of isoschizomers like HpaII and MspI, which recognize the same restriction site (CCGG), but differ in their sensitivity to methylated cytosine residues [23]. MSAP is also relatively cost-effective, since no sequencing is involved, but it has low resolution and does not give information about cytosine methylation at individual nucleotide level.
In wheat, several reports of the study of differential DNA methylation using each of the above three approaches are available, both at the whole genome level and at specific gene level. For instance, MSAP was used in at least three studies in wheat [24,25,26]. One of these studies also reported methylation pattern due to two Lr genes, namely Lr19 and Lr41 [25]. MeDIP has also been used for exploring the epigenomic landscape along chromosome 3B under the Epi3B project that was launched at INRA in France [Joint EUCARPIA Cereal Section & International Triticeae Mapping Initiative (I·T·M·I) Conference, International Wheat Genome Sequencing Consortium (IWGSC) workshop 29-06-2014)]; the results of this work on Epi3B were not published (personal communication). BiS-Seq has been used in at least three studies to examine differential methylation at the whole genome level [25, 27, 28]. In view of the large genome of wheat, sometimes genome complexity reduction techniques such as “sequence capture” was also used in combination with Methyl-Seq, a method based on ‘Agilent SureSelect Methyl-Seq’ [29]. Genome-wide differential methylation patterns in wheat have been examined for a variety of traits including resistance to powdery mildew [30], vernalization [31] and seed germination [32]. Studies on differential methylation of individual genes were also conducted, as in case of photoperiod insensitivity gene Ppd-B1a [33]. Studies were also conducted on methylation patterns due to aneuploidy/alien chromatin introgression [34, 35] and polyploidization [36, 37]. Obviously, interest in DNA methylation studies has increased in recent years.
The gene Lr28 is an alien gene from Aegilops speltoides Tausch that confers resistance against the most prevalent Indian leaf rust pathotype 77-5. Therefore, this gene has been deployed for developing several leaf rust resistant Indian wheat cultivars (e.g. MACS6145), although the resistance imparted by Lr28 has recently been overcome by a new pathotype (121R60-1 = 77−10) [38]. However, relatively fewer studies have been conducted to understand the molecular mechanism underlying leaf rust resistance due to Lr28 [39,40,41,42,43]. In a recent study, the role of histone acetylation in regulating the expression of six genes during pathogen attack in the presence of Lr28 was also examined [44]. However, no studies on the role of DNA methylation in defence pathway due to Lr28 are available. The earlier methylation study on leaf rust involving two Lr genes (Lr19 and Lr41) was based on low-resolution MSAP approach [25]. In the present study, we examined differential methylation patterns associated with leaf rust resistance gene Lr28. For this purpose, differential gene expression due to DNA methylation was examined in a wheat cv. HD2329 and its resistant NIL (HD2329+Lr28). The study provides useful information about the role of differential methylation in regulation of gene expression in wheat-leaf rust pathosystem.
Material and methods
Origin of plant material
Initially the leaf rust resistance gene Lr28 was accidentally transferred from Aegilops speltoides into hexaploid wheat cv. Chinese Spring (CS) during attempts to transfer the gene Yr8 (a yellow rust resistance gene) from Aegilops comosa to common wheat [45]. Ae. speltoides was used only for the purpose of inducing homoeologous recombination between the wheat and Ae. comosa chromosomes (Ae speltoides suppresses the gene Ph1 and thus induces pairing between homoeologous chromosomes). A segment of chromosome 7S#2S of Ae. speltoides carrying Lr28 was translocated onto wheat chromosome 4AL resulting in a translocated chromosome T4AS.4AL-7S#2S [46]. Later, a donor stock CS2A/2M 4/2 containing Lr28 from Ae. speltoides and Yr8/Sr34 from Ae. comosa was used to transfer Lr28 into several Indian wheat cultivars (including HD2329) by Dr. M.K. Menon, IARI, Regional Station, The Nilgiris, Tamil Nadu, India [47, 48]. The resistant NIL HD2329+Lr28 was developed following seven backcrosses to a susceptible recurrent parent cv. HD2329. The susceptible cv. HD2329 and its resistant NIL (HD2329+Lr28) were used in the present study.
Inoculation of seedlings with P. triticina inoculum
Inoculation of seedlings with P. triticina inoculum was carried out following Sharma et al. [39]. For this purpose, the seedlings of the susceptible cv. HD2329 and resistant NIL HD2329+Lr28 were raised at the National Phytotron Facility, ICAR-Indian Agricultural Research Institute (ICAR-IARI), New Delhi, in growth-chambers under controlled conditions of 16 h day (25 °C) and 8 h night (18 °C) with 240 µmol m− 2 s− 1 light. P. triticina inoculum (urediniospores and talcum powder in 1:10 ratio) suspended in water (30 urediniospores per 10X microscopic area) was evenly spread on moistened open leaves of each of the 14-day old seedlings (two-leaf stage) with the help of a camel hair brush (0 size) (Michigan Brush Manufacturing Company Inc., New Delhi, India). The pots carrying the seedlings were well-watered and covered with water saturated polythene to maintain the desired level of relative humidity (RH > 95%). After inoculation, the seedlings were incubated in a humid chamber maintained at 23 ± 2 °C in the dark for a period of 36 h; normal conditions were restored thereafter
DNA isolation
Leaf samples for DNA isolation were collected from 14-day old seedlings of the two NILs at 0 hbi and 96 hai. The 96 hai stage was chosen on the basis of our earlier findings showing pale halo islands at the sites of infection on the leaves of the above susceptible cultivar and also due to our earlier observation of the abundance of differentially expressed transcripts between the susceptible cultivar and resistant NIL at 96 hai [40]. Genomic DNA was isolated using modified CTAB method [49] from leaf samples collected from two biological replicates of the following four treatments: (i) HD2329 (susceptible cultivar) at 0 hbi (S0); (ii) HD2329 (susceptible cultivar) at 96 hai (S96); (iii) HD2329+Lr28 (resistant NIL) at 0 hbi (R0), and (iv) HD2329+Lr28 (resistant NIL) at 96 hai (R96).
Methylation sensitive amplified polymorphism (MSAP)
MSAP analysis was carried out using two pairs of restriction endonucleases (EcoRI + HpaII and EcoRI + MspI) (New England Biolabs Inc.) for restriction digestion of DNA of the above four samples. HpaII and MspI represent a pair of isoschizomers; both can cleave the un-methylated restriction site 5′-CCGG-3′; HpaII also cleaves hemi-methylated external cytosine, mCCGG [50, 51], while MspI cleaves hemi- or fully methylated internal cytosine (CmCGG) but not hemi- and fully methylated external cytosine (mCCGG) or both external and internal cytosines (mCmCGG) of the restriction site CCGG.
Aliquots (500 ng) of DNA were digested for 2 h at 37 °C with 10 U each of EcoRI + MspI and EcoRI + HpaII to ensure complete digestion of DNA. This was followed by addition of 5 pmol EcoRI and 50 pmol of HpaII/MspI adapters (Xcelris Genomics, India) to the ligation mixture containing 1 unit of T4 DNA ligase and 1 × ligation buffer (New England Biolabs Inc.). The mixture was incubated at 37 °C for 3 h. The ligation mixture was then diluted 1:10 with TE buffer, and was used as a template for the pre-amplification with single nucleotide selective EcoRI-A primer (GACTGCGTACCAATTCA) and MspI/HpaII-T primer (ATCATGAGTCCTGCTCGGT). The PCR reaction was performed for 30 cycles of 45 s denaturation at 94 °C, 40 s annealing at 56 °C, and 40 s extension at 72 °C. The PCR amplified products were further diluted to 20-fold with TE buffer to use it as a template for selective amplification with each of the 27 pairs of selective primer combinations carrying three selective nucleotides (Supplementary Table 1). Subsequently, a mix of Gene scan 500ROX (range 35–500 bp; used as size standard) was added to the selective amplification product. The resulting mixture was used for capillary electrophoresis on ABI3730 DNA analyzer, Applied Biosystems, Life Technologies Pvt. Ltd., California, USA for resolving the amplified DNA fragments as peaks in the electropherograms. The results were analysed for four different MSAP profiles (Types I to IV) that are expected from the EcoRI + HpaII and EcoRI + MspI combinations (Table 1).
Scoring and analysis of electropherograms following MSAP
The amplified DNA fragments were visualized as peaks on electropherograms; fragment size in each case was determined with the help of the size standards using the software GeneMarker version 1.80. The presence and absence of peaks (amplified fragments) in each treatment were scored as 1 (for presence of peak) and 0 (for absence of peak) and methylation patterns of the restriction site 5′-CCGG-3′ were inferred as shown in Table 1 [51]. Whole genome level methylation was inferred on the basis of the total number of peaks (fragments) obtained in each treatment. The differential 5-methylcytosine peaks in two different treatment combinations were inferred on the basis of presence/absence of peaks.
Methylated DNA immunoprecipitation (MeDIP)
For MeDIP analysis, DNA of two biological replicates was pooled in equal concentrations for each of the four treatments as detailed above i.e. S0, S96, R0 and R96. MeDIP sequencing of the pooled immunoprecipitated DNA of each sample was undertaken to decipher the genome-wide DNA methylome. The sequencing data was obtained through outsourcing the sequencing work to SciGenome Pvt. Ltd., India. The protocol adapted by Li et al. [52] with few modifications was followed for preparing MeDIP-Seq libraries. Briefly, the following steps were involved in MeDIP sequencing: (i) 1 µg of the genomic DNA was sheared independently for each of the four treatments and end repaired to obtain sheared fragments with blunt ends. (ii) A single ‘A’ nucleotide was then added to the 3′ ends of the blunt fragments to prevent them from ligating to one another during the adapter ligation reaction. A corresponding single ‘T’ nucleotide on the 3′ end of the adapter provides a complementary overhang for ligating the adapter to the fragment. This strategy ensures a low rate of chimera (concatenated template) formation. (iii) The multiple indexing adapters were then ligated to the ends of the DNA fragments to prepare them for hybridization on to the flow cell. (iv) This was followed by immunoprecipitation using MeDIP buffer and 5mC specific monoclonal antibody (Methylamp Methylated DNA capture kit, Epigentek, USA). This immunoprecipitated DNA is supposed to be enriched for methylated DNA fragments, which had adapter molecules on both ends for amplification of the DNA in the library. (v) The PCR was performed with the PCR Primer Cocktail that annealed to the ends of the adapters. The number of PCR cycles was kept to the minimum to retain library representation. (vi) Qubit concentration of library was checked followed by quality check of the library; this was done by loading 1 µl of sample on an Agilent Technologies 2100 Bioanalyzer using a DNA-specific chip (e.g., Agilent DNA 1000 or DNA HS chip) and (vii) MeDIP library obtained thus was sequenced and the data were obtained in the form of FASTA files and reads quality checked using the following parameters: Base/sequence quality score: Phred score > 30; average base content per read: 100 bp; GC distribution: > 40% and over-represented sequences. Based on the quality of sequence reads, trimming was done, wherever necessary, to retain only high-quality sequence.
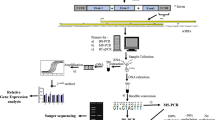
The pre-processed reads were aligned to the reference genome of wheat (Triticum aestivum) downloaded from https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Assemblies/ v1.0/. The alignment was performed using Bowtie program V.2 with default parameters. These alignment files (*.bam file) were used as input (i) for identification of methylated peaks using MACS2.0 (Model Based Analysis of ChIP-Seq data) software [21, 53] in all the four individual treatments (S0, R0, S96 and R96) and (ii) for extracting the differential methylated regions (DMRs) in the four different pairs of treatments (S0 vs S96, R0 vs R96, S0 vs R0 and S96 vs R96) using MEDIPS (R) package [21, 54, 55]. In all these four treatment comparisons, the second treatment was compared to first. The above-mentioned wheat genome reference sequence along with the bam files were used in MEDIPS to calculate the differential methylation between the two treatments of a pair at a genome-wide window of regular width of 500 bps. The DMRs following FDR < 0.05 and FC ≥ 2 or ≤ −2 (log FC ≥ 1 or ≤ −1) [21, 54] were considered to be differentially methylated and were used for further detailed analysis. The methylated peaks in individual treatments and also the DMRs between the four different pairs of treatments were assigned to different genomic regions [introns, exons, promoters (− 1 kb to + 100 bp), transcription termination sites (TTS) (− 100 bp to + 1 kb) and intergenic regions] using Homer software [56] and latest version of wheat reference sequence annotation v1.1 available at IWGSC (https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Annotations /v1.1/). The pipeline followed for analysis of data is available in Supplementary Fig. 1.
Gene ontology (GO) analysis and identification of transposable elements
GO analysis was carried out using Blast2GO [57] and differentially methylated genes (DMGs) (introns, exons, promoters and TTS) were assigned to cellular localisations, molecular functions and biological processes. For identifying transposable elements (TEs) in differentially methylated regions (DMRs), the nucleotide sequences of all the DMRs distributed in different genomic regions (exons, introns, promoters, TTS and intergenic) were used as input in the online tool Censor [58] (https://www.girinst.org/censor/). The TEs (including LTR = long terminal repeats and non-LTRs) and simple repeats were identified in the DMRs of all the four treatment pairs.
Relationship between methylation and gene expression
The DMGs identified using MeDIP analysis were compared with differentially expressed genes (DEGs) to identify the genes undergoing both methylation and differential expression and to establish the relationship between them. For this purpose, RNA-Seq data were available for all the four treatment pairs; differentially expressed genes were also available for the four treatment pairs that were used in MeDIP. The fold changes in methylation of the genic regions (intron, exon, promoter and TTS) of DEGs were compared to the fold changes in expression of the corresponding DEGs. The fold change in methylation levels of genes showing differential methylation in more than one out of the above four genic regions were merged; average fold change in methylation was calculated in each case for comparison with corresponding fold change in expression that was available from RNA-Seq data.
RNA isolation and qRT-PCR analysis: validation of DMGs
Quantitative real time PCR (qRT-PCR) analysis was performed for comparing abundance of gene-specific transcripts (based on RNA-Seq data) with methylation patterns. For this purpose, first, total RNA was isolated from the 14-days old seedlings of susceptible cv. HD2329 and its resistant NIL (HD2329+Lr28) at 0 hbi and 96 hai using the TRIzol Reagent (Ambion) as per the manufacturer’s specifications. Then, cDNA synthesis was performed from the total RNA using Verso cDNA Synthesis Kit (Thermo Fisher Scientific, USA) with RT enhancer, to remove any contaminating DNA, according to the manufacturer’s instructions.
For conducting qRT-PCR, 18 DMGs were selected, which included 10 DMGs for which RNA-Seq data was also available (Table 2); these 18 genes were selected such that they contained domains involved in biotic/abiotic stress tolerance and they had more than twofold difference in methylation level. Primers for these DMGs were designed using Primer Express (ver. 3.0) software (Applied Biosystems; for details of primers see Supplementary Table 2). The qRT-PCR was performed using ABI 7500 Real-Time PCR System (Applied Biosystems). All reactions were performed using three biological and three technical replicates and the target gene expression (fold change) between two treatments was calculated using 2∆∆CTCT method by using actin as a reference gene for normalisation [59]. Heat maps were generated using online tool Clustvis (http://biit.cs.ut.ee/clustvis/) and were used to depict the fold changes in qRT-PCR results and differential methylation patterns following MeDIP analysis.
Results
MSAP analysis
Global methylation patterns using MSAP
The number of MSAP fragments amplified with 27 individual primer pairs in four individual treatments ranged from 4 (E-AGG/H-TTC) to 24 (E-ACG/H-TCA); the size of fragments ranged from 61 to 294 bp. The details of amplified fragments belonging to four different methylation patterns (Types I to IV in Table 1) are summarized in Table 3 (for details of primer pairs, see Supplementary Table 1).
Differential methylation detected using MSAP
Four treatments were compared in pairs (S0 vs S96; R0 vs R96; S0 vs R0; S96 vs R96) using four MSAP types (Type I to Type IV). A summary of this comparison is available in Table 4, where differences in methylation patterns with passage of time (0 hbi to 96 hai) and also between susceptible cultivar and its resistant NIL at the same time can be noticed. The results suggest that the number of differentially methylated fragments is almost the same (> 200) in all the four comparisons, suggesting that there are large-scale differences in methylation patterns in different pairs of comparisons. The data also suggest that there are differences in methylation patterns not only over time (0–96 h), but also due to presence or absence of Lr28 gene at the same time point. For instance, in the susceptible cultivar, presence of more sites show demethylation in S96, suggesting a general pattern that many more genes are activated (demethylated) than the number of genes, which may undergo repression during progression of infection in the susceptible cultivar. The trend seems to be opposite in the resistant NIL carrying Lr28 (R0 vs R96), suggesting that in resistant NIL, methylation increases due to the presence of Lr28, thus bringing about repression of many genes in the resistant NIL. However, the sites of methylation reduction in S96 and R0 with respect to S0 are different, suggesting that the genes that are activated in suscpetible and resistant lines should be different.
MSAP data also give some information about the methylation context, as evident from differences between T1 and T2 shown in Table 4. Major differences include mCCG to mCG in S0 vs S96 and in S0 vs R0 and mCG to mCCG in R0 vs R96 and S96 vs R96.
In summary, infection seems to be facilitated by demethylation (activation of more genes) and increased mCG, while resistance is facilitated by methylation (repression of gene activity) associated with increased mCCG.
MeDIP analysis
MeDIP sequence data and methylated regions in four different treatments
The total number of sequence reads was 504, 428, 562; the proportions of sequences aligned to reference genome of wheat ranged from 80.12% (R0) to 93.12% (S96) with an overall value of 87.94% (Supplementary Table 3). Peak-calling gave 100,452 peaks with the minimum number of peaks (18,249) in R0, and the maximum number of peaks (32,722) in S96 (Table 5). The number of peaks on 21 individual chromosomes also varied (Fig. 1), but the pattern in four treatments seems to be the same (Fig. 1). The maximum number of peaks were available in the intergenic regions followed by promoters, TTSs and either exons or introns (for details see Table 5). A large number of peaks could not be assigned to any of the 21 chromosomes. The MeDIP raw reads of susceptible cv. HD2329 and resistant NIL HD2329 + Lr28 at 0 hbi and 96 hai were deposited in Sequence Read Archive (SRA), National Center for Biotechnology Information (NCBI) and can be accessed through Bioproject Database under the accession number PRJNA517568.

Variation in the number of methylated peaks identified in four different treatments and mapped to 21 chromsomes (chr1A to chr7D) of wheat using MACS 2.0 software. A large number of peaks could not be assigned to chromosomes; these are designated as chrUn (Un for unassigned). S0 = HD2329 at 0 hbi; S96 = HD2329 at 96 hai; R0 = HD2329+Lr28 at 0 hbi; R96 = HD2329+Lr28 at 96 hai
Differentially methylated regions (DMRs)
The data on DMRs are summarised in Fig. 2. It s apparent that the frequency of hypomethyalted DMRs increases with passage of time after inoculation both in susceptible cultivar and its resistant NIL, as apparent from comparison of S96 with S0 and that of R96 with R0. However, the frequency of hypermethylated DMRs is higher in resistant NIL relative to suscpetible cultivar at both the time-points (0 hbi, i.e. S0 vs. R0 and 96 hai, i.e. S96 vs. R96 ) (Fig. 2).

Variation in the number of diferentially methylated regions (DMRs) identified using MeDIP in four different comparisons (S0 vs S96, S0 vs R0, R0 vs R96 and S96 vs R96). DMRs were identified following the criteria FDR < 0.05 and FC ≥ 2 or ≤ − 2 (log FC ≥ 1 or ≤ − 1) (Wang et al. [21]; Hu et al. [54]) where FC ≥ 2 indicates hypermethylated (hyper) regions and FC ≤ − 2 indicates hypomethylated (hypo) regions. S0 = HD2329 at 0 hbi; S96 = HD2329 at 96 hai; R0 = HD2329+Lr28 at 0hbi; R96 = HD2329+Lr28 at 96 hai (in each case, the second treatment is compared with the first)
The number of unique DMRs were substantially higher in S0 vs R0 (1467) and S96 vs R96 (1566) when compared to the remaining two treatment pairs. It is also apparent that there are more unique DMRs in susceptible cultivar (737) than in the resistant NIL (361) (Fig. 3).

Venn diagram showing distribution of the number of DMRs in four pairs of different treatments (S0 vs S96; R0 vs R96, S0 vs R0 and S96 vs R96). S0 = HD2329 at 0 hbi; S96 = HD2329 at 96 hai; R0 = HD2329+Lr28 at 0 hbi; R96 = HD2329+Lr28 at 96 hai (in each case, the second treatment is compared with the first)
Identification of genes in DMRs and distribution of methylated regions in coding and non-coding regions
DMRs were subjected to identification of individual genes; 1320 genes (common genes between different treatment pairs were counted only once) were identified. These genes belonged to diverse classes and encoded proteins including the following: proteins with NBS-LRR domain, protein kinases, serine threonine kinases, NADH ubiquitine/quinone reductase, glutathione-S-transferase, ferredoxin, cytochrome b6/f, transcription factors like WAK, Myb; transporters like citrate transporter, ABC transporter, methyltransferases like SAM dependent carboxyl methyltransferase, etc. Some of the genes were unique to an individual pair of treatments; others occurred in more than one treatment (Supplementary Table 4).
The DMRs and individual genes were also subjected to study of methylation in five different components of the genomic regions (introns, exons, promoters, TTS and intergenic regions). The results of this analysis are presented in Table 6 and further details are provided in Supplementary Tables 5–12. The pattern of methylation in DMRs did not differ, when examined at the level of individual genes. These results also corresponded with the overall genome-wide representation of hypermethylated and hypomethylated DMRs (Fig. 2).
Transposable elements and SSRs in DMRs
Transposable elements (TEs) and simple sequence repeats (SSRs) were also identified in the DMRs. TEs mainly included DNA transposons, EnSpm/CACTA, Harbinger, Mariner/Tc1, Hat, MuDR, LTR and non-LTR transposons (Table 7). Highest number of TEs and SSRs were identified in the DMRs of S96 vs R96, and the least were identified in R0 vs R96, suggesting that in the resistant NIL, the frequency of TEs in DMRs decreased with passage of time.
Functional annotation of genes associated with DMRs
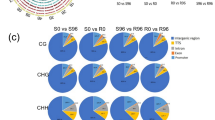
GO terms were available for 1130 of 1320 genes and were classified into cellular components, molecular function and biological processes. Proteins encoded by some of these genes also belonged to photosynthetic membranes, membrane protein complexes and cell organelles. When all the three categories of GO terms were compared, maximum GO terms were found for biological processes. In molecular functions, protein binding, heterocyclic ion binding and organic cyclic compound binding were the most predominant terms (Figs. 4 and 5).

Bar diagram showing results of GO annotation in the form of proportions of differentially hypermethylated genes (DMGs) in four different comparisons shown in the form of bars with four different colours (S0 vs S96 hyper, etc.) belonging to three catgories of functions: cellular componenet, molecular functions and bilogical proceses of hypermethylated genes (in each case, the second treatment was compared with first treatment) in four different treatment pairs. S0 = HD2329 at 0hbi; S96 = HD2329 at 96 hai; R0 = HD2329+Lr28 at 0hbi; R96 = HD2329+Lr28 at 96 hai (in each case, the second treatment is compared with the first)

Bar diagram showing results of GO annotation in the form of proportions of differentially hypomethylated genes (DMGs) in four different comparisons shown in the form of bars with four different colours (S0 vs S96 hyper, etc.) belonging to three catgories of functions (cellular component, molecular function and biological process) (in each case the second treatment is compared to first treatment). S0 = HD2329 at 0 hbi; S96 = HD2329 at 96 hai; R0 = HD2329+Lr28 at 0 hbi; R96 = HD2329+Lr28 at 96 hai (in each case, the second treatment is compared with the first)
Relationship of gene expression and methylation
The MeDIP samples were also used for RNA-Seq, the data of which was used for a comparison with DMRs. Only 214 of the 1320 genes identified from DMRs were available in RNA-Seq data for comparison (Table 8). A proportion of these 214 genes were exclusively methylated in exons (7), introns (3), promoters (75) and TTS (46), whereas the remaining genes were methylated in more than one genomic region.
Out of the 214 genes, methylation in 88 genes showed negative relationship with gene expression, whereas remaining 126 genes showed positive relationship. A representative heat map showing the relationship of gene expression with methylation in S96 vs R96 is shown in Fig. 6; data for all the four treatment pairs is given in Supplementary Tables 13–16. Genes in four treatment pairs encoded mainly for the following proteins: ribosomal proteins, photosystem genes, cytochrome genes, ATPase, and NADH oxidoreductase, heat shock proteins, etc.

Heatmap showing the relationship between methylation and gene expression for some important genes (relevant to disease resistance) in S96 vs R96 (for each row, name of genes given on the right of the heatmap). The scale on the extreme right shows the levels of methylation/gene expression (dark red shade indicates maximum change in expression/methylation whereas dark blue shade indicates minimum change in methylation/gene expression). Both for methylation and gene expression, DMGs and DEGs showing FC ≥ 2 (log FC ≥ 1) were considered as hypermethylated/upregulated and FC ≤ − 2 (log FC ≤ − 1) were considered as hypomethylated/downregulated, respectively. The letters mentioned in parenthesis at the end of the name of each gene indicates genomic regions as follows: P promoter, TTS transcription termination site, E exon, I intron. S96 = HD2329 at 96 hai; R96 = HD2329+Lr28 at 96 hai.
Quantitative real time PCR analysis
QRT-PCR was conducted for 18 selected DMGs, which differed in methylation patterns in different treatments as detailed in Table 1. For 10 of these 18 DMGs, RNA-Seq data was also available. qRT-PCR results for only four of these 10 DMGs matched the expression pattern inferred from RNA-Seq data; these genes included genes containing domains for MADS box, NADH-quinone oxidoreductase, cytochrome b6/f complex and photosystem I. These genes showed abundance in RNA-Seq as well as qRT-PCR data and were hypomethylated. The remaining six genes showed opposite pattern during qRT-PCR and RNA-Seq (Fig. 7).

Heatmap showing the methylation and gene expression changes in a set of genes in all the four treatment pairs (S0 vs S96, S0 vs R0, R0 vs R96 and S96 vs R96). The genes indicated with RNA-Seq are the common differentially expressing genes available in transcriptome data and Up and Down indicate the change in the level of expression i.e. upregulated and downregulated. The scale shown at the top left corner indicates the methylation/expression fold changes. The heat map is drawn using online tool Clustvis with each row subjected to univariate scaling in order to represent the fold changes in each treatment pair. In each treatment pair, the expression/methylation fold change is indicated in second treatment relative to first treatment
For the remaining eight of the 18 DMGs, for which RNA-Seq data was not available, qRT-PCR results were compared with only methylation patterns. For only three of these eight DMGs, qRT-PCR results matched with methylation patterns assuming that methylation leads to repression of gene expression; these three genes included genes encoding ATPase, UDP glucosyl transferase and NBS-LRR (Fig. 7)
Discussion
Molecular mechanism involved in disease resistance in plants (including leaf rust in wheat) has been a subject of intensive study. A two-layer system of plant immunity involving PTI and ETI has been widely accepted. Several earlier studies and our own studies (including the present study) involving Lr28 gene for leaf rust resistance in wheat suggest that the presence of the pathogen is perceived by the host cells with the help of receptor molecules of some kind [38, 39, 41]. This is apparently followed by activation of many genes and repression of perhaps fewer genes, which start operating in more than one defence pathways that are interconnected in a complex manner to provide protection. Among these genes, the most extensively studied genes are R genes which generally encode proteins containing nucleotide-binding site leucine-rich repeat i.e. NBS-LRR or nod-like receptors (NLR) domains, although a variety of following kinases are also known to be involved: receptor like kinases (RLKs), mitogen associated protein (MAP) kinases, wall associated kinases (WAK), etc. Calcium ion fluxes, generation of ROS and oxidative burst have also been suggested to be involved to enforce physical barriers by cell wall appositions and causing hypersensitive response (HR). An oligomerization of NLR with other proteins leading to the formation of a resistosome/inflammasome has also been suggested in a recent study [11]. More recently, presence of NLR-TIR domain carrying NADase activity for cleavage of NAD+ has also been suggested to play a role in cell death and HR [12, 13]. Expression of all these genes/protein domains needs to be regulated in an intricate but precise manner to provide defence on pathogen infection. We believe that this regulation is partly achieved through epigenetic modifications including reversible DNA methylation and demethylation, histone modifications and non-coding RNAs. The present study is focussed on DNA methylation as a component of epigenetic modifications, which was examined using MSAP and MeDIP.
Resolution of DNA methylation using MSAP
MSAP allowed us to analyse differentially methylated sites (DMSs) and also provided some information about methylation at CG and CCG (CHG) contexts; a similar analysis was earlier conducted in tobacco [60]. MSAP results also allowed us to study changes in methylation contexts from mCG to mCCG and vice versa in both S and R lines during 96 hai. Alteration of mCCG to mCG seems to be the most conspicuous change followed by the reverse methylation contexts, i.e. mCG to mCCG (Table 4). This is also in agreement with earlier study in wheat where CG methylation was reported to be most abundant followed by CHG (H = C in the present study) and CHH methylation [24]. Methylation in intergenic regions was particularly observed in TEs (LTR and non-LTR retrotransposons, DNA transposons) and SSRs (Table 7). In earlier studies, the role of non-CG methylation in addition to CG methylation was also observed in case of Arabidopsis [61], rice [62] and wheat [24]. In Arabidopsis, it was also shown that gene body methylation mainly occurs in CG, although it also occurs at CHH and CHG in intergenic regions.
One of the limitations of MSAP is that exact methylation changes (methylation or demethylation) cannot be inferred due to the difficult interpretation of Type IV methylation pattern [63, 64]. Some researchers earlier considered type IV pattern as informative [65, 66], while others considered it to be uninformative and hence ignored it for inferring methylation from MSAP [63, 64]. However, in the present study, type IV was most abundant followed by type II, type III and type I patterns in that order (Table 3). Therefore, type IV results were critically examined. Further analysis revealed that during the passage of time after inoculation, type IV was changed to type II in majority of CCGG sites, suggesting demethylation of external cytosine or conversion of mCCGG to CmCGG; this change occurred in S cultivar during 96 hai (Table 4). A reverse situation leads to methylation of at least some CCGG sites or conversion of CmCGG to mCCGG (Table 4). Based on the available information of four different methylation patterns, changes from types III, II and IV to type I or type IV to type III can be classified as demethylation and the opposite situations may be classified as methylation. This indicated occurrence of demethylation during the passage of 96 hai both in S (in S0 vs S96) and R lines (in S96 vs R96) (Table 4). Similar pattern in S cultivar was also observed with MeDIP, which also helped us to identify the DMRs and the corresponding genes (DMGs), which are involved in susceptibility or resistance. Methylation changes due to both the techniques are dicussed later in this “Discussion” section.
Resolution of DNA methylation using MeDIP
MeDIP allowed us to identify differentially methylated regions (DMRs). These DMRs also included those in the body of the genes, which are then designated by us as differentially methylated genes (DMGs); we also assume that in most DMRs/DMGs, methylation should be reversible thus playing a role in regulation of gene expression. However, it was not possible to identify individual methylated cytosines in specific genes, which requires bisulphite-sequencing [67]. In the present study, the distribution of methylation in different genomic regions (like introns, exons, promoter, TTS, intergenic) was also found to be important, since the effect of methylation in promoter sequences differs from the effect of methylation in other parts of the genome. In the present study, 75–94% DMRs were present in regions outside the promoter regions (1 kb upstream of TSS). Similar results were reported earlier in the Arabidopsis-Pseudomonas syringae pathosystem [68]. Such DMRs have also been shown to have a role in limiting transcription and in providing stability to the TEs [69]. Hypermethylated DMRs containing TEs were particularly conspicuous in R NIL relative to S cultivar (S96 vs R96) (Table 7), which may be associated with the repression of neighbouring genes, as also reported earlier [70]. Differential methylation in promoters and TTS in the present study also seem to be in agreement with the results of earlier studies in maize [71] and rice [21].
Methylation versus gene expression
The DMGs were also compared with differentially expressed genes (DEGs) identified in the RNA-Seq data. Only 16% (214 out of 1320 genes) DMGs were available in RNA-Seq data indicating only a partial role of DNA methylation in regulation of genes involved in defense against leaf rust (Table 8). Obviously, several other genetic and epigenetic factors should be involved in regulating leaf rust resistance in wheat. Further comparison revealed that for 88 of the 214 DMGs (Supplementary Tables 13–16), the expression pattern matched with methylation results assuming that hypermethylation was associated with repression of gene expression and hypomethylation was associated with enhanced gene expression. In 20 of these 88 genes, exclusive methylation of the promoter region suppressed the gene expression, which is in agreement with earlier observations [72, 73]. In the remaining 68 DMGs, besides the promoters, specific regions of gene body were also differentailly methylated. Therefore, specific role of the promoter methylation in regulation of gene expression seems to be limited.
For the remaining 126 of the 214 DMGs, hypermetylation was associated with higher gene expression, while hypomethylation was associated with repression of gene expression. This was contrary to the generally expected pattern. Similar results, however, were also reported in rice under drought stress [21], in maize under salt stress [71] and in other plant systems like Populas trichocarpa [74] and apple [75]. Overall, we conclude that resistance to leaf rust is influenced by expression of a number of genes partly mediated by DNA methylation. This methylation may occur exclusively in promoter region or in promoter as well as other genomic regions (introns. exons and TTS).
On the basis of a comparison of RNA-Seq data (earlier data and the data obtained in the present study) and the methylation data obtained from MeDIP samples used in the present study, we believe that the widely accepted belief that hypermethylation leads to repression and hypomethylation leads to activation of genes is an oversimplification, and that this generalization is applicable only in a small fraction of DMGs. qRT-PCR results also support this observation since only four out of the eight DMGs showed the expected negative relationship of methylation with gene expression. Role of some of these DMGs in response to biotic stresses in wheat or other cereals is known from published literature (summarized later in Table 9).
The details of the complexity associated with infection and immunity processes and involvement of DNA methylation in these processes have also been widely discussed in published literature and are summarized in a recent review [76]. We believe that differential methylation of a number of genes in the two lines used in the present study, is largely due to the presence or absence of Lr28 gene, although some differences in the genetic background between the two lines and the associated downstream pathways may also be responsible. However, we believe that the leaf rust resistance gene Lr28 had no harmful effect due to possible linkage drag as revealed in an earlier report where the two lines used in the present study were compared for yield related and bread making quality traits and no deleterious effects due to the resistance gene Lr28 were observed [77]. A summary of the earlier studies that are relevant to the discussion is presented in Table 9; the results of the present study are discussed in the following text.
In the past a role of differential methylation in incompatible/compatible interactions has been reported in different host-pathogen systems. First, hypomethylation in the presence of powdery mildew resistance gene was reported in wheat [30]. Second, MSAP revealed a role of hyper-methylation in bacterial blight adult plant resistance in rice [15]. Third, in Arabidopsis, hyper/hypomethylation was shown to be associated with compatible/incompatible interactions involving a number of pathogens including P. syringae, Plectsphaerella cucumerina, Alternaria brassicicola, Hyaloperonospora arabidopsidis and Fusraium oxysporium [78, 79]; for review, see [76] Deleris et al.).
Hypomethylation of many and hypermethylation of few but different sets of genes involved in susceptibility (S0 vs S96) and resistance (R0 vs R96)
The results of the present study seem to suggest that both hypomethylation and hypermethylation of different sets of genes occur during S0 to S96 and R0 to R96. Our results also suggest that in both susceptible cultivar and resistant NIL, many genes get hypomethylated during passage of time after inoculation. Interestingly, only eight DMGs were common between the susceptible cultivar and resistant NIL indicating that resistance and susceptibility are mainly governed by activation of different sets of unique genes (160 in resistant NIL and 56 in susceptible cultivar). Assuming that hypermethylation leads to repression and hypomethylation leads to activation of genes, these results can be used to infer that activation of different sets of unique genes is involved in resistant and susceptible reactions. However, this inference needs to be treated with caution as hypomethylation does not always lead to gene activation and similarly, hypermethylation does not always leads to suppression of gene expression as reported earlier and also revealed by comparison of methylation with gene expression during the present study [21, 71, 74, 75].
Unique hypomethylated genes in susceptible S0 vs S96 and resistant R0 vs R96 reactions
The unique DMGs in the susceptible cultivar perhaps facilitate pathogen infection (assuming hypomethylation leads to gene activation) (Supplementary Table 4). Except PR6 (Protease inhibitor Birk I)and dirigent protein coding genes, which showed up-regulation in expression data, no other gene was available in differential RNA-Seq expression data. Some of these unique DMGs were earlier reported to show induced expression in susceptible genotypes and thus help the pathogen for survival and multiplication on the host. For instance, PR6 (protease inhibitor) was highly expressed in susceptible wheat cultivars when infected with Fusarium graminearum [80]. Similarly, a dirigent protein coding gene had high expression during compatible interaction between Vitis vinifera and Erysiphe necator, causing powdery mildew [81]; dirigent proteins are also widely known to be associated with resistance against biotic and abiotic stresses [82, 83]. Overexpression of the gene encoding WRKY7 TF was also shown to repress the expression of defence related genes encoding PR1 proteins during compatible intercation of P. syringaea in Arabidopsis [84].
The unique DMGs which were identified in the resistant NIL with progress of infection are perhaps involved in the defence response in the resistant NIL. The details of such unique DMGs are provided in Supplementary Table 4. Some of these DMGs like ribosomal protein L2, photosystem I and ATPase were also available in RNA-Seq data where they showed upregulation. Some DMGs which showed hypomethylation mediated up-regulation as examined by qRT-PCR analysis included cytochrome C oxidase, photosystem I, and cytochrome b6/f complex. Some of these genes were earlier reported to be involved in defence response against different biotic stresses. Following are two such examples: (i) In Arabidopsis, Attrxh5 which is a type of cytosolic thioredoxin was shown to be induced due to wounding, abscission, senescence as well as during incompatible interaction between Arabidopsis and bacterial pathogen P. syringeae [85]. The role of thioredoxins in oxidative stress mediated redox signalling in plant immunity is reviewed in detail by Perez and Spoel [86]. (ii) Bulb type lectin domain containing genes (B-Lec) belong to a class of lectin like receptor kinases (Lec-RLKs) which are known to play a critical role during stress conditions, besides plant growth and development. In wheat, a number of different types of Lec-RLKs were characterised and about 120 different types of B-Lec encoding genes were characterised and their expression was found to be affected due to inoculation with Blumeria graminis and Puccinia striiformis [87]. The roles of some other unique DMGs in resistance or susceptibility are summarised in Table 9.
Unique hypermethylated genes in susceptible cultivar (S0 vs S96) and resistant NIL (R0 vs R96)
Assuming that the hypermethylation leads to repression of gene activity, it seems that the genes which get hypermethylated in susceptible cultivar or resistant NIL perhaps negatively regulate susceptibility or resistance. Majority of the hypermethylated DMGs in susceptible cultivar and resistant NIL were unique (28 in S0 vs S96 and 43 in R0 vs R96) to either of the two treatment pairs except four DMGs, which were common to both. At least some of these genes (for details, refer to Supplementary Table 4) are known to provide resistance when over expressed indicating that their repression will lead to susceptibility. For instance, (i) RRBI2-3 which is a type of Bowman Birk trypsin inhibitor when overexpressed in rice, provided resistance against fungal pathogen Pyricularia oryzae indicating that its repression would lead to susceptibility in rice [88]. (ii) In Arabidopsis, the mutants of MAX2 (a type of F-box protein), showed increased susceptibility to bacterial pathogen like Pectobacterium carotovorum, and fungal hemi-biotrophic pathogen like P. syringeae [89].
Similarly, the unique hypermethylated genes in the resistant reaction (Supplementary Table 4) which were earlier reported to induce resistance through negative regulation include genes involved in photosynthesis like cyt b6/f and photosystem II which exhibited decreased expression in response to P. syringeae infection in soybean leading to hypersensitive reaction associated resistance. This resistance was induced due to damage in the photosynthesis PSII system which leads to oxidative burst leading to expression of other defence related genes [90]. Many of the domains contained in hypermethylated genes are similar to the domains of the genes which underwent hypomethylation, indicating similar function although the actual genes differ in two cases
Hypermethylation of many and hypomethylation of few genes due to the presence of Lr28 (S0 vs R0 and S96 vs R96)
Our results also suggest that methylation status of the DMRs differ between resistant NIL and susceptible cultivar merely due to the presence of Lr28 in the resistant NIL (Fig. 2). Thus, a large number of DMGs showed hypermethylation (423 in S0 vs R0 and 298 in S96 vs R96) and few DMGs showed hypomethylation (9 in S0 vs R0 and 167 in S96 vs R96) in the resistant NIL. Further, between the two treatment pairs, there were 167 common hypermethylated DMGs and only one common hypomethylated DMG in the resistant NIL.
Hyper-methylated genes due to the presence of Lr28
The hypermethylated DMGs perhaps may act as negative regulators of leaf rust resistance assuming that they get repressed due to the presence of Lr28. The 177 common hypermethylated DMGs involving the treatment pairs S0 vs R0 and S96 vs R96 as well as the unique DMGs identified in R0 (relative to S0) may be perhaps involved in preparing the host for future attack by pathogen; these may act as constitutive barrier as also reported earlier in wheat involving powdery mildew resistance gene [91]. Ninety six DMGs identified in resistant NIL in the absence of infection (R0) also showed association with DEGs in RNA-Seq data. Some of these genes contained domains for tetratricopeptide repeat 1, ribosomal proteins L23/L25, ATPase, ATP synthase, clp protease, etc. (Supplementary Table 4).
The unique DMGs that are hypermethylated in resistant NIL in the treatment comparison S96 vs R96 may be actually involved in defence response against leaf rust disease. These DMGs contain the domains for lipid transfer protein, AP2/ERF, clp protease proteolytic subunit B3 domain binding domain, a cysteine peptidase, wall associated receptor kinase, etc. (Supplementary Table 4).These DMGs perhaps act as negative regulators of leaf rust resistance assuming that hypermethylation of these genes leads to silencing of genes. For instance, (i) in rice, the loss of function mutant of WAK112d was shown to induce resistance against M. oryzae [92] (Table 9); (ii) in rice, RNAi knockdown of TFs with AP2/ERF domain (e.g. OsERF109) resulted in increased drought tolerance [93]; this effect seems to be due to altered ethylene biosynthesis. In Arabidopsis also, AtERF014 negatively regulate resistance against necrotropic fungus Botrytis cineria where the knockdown of this gene gave increased resistance [94]; (iii) in wheat, a lipid transfer protein (TaDIR1-2) was shown to confer resistance against Puccinia striiformis, since knockdown of this gene has been reported to enhance disease resistance by modulating ROS and/or SA signalling [95].
Hypomethylated genes due to the presence of Lr28
There were eight DMGs in R0 but 39 DMGs in R96; only one DMG being common. Some unique genes in R96 which also showed upregulation in RNA-Seq data include those encoding glutathione transferase or GST, glutaredoxin, NBS-LRR (NLR), ABC transporter, lectin domain containing protein kinase, UDP-glucosyltransferase, etc. (Supplementary Tables 13–16). Activation of these genes also seems to provide enhanced resistance. Several examples are available from published literature, where some of these genes were shown to be involved in providing resistance against plant pathogens. Following are some examples: (i) the role of GSTs in providing resistance against bacterial, fungal and viral diseases is widely known (for details see review by Gullner et al. [96]; also see Table 9). GSTs perhaps act as ROS scavenging enzymes and thus help in enhancing Lr28 mediated leaf rust resistance. (ii) Glutaredoxins represent another group of oxidoreductases that are involved in controlling ROS. Genes from both these classes exhibited higher expression in the incompatible interaction in our earlier study involving transcriptome analysis [39], thus indicating that ROS homeostasis is an important mechanism imparting leaf rust resistance in wheat. (iii) Genes encoding proteins with LRR repeats are also important components of receptor like kinases and are reported to exhibit higher expression during incompatible interaction involving the wheat-leaf rust pathosystem involving Lr28 [11, 39, 41, 42]. (iv) Adult plant leaf rust resistant gene Lr34 is also known to encode for ABC transporter [97]. (v) Genes encoding proteins containing lectin domain were found to be strongly upregulated in resistant genotypes for fusarium head blight [98]. Thus, although the role of all these genes in imparting disease resistance was known, but perhaps it is for the first time, that evidence of regulation of the expression of all these genes through hypomethylation is being reported in the present study.
DMGs involved in biochemical and physiological pathways
We discussed above the differential methylation of some specific genes that are apparently involved in resistance (F-box, glutaredoxin, kelch repeats, S/TPK, etc.). However, there were other differentially methylated genes that are known to be involved in physiological (photosynthesis and electron transport) and biochemical pathways (oxidative stress, growth hormone response) and also in plant development. For instance, genes encoding PS1, PSII and NADH- Ubiquitone Plasto Quinone Oxidoreductase that were found to be differentially methylated are known to be involved in photosynthesis and electron transport. In earlier studies, expression of these genes was shown to be drastically affected during biotic stresses [99] but also during abiotic stresses like cold, salt, etc. [100]. In case of rice also, many genes involved in photosynthesis, oxidative stress, transport and other metabolic processes have been reported to be differentially methylated due to drought stress [21]. Similarly, oxidative stress response genes like glutathione S transferase encoding gene was also differentially methylated in resistant reaction during the present study. This gene helps in the protection of cells from oxidative damage in plants during various abiotic (reviewed in [101]) and biotic stresses (reviewed in [96]). Examples of methylation of genes involved in plant development were also available. For instance, a hormone regulated gene KNOX1, that is known to be involved in leaf development [102] was hypermethylated during incompatible interaction, so that its silencing may perhaps impair plant development, thereby rendering the plant susceptible.
Some specific features related to resistance due to Lr28
Overall, the results of DNA methylation in resistant NIL (R) and susceptible cultivar (S) suggest some specific features which may be mainly related to resistance conferred by Lr28 gene. Some of these specific features include the following: (a) Alteration of mCG to mCCG which is more conspicuous during resistance as revelaed through MSAP analysis in treatment pairs R0 vs R96 and S96 vs R96. (b) Hypomethylation of different sets of genes seems to be involved in imparting susceptibility and resistance. Some important DMGs which are specifically involved in imparting leaf rust resistance are those carrying domains for ATP synthase, ATPase, NBS-LRR, bulb type lectin, clp protease, cytochrome c oxidase, isoprenoid synthase, cytochrome C oxidase, photosystem I, and cytochrome b6/f complex, etc. (Supplementary Table 4). Other DMGs that may also be responsible for resistance include those which get uniquely hypermethylated in resistant NIL in the treatment pair S96 vs R96 which include genes containing domains for ABC transporter, glutathione S transferase, glutaredoxin, lectin domain containing receptor kinases, etc. (Supplementary Table 4). Even the hypermethylated DMRs containing SSRs and transposable elements are more abundant in R96 (relative to S96) (Table 7).
Conclusions
The following important conclusions were made, based on the present study: (i) Both MSAP and MeDIP approaches can be used to identify specific DMRs involved in wheat-leaf rust pathosystem. However, complete information about the role of different methylation contexts (CG, CHG and CHH) could not be deciphered, although MSAP provided some information about CCG and CG contexts. Only future studies involving bisulfite sequencing (BiS-Seq) may give better insights into the role of different methylation contexts in wheat-leaf rust interaction. BiS-Seq may also provide validation for the role of differentially methylated genes that were related with rust resistance during the present study (such BiS-Seq using the same genotypes is currently underway in our lab). (ii) Only some DMGs could be related with resistance/susceptibility, since only 16% of DMGs were available in expression data (RNA-Seq). Some of these genes seem to have a direct role in disease resistance, while some others were apparently involved in physiological or biochemical pathways that may indirectly influence disease resistance/susceptibility. (iii) Differential methylation was identified in both intergenic and intragenic regions, but methylation in intergenic region is more abundant followed by promoter/TTS/intron and exon. (iv) Since only 16% DMGs match the RNA-Seq based gene expression data, it was concluded that other epigenetic factors like histone modifications, non-coding RNA (miRNA and lncRNA) may also be involved in partial epigenetic control of leaf rust infection. (v) Since DMRs carried a large number of TEs, it is possible that a large fraction of DMRs be involved in maintaining genome stability, probably through silencing the TEs. (vi) Since DNA methylation was found to be associated with positive (unexpected) as well as negative (expected) gene expression, we inferred that DNA methylation may not always repress gene expression during leaf rust resistance
References
Ray DK, Mueller ND, West PC, Foley JA (2013) Yield trends are insufficient to double global crop production by 2050. PLoS ONE 8:e66428
Curtis T, Halford NG (2014) Food security: the challenge of increasing wheat yield and the importance of not comprising food security. Ann Appl Biol 164:354–372
Draz I, Abou-Elseoud MS, Kamara AEM, Alaa-Eldein OA, El-Bebany A (2015) Screening of wheat genotypes for leaf rust resistance along with grain yield. Ann Agric Sci 60:29–39
McIntosh RA, Dubcovsky J, Rogers WJ, Morris C, Xia XC (2017) Catalogue of gene symbols for wheat: 2017 supplement. [Verified 12 April 2018] 36:103–110. https://shigen.nig.ac.jp/wheat/komugi/genes/macgene/supplement2017.pdf
Feuillet C, Travella S, Stein N, Albar L, Nublat A, Keller B (2003) Map-based isolation of the leaf rust disease resistance gene Lr10 from the hexaploidy wheat (Triticum aestivum L.) genome. Proc Natl Acad of Sci USA 100:15253–15258
Huang L, Brooks SA, Li W, Felers JP, Trick HN, Gill BS (2003) Map-based cloning of leaf rust resistance gene Lr21 from the large and polyploid genome of bread wheat. Genetics 164:655–664
Cloutier S, McCallum BD, Loutre C, Banks TW, Wicker T et al (2007) Leaf rust resistance gene Lr1, isolated from bread wheat (Triticum aestivum L.) is a member of the large psr567 gene family. Plant Mol Biol 65:93–106
Krattinger SG, Lagudah ES, Spielmeyer W, Singh RP, Huerta-Espino J, McFadden H et al (2009) A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 323:1360–1363
Salcedo A, Rutter W, Wang S, Akhunova A, Bolus S, Chao S et al (2017) Variation in the AvrSr35 gene determines Sr35 resistance against wheat stem rust race Ug99. Science 358:1604–1606
Chen J, Upadhayay NM, Ortiz D, Sperschneider J, Bouton C, Breen S et al (2017) Loss of AvrSr50 by somatic exchange in stem rust leads to virulence for Sr50 resistance in wheat. Science 358:1607–1610
Wang J, Hu M, Wang J, Ki J, Han Z, Wang G, Qi Y, Wang HW, Zhou JM, Chai J (2019) Reconstitution and structure of a plant NLR resistosome conferring immunity. Science 364:eaav5870
Horsefield S, Burdett H, Zhang X, Manik MK, Shi Y, Chen J, Qi T et al (2019) NAD+ cleavage activity by animal and plant TIR domains in cell death pathways. Science 365:793–799
Wan L, Essuman K, Anderson RG, Sasaki Y, Monteiro F, Chung EH, Nishimura EO et al (2019) TIR domains of plant immune receptors are NAD+-cleaving enzymes that promote cell death. Science 365:799–803
Zhang M, Kimatu JN, Xu K, Liu B (2010) DNA cytosine methylation in plant development. J Genet Genomics 37:1–12
Sha AH, Lin XH, Huang JB, Zhang DP (2005) Analysis of DNA methylation related to rice adult plant resistance to bacterial blight based on methylation-sensitive AFLP (MSAP) analysis. Mol Genet Genomics 273:484–490
Kim MY, Zilberman D (2014) DNA methylation as a system of plant genomic immunity. Trends Plant Sci 19:320–326
Zhang H, Lang Z, Zhu JK (2018) Dynamics and function of DNA methylation in plants. Nat Rev Mol Cel Biol 19:489–506
Paszkowski J, Whitham SA (2001) Gene silencing and DNA methylation processes. Curr Opin Plant Biol 4:123–129
Maunakea AK, Chepelev I, Cui K, Zhao K (2013) Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res 23:1256–1269
Yan X, Dong X, Liu L, Yang Y, Lai J, Guo Y (2016) DNA methylation signature of intergenic region involves in nucleosome remodeler DDM1-mediated repression of aberrant gene transcriptional read-through. J Genet Genomics 43:513–523
Wang W, Quin Q, Sun F, Wang Y, Xu D, Li Z et al (2016) Genome-wide differences in DNA methylation changes in two contrasting rice genotypes in response to drought conditions. Front Plant Sci 7:1675
Lim YC, Li J, Ni Y, Liang Q, Zhang J et al (2017) A complex association between DNA methylation and gene expression in human placenta at first and third trimesters. PLoS ONE 12:e0181155
Reyna-Lopez GE, Simpson J, Ruiz-Herrera J (1997) Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol Gen Genet 253:703–710
Wang M, Qin L, Xie C, Li W, Yuan J, Kong L, Yu W, Xia G, Liu S (2014) Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol 55:1354–1365
Fu SJ, Wang H, Feng LN, Sun Y, Yang WX, Liu DQ (2009) Analysis of methylation sensitive amplified polymorphism in wheat genome under the wheat leaf rust stress. Yi Chuan 31:297–304
Wang D, Zhao J, Bai Y, Ao Y, Guo C (2017) The variation analysis of DNA methylation in wheat carrying gametocidal chromosome 3C from Aegilops triuncialis. Int J Mol Sci 18:1738
Gardiner LJ, Tulloch MQ, Olohan L, Price J, Hall N, Hall A (2015) A genome-wide survey of DNA methylation in hexaploid wheat. Genome Biol 16:273
Kumar S, Beena AS, Awana M, Singh A (2017) Salt-induced tissue specific cytosine methylation downregulates expression of HKT genes in contrasting wheat (Triticum aestivum) genotypes. DNA Cell Biol 36:283–294
Olohan L, Gardiner LJ, Lucaci A, Steuernagel B, Wulff B, Kenny J et al (2018) A modified sequence capture approach allowing standard and methylation analyses of the same enriched genomic DNA sample. BMC Genomics 19:250
Pan L, Liu X, Wang Z (2012) Comparative DNA methylation analysis of powdery mildew susceptible and resistant near-isogenic lines in common wheat. Life Sci J 10:2073–2083
Sherman JD, Talbert LE (2002) Vernalisation induced changes of the DNA methylation pattern in winter wheat. Genome 45:253–260
Meng FR, Li YC, Yin J, Liu H, Chen XJ, Ni ZF et al (2012) Analysis of DNA methylation during the germination of wheat seeds. Biol Plant 56:269–275
Sun H, Guo Z, Gao L, Zhao G, Zhang W, Zhou R et al (2014) DNA methylation pattern of photoperiod-B1 is associated with photoperiod insensitivity in wheat (Triticum aestivum). New Phytol 204:682–692
Gao L, Diarso M, Zhang A, Zhang H, Dong Y, Liu L, Lv Z, Liu B (2016) Heritable alteration of DNA methylation induced by whole-chromosome aneuploidy in wheat. New Phytol 209:364–375
Yong Z, Zhao HL, Cheng L, ZuJun Y, JinHua P, JiangPing Z et al (2008) Analysis of DNA methylation variation in wheat genetic background after alien chromatin introduction based on methylation-sensitive amplification polymorphism. Chin Sci Bull 53:58–69
Shaked H, Kashkush K, Ozkan H, Feldman M, Levy AA (2001) Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 13:1749–1759
Levy AA, Feldman M (2004) Genetic and epigenetic reprogramming of the wheat genome upon allopolyploidization. Biol J Linn Soc 82:607–613
Bhardwaj SC, Prashar M, Jain SK, Kumar S, Datta D (2010) Virulence of Puccinia triticina on Lr28 in wheat and its evolutionary relation to prevalent pathotypes in India. Cereal Res Commun 38:83–89
Sharma C, Saripalli G, Kumar S, Gautam T, Kumar A, Rani S et al (2018a) A study of transcriptome in leaf rust infected bread wheat involving seedling resistance gene Lr28. Funct Plant Biol 45:1046–1064
Dhariwal R, Vyas S, Govindraj RB, Jha SK, Khurana JP, Tyagi AK, Prabhu KV, Balyan HS, Gupta PK (2011) Analysis of differentially expressed genes in leaf rust infected bread wheat involving seedling resistance gene Lr28. Funct Plant Biol 38:479–492
Singh D, Kumar D, Satapathy L, Pathak J, Chandra S, Riaz A, Bhaganangre G, Dhariwal R, Kumar M, Prabhu KV, Balyan HS, Gupta PK, Mukhopadhyay K (2017) Insights of Lr28 mediated wheat leaf rust resistance: transcriptomic approach. Gene 637:72–89
Chandra S, Singh D, Pathak J, Kumari S, Kumar M, Poddar R, Balyan HS, Gupta PK, Prabhu KV, Mukhopadhyay K (2016) De novo assembled wheat transcriptomes delineate differentially expressed host genes in response to leaf rust infection. PLoS ONE 11:e0148453
Satapathy L, Singh D, Ranjan P, Kumar D, Kumar M, Prabhu KV, Mukhopadhyay K (2014) Transcriptome-wide analysis of WRKY transcription factors in wheat and their leaf rust responsive expression profiling. Mol Genet Genomics 289:1289–1306
Sharma C, Kumar S, Saripalli G, Jain N, Raghuvanshi S, Sharma JB, Prabhu KV, Balyan HS, Gupta PK (2018b) H3K4/K9 acetylatatin and Lr28-mediated expression of six leaf rust responsive genes in wheat (Triticum aestivum). Mol Genet Genomics 94:227–241
Riley R, Chapman V, Johnson R (1968) Introduction of yellow rust resistance of Aegilops comosa into wheat by genetically induced homoeologous recombination. Nature 217:383–384
Friebe B, Jiang J, Raupp W, McIntosh RA, Gill BS (1996) Characterisation of wheat-alien translocations conferring resistance to disease and pests: current status. Euphytica 91:59–87
Tomar SMS, Menon MK (1998) Adult plant response to near-isogenic lines and stocks of wheat carrying specific Lr genes against leaf rust. Indian Phytopathol 51:61–67
Naik S, Gill KS, Prakasa Rao VS, Gupta VS, Tamhankar SA, Pujar S, Gill BS, Ranjekar PK (1998) Identification of a STS marker linked to the Aegilops speltoides-derived leaf rust resistance gene Lr28 in wheat. Theor Appl Genet 97:535–540
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Perez-Figueroa (2013) Msap: a tool for the statistical analysis of methylation-sensitive amplified polymorphism data. Mol Ecol Resour 13:522–527
Fulnecek J, Kovarik A (2014) How to interpret methylation sensitive amplified polymorphism (MSAP) profiles? BMC Genet 15:2
Li N, Ye M, Li Y, Yan Z, Butcher LM, Sun J, Han X, Chen Q, Zang X, Wang J (2010) Whole genome DNA methylation based on high throughput DNA technology. Methods 52:203–212
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE et al (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9:R137
Lienhard M, Grimm C, Morkel M, Herwig R, Chavez L (2014) MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 30:284–286
Hu J, Chen X, Zhang H, Ding Y (2015) Genome-wide analysis of DNA methylation in photoperiod- and thermo-sensitive male sterile rice Peiai 64S. BMC Genomics 16:102
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P et al (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38:576–589
Conesa A, Gotz S (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008:619832
Kohany O, Gentles AJ, Hankus L, Jurka J (2006) Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform 7:47
Livak KJ, Schmittgen (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408
Wada Y, Miyamoto K, Kusano T, Sano H (2004) Association between up-regulation of stress-responsive genes and hypomethylation of genomic DNA in tobacco plants. Mol Genet Genomics 271:658–666
Zhang YU, Harris CJ, Liu Q, Liu W, Ausin I et al (2018) Large scale comparitive epigenomics reveals hierarchial regulation of non-CG methylation in Arabidopsis. Proc Natl Acad Sci USA 115:E1069–E1074
Garg R, Chevala VVSN, Shankar R, Jain M (2015) Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci Rep 5:14922
Zhong L, Xu Y, Wang J (2009) DNA methylation changes induced by salt stress in wheat Triticum aestivum. Afr J Biotechnol 22:6201–6207
He S, Xu W, Li F, Wang Y, Liu A (2017) Intraspecific DNA methylation polymorphism in the non-edible oilseed plant castor bean. Plant Divers 39:300–307
Karan R, DeLeon T, Biradar H, Subudhi PK (2012) Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS ONE 7:e40203
Marconi G, Pace R, Traini A, Raggi L, Lutts S et al (2013) Use of MSAP markers to analyse the effects of salt stress on DNA methylation in rapeseed (Brassica napus var. oleifera). PLoS ONE 8:e75597
Laird M (2010) Principles and applications of genome wide DNA methylation analysis. Nat Rev Genet 11:191–203
Dowen RH, Pelizzola M, Schmitz RJ, Lister R, Dowen JM, Nery JR, Dixon JE, Ecker JR (2012) Widespread DNA methylation in response to biotic stress. Proc Natl Acad Sci USA 109:E2183:E191
Takuno S, Gout BS (2012) Body methylated genes in Arabidopsis thaliana are functionally important and evolve slowly. Mol Biol Evol 219:227
Hollister JD, Gaut BS (2009) Epigenetic silencing of transposable elements: a trade-off between reduced transposition and deleterious effects on neighbouring gene expression. Genome Res 19:1419–1428
Sun L, Miao X, Cui J, Deng J, Wang X, Wang Y et al (2018) Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across different salt stress in maize (Zea mays L.) Euphytica 214:25
Curradi M, Izzo A, Badaracco G, Landsberger N (2002) Molecular mechanisms of gene silencing mediated by DNA methylation. Mol Cell Biol 22:3157–3173
Mette MF, Aufsatz W, Winden J, Matzke MA, Matzke AJM (2000) Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J 19:5194–5201
Liang D, Zhang D, Wu H, Huang C, Shuai P et al (2014) Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet 15:S9
Xu J, Zhou S, Gong X, Song Y, van Nocker S, Ma F, Guan Q (2018) Single base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol J 16:672–687
Deleris A, Halter T, Navarro L (2018) DNA methylation and demethylation in plant immunity. Annu Rev Phyto Pathol 54:579–603
Kumar AA, Raghavaiyya P (2004) Effect of the leaf rust resistance gene Lr28 on grain yield and bread making quality of wheat. Plant Breed 123:35–38
Pavet V, Quintero C, Cecchini NM, Rosa AL, Alvarez ME (2006) Arabidopsis displays centromeric DNA hypomethylation and cytological alterations of heterochromatin upon attack by Pseudomonas syringae. Mol Plant Microbe Interact 19:577–587
López Sánchez A, Stassen JH, Furci L, Smith LM, Ton J (2016) The role of DNA (de)methylation in immune responsiveness of Arabidopsis. Plant J 88:361–374
Shi-wen Wu Hong-wei, Zai-dong W, Ling-rang Y K (2017) Expression comparisons of pathogenesis-related (pr) genes in wheat in response to infection/infestation by Fusarium, ydv aphid-transmitted and hessian fly. J Integr Agric 13:926–936
Borges AF, Ferreira RB, Monteiro S (2013) Transcriptomic changes following the compatible interaction Vitis viniferae-Erysiphe necator. Paving the way towards an enantioselective role in plant defence modulation. Plant Physiol Biochem 68:71–80
Liao Y, Liu S, Jiang Y, Hu C et al (2017) Genome-wide analysis and environmental response profiling of dirigent family genes in rice (Oryza sativa). Genes Genom 39:47–62
Paniagua C, Bilkova A, Jackson P, Dabravolski S et al (2017) Dirigent proteins in plants: modulating cell wall metabolism during abiotic and biotic stress exposure. J Exp Bot 68:3287–3301
Kim KC, Fan B, Chen Z (2006) Pathogen-induced Arabidopsis WRKY7 is a transcriptional repressor and enhances plant susceptibility to Pseudomonas syringae. Plant Physiol 142:1180–1192
Laloi C, Mestres-Ortega D, Marco Y, Meyer Y, Reichheld JP (2004) The Arabidopsis cytosolic thioredoxin h5 gene induction by oxidative stress and its W-box-mediated response to pathogen elicitor. Plant Physiol 134:1006–1016
Perez CM, Spoel SH (2019) Thioredoxin mediated redox signalling in plant immunity. Plant Sci 279:27–33
Shumayla SS, Kumar R, Mendu V, Singh K, Upadhyay SK (2016) Genomic dissection and expression profiling revealed functional divergence in Triticum aestivum leucine rich repeat receptor like kinases (TaLRRKs). Front Plant Sci 7:1374
Qu LJ, Chen J, Liu M et al (2003) Molecular cloning and functional analysis of a novel type of Bowman-Birk inhibitor gene family in rice. Plant Physiol 133:560–570
Piisila M, Keceli MA, Brader G, Jakobson L, Joesaar I, Sipari N, Kollist H, Palva ET, Kariola T (2015) The F-box protein MAX2 contributes to resistance to bacterial phytopathogens in Arabidopsis thaliana. BMC Plant Biol 15:53
Zhou J, Rodriguez-Zas S, Aldea M, Li M, Zhu J, Gonzalez DO, Vodkin LO, DeLucia E, Clough SJ (2005) Expression profiling soybean response to Pseudomonas syringae reveals new defense-related genes and rapid HR-specific downregulation of photosynthesis. Mol Plant Microbe Interact 18:1161–1174
Xin M, Wang X, Peng H, Yao Y, Xie C, Han Y, Ni Z, Sun Q (2012) Transcriptome comparison of susceptible and resistant wheat in response to powdery mildew infection. Genom Proteom Bioin 10:94–106
Delteil A, Gobbato E, Cayrol B, Estevan J, Michel-Romiti C, Dievart A, Kroj T, Morel JB (2016) Several wall-associated kinases participate positively and negatively in basal defense against rice blast fungus. BMC Plant Biol 16:17
Yu Y, Yang D, Zhou S, Gu J, Wang F, Dong J, Huag R (2017) The ethylene response factor OsERF109 negatively affects ethylene biosynthesis and drought tolerance in rice. Protoplasma 254:401–408
Zhang H, Hing Y, Huang L, Li D, Song F (2016) Arabidopsis AtERF014 acts as a dual regulator that differentially modulates immunity against Pseudomonas syringae pv. Sci Rep 6:30251
Ahmed SA, Liu P, Xue Q, Ji C, Qi T, Guo J, Guo J, Kang Z (2017) TaDIR1-2 a wheat ortholog of lipid transfer protein AtDIR1 contributes to negative regulation of wheat rust resistance against Puccinia striiformis f.sp.tritici. Front Plant Sci 8:521
Gullner G, Komives T, Kiraly L, Schroder P (2018) Glutathione S-transferase enzymes in plant-pathogen interactions. Front Plant Sci 9:1836
Krattinger SG, Lagudah ES, Wicker T, Risk JM, Ashton AR, Selter LL, Matsumoto T, Keller B (2011) Lr34 multi-pathogen resistance ABC transporter: molecular analysis of homoeologous and orthologous genes in hexaploid wheat and other grass species. Plant J 65:392–403
Pan Y, Liu Z, Rocheleau H, Fauteux F, Wang Y, McCartney C, Ouellet T (2018) Transcriptome dynamics associated with resistance and susceptibility against fusarium head blight in four wheat genotypes. BMC Genomics 19:642
Rojas CM, Senthil-Kumar M, Tzin V, Mysore KS (2014) Regulation of primary plant metabolism during plant-pathogen interactions and its contribution to plant defense. Front Plant Sci 5:17
Amaral MN, Arge LWP, Benitez LC, Danielowski R (2016) Comparative transcriptomics of rice plants under cold, iron and salt stresses. Funct Integr Genomics 16:567–579
Kumar S, Trivedi PK (2018) Glutathione S-Transferases: Role in combating abiotic stresses including arsenic detoxification in plants. Front Plant Sci 9:751
Hake S, Smith HM, Holtan H, Magnani E, Mele G, Ramirez J (2004) The role of knox genes in plant development. Ann Rev Cell Dev Biol 20:125–151
Muthusamy SK, Dalal M, Chinnusamy V, Bansal CK (2016) Differential regulation of genes coding for organallel and cytosolic ClpATPases under biotic and abiotic stresses in wheat. Front Plant Sci 7:929
Pulido P, Llamas E, Rodriguez-Concepcion M (2017) Both Hsp70 chaperone and Clp protease plastidial systems are required for protection against oxidative stress. Plant Signal Behav 12:e1290039
Kovacs V, Pal M, Vida G, Szalai G, Janda T (2011) Effect of powdery mildew infection on the antioxidant enzyme activities in different lines of Thatcher based wheat. Acta Biol Szeged 55:99–100
Sharma R, Sahoo A, Devendran R, Jain M (2014) Over-expression of a rice tau class glutathione s-transferase gene improves tolerance to salinity and oxidative stresses in Arabidopsis. PLoS ONE 9:e92900
Zhou SM, Kong XZ, Kang HH, Sun XD, Wang W (2015) The involvement of wheat F-box protein gene TaFba1 in the oxidative stress tolerance of plants. PLoS ONE 10:e0122117
Kong X, Zhou S, Yin S, Zhao Z, Han Y, Wang W (2016) Stress inducible expression of an F-box gene TaFBA1 from wheat enhanced the drought tolerance in transgenic tobacco plants without impacting plant growth and development. Front Plant Sci 7:1295
Li Q, Wang W, Wang W, Zhang G, Liu Y, Wang Y, Wang W (2018) Wheat F-box protein gene TaFBA1 is involved in plant tolerance to heat stress. Front Plant Sci 9:521
Powell JJ, Carere J, Fitzgerald TL, Stiller J, Covarelli L, Xu Q et al (2017) The Fusraium crown rot pathogen pseudo graminearum triggers a suite of transcriptional and metabolic changes in bread wheat. Ann Bot 119:853–867
Chen X, Chen H, Yuan JS, Kollner TG, Chen Y, Guo Y et al (2018) The rice terpene synthase gene OsTPS19 functions as an (S)-limonene synthase in planta, and its overexpression leads to enhanced resistance to the blast fungus Magnaporthe oryzae. Plant Biotechnol J 16:1778–1787
Yadav IS, Sharma A, Kaur S, Nahar N, Bhardwaj SC, Sharma TR, Chhuneja P (2016) Comparative temporal transcriptome profiling of wheat near isogenic line carrying Lr57 under compatible and incompatible interactions. Front Plant Sci 7: 1943
Cao C, Xing L, Wang X, Yang X, Wang W, Sun Y et al (2011) Serine/threonine kinase gene Stpk-V, a key member of powdery mildew resistance gene Pm21, confers powdery mildew resistance in wheat. Proc Natl Acad Sci USA 108:7727–7732
Liu X, Yang L, Zhou X, Zhou M, Lu Y, Ma L, Ma H, Zhang Z (2013) Transgenic wheat expressing Thinopyrum intermedium MYB transcription factor TiMYB2R-1shows enhanced resistance to the take-all disease. J Exp Bot 64:2243–2253
Acknowledgements
Financial assistance from the Department of Biotechnology (Grant Number BT/PR3337/AGR/2/819/2011), Government of India for carrying out this study is gratefully acknowledged. GS, CK, TG and KS were each awarded JRF/SRF/RA by the Department of Biotechnology, Government of India. PKG and HSB were each awarded the position of Senior Scientist by Indian National Science Academy, New Delhi. PKG was also awarded a National Academy of Sciences India (NASI) Senior Scientist Platinum Jubilee Fellowship during the tenure of this research work. Facilities available in DBT-sponsored Bioinformatics Infrastructure Facility (BIF) were also utilized for this work.
Author information
Authors and Affiliations
Contributions
PKG and HSB conceived the experiment. GS with the help of CS, KS and TG conducted data analysis. GS and CS also prepared the first draft of the manuscript which was edited and finalised by PKG, HSB and PKS. PP provided inoculum for leaf rust pathotype 77-5 and NJ, KVP and JBS raised and provided the seedlings tissues. JKR assisted GS and CS in conducting MSAP analysis.
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that there is no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Saripalli, G., Sharma, C., Gautam, T. et al. Complex relationship between DNA methylation and gene expression due to Lr28 in wheat-leaf rust pathosystem. Mol Biol Rep 47, 1339–1360 (2020). https://doi.org/10.1007/s11033-019-05236-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-019-05236-1