
Abstract
Functional specialization of cells is among the most fundamental processes of higher organism ontogenesis. The major obstacle to studying this phenomenon in plants is the difficulty of isolating certain types of cells at defined stages of in planta development for in-depth analysis. A rare opportunity is given by the developed model system of flax (Linum usitatissimum L.) phloem fibres that can be purified from the surrounding tissues at the stage of the tertiary cell wall deposition. The performed comparison of the whole transcriptome profile in isolated fibres and other portions of the flax stem, together with fibre metabolism characterization, helped to elucidate the general picture of the advanced stage of plant cell specialization and to reveal novel participants potentially involved in fibre metabolism regulation and cell wall formation. Down-regulation of all genes encoding proteins involved in xylan and lignin synthesis and up-regulation of genes for the specific set of transcription factors transcribed during tertiary cell wall formation were revealed. The increased abundance of transcripts for several glycosyltransferases indicated the enzymes that may be involved in synthesis of fibre-specific version of rhamnogalacturonan I.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Higher plant organism consists of approximately 40 functionally different cell types (Carpita and McCann 2000). Their specialization is among the major processes of plant ontogenesis. The study of plant cell functional specialization is usually hampered by the difficulty or even impossibility of isolating plant cells of certain types and at certain stages of development in quantities sufficient for in-depth characterization. Because plant samples usually contain many different cell types, it is difficult to characterize tissue (cell)-specific processes. Some steps of cell specialization, such as the formation of tracheary elements, have been effectively studied in plant cell cultures (Fukuda 1992; Devillard and Walter 2014). However, many developmental processes are not reproducible in vitro. Moreover, the presence of organismal control that may have a significant impact on the regulation of cell development is excluded in experiments with cell culture.
One of the rare experimental systems that permits the study of in planta specialization of certain cell type is the developing of primary phloem fibres of flax (Linum usitatissimum L.). On the basis of their compact (within bundles) location in the stem and the presence of a thick cell wall, developing phloem fibres can be effectively purified from the surrounding tissues and used for various types of analysis (Hotte and Deyholos 2008; Mokshina et al. 2014). Primary phloem fibres of flax originate from procambium, close to the very stem tip (Esau 1977). Older fibres, which have reached a more advanced stage of fibre specialization, are found lower on the stem. Stem parts containing phloem fibres at different developmental stages are well characterized (Gorshkova et al. 2003; Ageeva et al. 2005). The possibility of isolating fibres at specific stage of specialization together with the published, annotated full genome sequence make flax a convenient model to study various aspects of plant fibre development.
The major function of mature flax phloem fibres is to provide strength and flexibility for a tall and narrow stem, which experiences severe mechanical stresses. Fibre metabolism at advanced stages of cell specialization includes deposition of a tertiary cell wall, which is a fibre-specific cellulose-enriched cell wall type that is also called the G-layer and is deposited after the secondary cell wall (S). The tertiary cell wall is quite distinct from the secondary cell wall, which consists of three main components, cellulose, xylan, and lignin, present in approximately equal amounts. Major peculiarities of the tertiary cell wall type, which have been summarized in several reviews (Gorshkova et al. 2010; Mellerowicz and Gorshkova 2012), are the overwhelming content of cellulose (80–90 %), high crystallinity of cellulose, very low angle of cellulose microfibrils, considerable thickness of the G-layer (up to 10 μm), and absence of xylan and lignin. Glucomannans and likely some xyloglucan (Morvan et al. 2003) are present in flax fibres as minor constituents, whereas the major non-cellulosic polymer is a tissue- and stage-specific version of rhamnogalacturonan I (RG-I) with long side chains made of β-(1→4)-galactan. This RG-I is synthesized in the Golgi apparatus and secreted outside the plasma membrane; active vesicular traffic is characteristic for this stage of tertiary cell wall development (Salnikov et al. 2008). The secreted RG-I is modified by an enzyme expressed specifically in fibres that form the tertiary cell wall, β-1,4-galactosidase, which partially trims off Gal residues (Roach et al. 2011). Such trimming leads to RG-I entrapment by laterally interacting cellulose microfibrils and is important for G-layer maturation (Roach et al. 2011; Gorshkova et al. 2015).
Fibres with G-layers are widely spread in the plant kingdom and can be located in various plant organs (tendrils, contractile roots, corms, peduncles) but are especially known for their presence in various fibre crops (ramie, hemp, flax) and in the tension wood of some deciduous trees (Gorshkova et al. 2012). In recent years, G-layers have attracted considerable attention due to enhanced suitability for saccharification: their low lignin, low pentosan, and high cellulose contents are key characteristics that are thought to facilitate the decomposition of cell walls by microbial enzymes (Muñoz et al. 2011; Healey et al. 2015).
Several genome-wide gene expression studies using microarrays and NGS technology have been performed on the samples that included fibres with G-layers. Such data were obtained for reaction wood of Populus and Eucalyptus species (Andersson-Gunnerås et al. 2006; Mizrachi et al. 2015; Gerttula et al. 2015) and for flax stem (Day et al. 2005; Roach and Deyholos 2007; Dash et al. 2014). Genes potentially involved in fibre development and G-layer formation were revealed. However, tissue mixtures were used as samples in all of these cases. Moreover, in earlier papers, there was no opportunity to rely on the full genome sequence, and the power of applied transcriptomic techniques was not sufficient to characterize the whole transcriptome. Methods to isolate flax phloem fibres at advanced stages of specialization were developed and used for proteomic analysis (Hotte and Deyholos 2008) and for the analysis of the expression of certain genes (Mokshina et al. 2014), but the general picture of the fibre transcriptome remained uncharacterized.
To enrich the understanding of highly specialized cell physiology and to obtain insights into mechanisms of fibre development, we elucidated the transcriptome landscapes within several parts of the flax stem. Comparison of gene expression data for fibres isolated at advanced stage of specialization and for other tissues, put on the basis of fibre metabolism characterization by analysis of label distribution after photosynthesis of intact plants with 14CO2, helped to obtain a general picture of the fibre’s physiology. The main participants, together with co-factors and helper proteins that have not yet appeared in the studies of fibre development and cell wall formation, were revealed. The obtained data provide a solid basis for further in-depth analysis of interplay between identified participants, including those involved in various regulatory systems.
Materials and methods
Plant materials
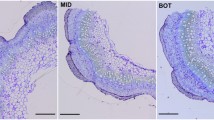
Flax (Linum usitatissimum L., cv. Mogilevsky) plants were grown in boxes with a soil layer of 50 cm in open air under natural daylight with daily watering. Plants were harvested at the period of rapid growth (4 weeks after sowing). Plant material was sampled according to a previously published scheme (Mokshina et al. 2014), taking the snap point (SP) as the reference. SP is an indicator of fibre developmental transition from intrusive growth to cell wall thickening; it is easily manually detected from the above stem region by the sharp increase in effort required to break the stem (Gorshkova et al. 2003). The following samples (Fig. 1) were collected for transcriptomic analysis (portions of five plants in each sample): TOP, 1 cm stem segment above the SP with phloem fibres in the process of elongation; MID, 1 cm stem segment below the SP containing fibres at early stages of tertiary cell wall thickening, and FIB, phloem fibres isolated from stem peels below SP further down the stem than the MID sample. Fibres were obtained by washing fibre-enriched stem peels in 80 % ethanol in a mortar several times, gently pressing with a pestle until removal of other tissues and chlorophyll. Fibres in the TOP portion contain only primary cell wall, whereas the MID and FIB samples have preformed primary and thin secondary layers and deposit tertiary cell wall (Gorshkova et al. 2003, 2010). All collected samples were frozen in liquid nitrogen and stored at -80 °C until analysis.

Scheme of sample collection. Segments of flax stem–top region (TOP), middle region (MID), and fibres isolated at an advanced stage of specialization (FIB)–were taken for total RNA extraction and used in transcriptome analysis (orange boxes), qRT-PCR (deep red boxes) and 14C-distribution experiments (green box, only FIB). Bars = 50 µm (TOP, MID) and 20 µm (FIB)
RNA and cDNA preparation
Collected samples were ground in liquid nitrogen. The total RNA from plant samples was isolated using a Trizol extraction method combined with an RNeasy Plant Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions. Residual DNA was eliminated by treatment with DNAse I using the DNA-free kit (Ambion, USA). RNA quantity and quality were confirmed with a NanoDrop ND-1000 spectrophotometer (USA) and 1 % agarose gel electrophoresis. Total RNA (1 μg) was converted to cDNA with M-MuLV Reverse Transcriptase (Fermentas, Lithuania) according to the standard protocol from the manufacturer.
RNA-Seq library preparation and sequencing
Total RNA (3 µg) extracted from TOP, MID and FIB was processed using the TruSeq Sample Prep Kit v2 (Illumina, USA) according to manufacturer instructions. Library quantification was performed using a Qubit fluorimeter (Invitrogen, USA). The obtained cDNA libraries were sequenced on a MiSeq platform (Illumina, USA) using MiSeq Reagent Kit v2 (Illumina, USA) following the manufacturer’s instructions; the length of single end (SE) reads was 75 nucleotides. Base calling and quality control was performed with Illumina software RTA (v1.18.54)/MiSeq Reporter (v2.5).
RNA-Seq data analysis
Raw RNA-Seq reads in FASTQ format obtained from the MiSeq System were analysed using the web-based platform Galaxy Project (https://usegalaxy.org/, Goecks et al. 2010). The obtained sequences were mapped to the L. usitatissimum L. genome sequence scaffolds (v1.0) using TopHat (v2.0.13) with default parameters (Kim et al. 2013) except for a minimum and maximum intron length, which was changed to 40 and 6000 bp, correspondingly (Wendel et al. 2002).
The reads were aligned to the reference genome, and transcript abundance was estimated using fragments per kilobase of exon per million mapped fragments (FPKM) metrics determined by Cufflinks according to a published protocol (Trapnell et al. 2012). Data were subjected to bias and multi-read correction to increase accuracy. The output GTF files were sent to Cuffmerge along with a reference GFF3 formatted annotation file downloaded from the Phytozome database (http://www.phytozome.net/). A merged GTF file along with the original alignment (.BAM) file produced by TopHat for each pool of reads was passed to Cuffquant that pre-calculates gene expression levels (Trapnell et al. 2012). Finally, the resulting (.CXB) files for further processing were sent to Cuffdiff to detect differentially expressed genes from pairwise comparison of three samples. The significance scores were adjusted for multiple testing using the Benjamini-Hochberg correction (Benjamini and Hochberg 1995). Differentially expressed genes (DEGs) were identified with a q-value (false discovery rate (FDR)) of < 0.05. RStudio and the R package CummeRbund v2.8.2 (Goff et al. 2013) were used to visualize the expression patterns of DEGs. Pseudocount of 0.001 was added to avoid division by zero when calculating fold changes (FC). Data of RNA-Seq analysis were interpreted using MapMan v3.6. (Thimm et al. 2004) adapted to flax.
Data regarding CDS and proteins lengths were extracted from Phytozome. Prediction of transmembrane domains was performed by the TMHMM program (http://www.cbs.dtu.dk/services/TMHMM/), of signal peptide by SignalP (http://www.cbs.dtu.dk/services/SignalP/) and of subcellular location by TargetP tools (http://www.cbs.dtu.dk/services/TargetP/). Functional annotation was obtained by Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Validation of transcriptome experiments using qRT-PCR
Twenty DE/non-DE genes identified by RNA-Seq were assayed by qRT-PCR. Real-time PCR validation was carried out on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, USA). Gene-specific primers for the analysed genes were designed using the Universal ProbeLibrary Assay Design Centre (http://lifescience.roche.com/) (Table S1). A cDNA dilution of 2.5 µl (1/15) was used for amplification. The PCR mixture (10 µl) also contained 0.4 µM of each forward and reverse gene-specific primer, 0.2 mM dNTPs, 1x SYBR Green (Sigma, USA), and 0.1 µl HS Taq Polymerase at 5 u/μl (Evrogen, Russia). The thermal cycling conditions were 95 °C for 5 min, 40 cycles at 95 °C for 15 s, and 60 °C for 1 min. A 60–95 °C melting curve was performed to confirm specificity of the products. From each of three biologically independent cDNA samples, two independent technical replications were performed and averaged for further calculations. Relative transcript abundance calculations were performed using the 2− ΔΔCt method (Livak and Schmittgen 2001). The genes of eukaryotic translation initiation factors 1 A, 5 A (LusETIF1, LusETIF5A) and glyceraldehyde-3-phosphate dehydrogenase (LusGAPDH) were used as the housekeeping genes (Table S1) (Huis et al. 2010). ΔΔCt values were generated using the TOP sample as a reference, and log2 ratios were calculated to show relative expression values (Livak and Schmittgen 2001).
Exposure of intact plants to 14CO2 and analysis of label distribution in fibres
To characterize the unperturbed metabolism in stem tissues, we used the approach described earlier with analysis of label distribution after photosynthesis of flax plants with 14CO2 (Gorshkova et al. 1997). Growing in soil intact plants of the same age and developmental stage as for RNA extraction were covered with an air-tight glass chamber connected with a gasholder containing 14CO2 (from 40 MBq Ba14CO3, Isotope, Russia). The volume of the gasholder was adjusted so that the final concentration of CO2 was close to 0.03 % and did not change significantly through the time of plant exposure to 14CO2. The airflow was provided by a built-in compressor. The plants were left to photosynthesize for 40 min of daylight. Then, the chamber was partitioned from the gasholder and removed from around the plants. The plants were harvested 24 h later. Fibres were isolated in the same way as for all other experiments. Cell walls were obtained from alcohol-insoluble residue (AIR) by sequential washings with 0.05 M potassium phosphate (pH 7.0) and acetone and overnight treatment with glucoamylase (Sigma, USA). An aliquot of suspended cell walls was used for radioactivity determination, and the rest of the cell walls were hydrolysed in 1 ml of 2 M TFA at 120 °C for 60 min to yield monosaccharides from matrix polysaccharides. Monosaccharide analysis was performed as described earlier (Mikshina et al. 2012) using high-performance anion-exchange chromatography on a CarboPac PA-1 column (4 mm × 250 mm, Dionex, USA) and pulse-amperometric detection (Dionex, USA). The protocol utilized does not permit the separation of xylose and mannose. For crystalline cellulose to be obtained, the remaining material was digested in acetic-nitric reagent for 1 h in a boiling water bath (Updegraff 1969). Radioactivity of all samples was determined by liquid scintillation counter Tri-Carb 2810 TR (Perkin Elmer, USA). The analysis was performed in four replicates with each sample containing fibres from four plants.
Results
General transcriptome landscape of isolated fibres
For the whole transcriptome of flax phloem fibres to be characterized at advanced stage of specialization, isolated fibres (FIB) as well as TOP and MID segments of stem were collected and used for RNA-Seq analysis (Fig. 1). A total of 66,841,618 raw reads were acquired, ranging from 19.3 to 24.2 million reads per sample (Fig. 2A). Approximately 95 percent of the total reads were successfully mapped to the reference sequence using the Cufflinks protocol. Gene expression level distributions and box plot analysis revealed that sets of sequencing data for analysed samples are comparable and suitable for downstream analysis (Figs. S1 and S2).

RNA-Seq analysis of three samples (TOP, MID, FIB) of flax stem. A Summary of RNA-Seq reads mapped to reference genes. B Venn diagram showing a number of specific, common and uniquely expressed genes in FIB (red circle), TOP (blue circle) and MID (yellow circle) samples among 31,250 genes with FPKM > 0. C Number of up- and down-regulated genes (q < 0.05) in the case of a pairwise analysis of MID, TOP and FIB samples of flax stem. D Comparison of gene expression values obtained by qRT-PCR and RNA-Seq analysis. Error bars show the standard error of the mean. The gene list is given in Table S1
In total, expression of 31,250 genes was detected across all three samples (FPKM > 1) (Fig. 2B), out of 43,486 protein-coding genes (Table S2) in whole-genome assembly of flax (Wang et al. 2012). The most abundant transcripts in fibres forming tertiary cell wall were those of the genes encoding metallothioneins (Lus10016546 and Lus10040811), lipid transfer proteins (Lus10015279, Lus10015278, Lus10039511 and Lus10017616), proteins involved in photosynthesis (ribulose bisphosphate carboxylase, photosystem II reaction centre W, chlorophyll A/B binding protein 1) and participants of protein synthesis and degradation, such as the translation process in ribosomes (Lus10037543, Lus10021532, Lus10016875 and Lus10015085) or ubiquitin-mediated degradation (Lus10002228). Some gene transcripts that were most abundant in flax fibres, such as Lus10026314, Lus10025868, and Lus10032860, had no annotated domains for corresponding protein and no orthologous genes in the Phytozome database (Table 2).
MapMan visualization of the general changes in fibre transcriptome as compared to the TOP sample (Fig. S3) indicated similar level of transcripts for many primary processes like photosynthesis or respiration. At the same time, the major colour of the map is blue, indicating down-regulation, which is partially explained by the mixture of several cell types in the TOP sample and the presence of the only one in the FIB sample. Up-regulation (red colour) is depicted for many genes of carbohydrate metabolism and for certain genes involved in other cellular processes. The latter are considered in the following sections.
Results of analysis using the algorithm Cufflinks/Cuffdiff have indicated that 804 unique genes (Fig. 2C and Fig. S4) had statistically significant differences (q < 0.05) in expression levels at values of FPKM > 16 according to recommendations of the Sequencing Quality Control project (SEQC/MAQC-III 2014). Pairwise comparison of samples revealed 652 DEGs in FIB compared with TOP (Fig. 2C).
To verify the results of RNA-Seq analysis, we used 20 transcripts as targets for quantitative real-time RT-PCR (qRT-PCR) analysis. These transcripts were selected to represent various functional categories and included 10 transcripts that were enriched according to RNA-Seq results in the stem part below the snap point (FIB and MID) and 10 transcripts that were enriched in the stem part above the snap point (TOP). The expression values calculated in RNA-Seq experiments were in good agreement with the data obtained by qRT-PCR (Fig. 2D).
Differentially expressed genes in flax phloem fibres at advanced stage of specialization
To profile differential gene expression in fibres at advanced stage of specialization, we compared transcriptomic data for fibres depositing tertiary cell wall (FIB) and for the TOP sample, which contains various tissues forming primary or (in some tissues) secondary cell walls. Phloem fibres in TOP are at the stage of intrusive elongation, enlarging their cell surface by deposition of primary cell wall (Ageeva et al. 2005). Such comparison permits the determination of both tissue- and stage-specific transcripts present in phloem fibres at advanced stage of specialization. Overall, we have found 156 genes with transcripts that were significantly enriched (cut-off log2FC > 2, q < 0.05) in FIB compared with TOP (Table 1). They mainly belonged to gene categories encoding enzymes of cell wall metabolism, ion transporters and transcriptional factors together with other proteins involved in regulation (Table 1).
The most up-regulated gene was Lus10037377 encoding xyloglucan endotransglucosylase/hydrolase (XET/XTH), the cell wall enzyme that modifies the most widely spread cross-linking glycan of primary cell walls (Fry et al. 1992). Up-regulation of XTH genes has also been demonstrated in eucalypt (Paux et al. 2005; Goulao et al. 2011) and poplar (Andersson-Gunnerås et al. 2006) during early tension wood formation that involves tertiary cell wall deposition in fibres. XET activity was found between S- and G-layers during tertiary cell wall formation in tension wood (Mellerowicz et al. 2008). It was hypothesized that XET modifies xyloglucan, which acts to staple S- and G-layers (Hayashi et al. 2010).
Among the genes with increased transcript abundance in fibres were those encoding β-galactosidase (Lus10028848), rhamnogalacturonate lyase-related proteins (Lus10004281, Lus10019231) and chitinase-related protein (Lus10003231). Up-regulation of these genes during tertiary cell wall formation in flax fibres was reported previously (Roach and Deyholos 2007; Hobson et al. 2010; Mokshina et al. 2014), validating RNA-Seq analysis.
Several glycosyltransferases that may be involved in cell wall polysaccharide synthesis had increased abundance of transcripts in the FIB sample (Table 1). In the CAZy database (http://www.cazy.org), they belong to different families; Lus10023057 belongs to GT2 and Lus10013790 belongs to GT47, whereas Lus10027753 appears as a non-classified GT. Additionally, up-regulated Lus10019809 is homologous to At4g15490, which encodes a GT1 family glycosyltransferase supposed to take part in glucosylation of various aromatic substrates, for example, in the course of flavonoid biosynthesis (http://www.cazy.org/GT1.html).
The list of transcription factors (TFs) for which gene expression was increased in fibres forming the tertiary cell wall was rather extended. According to PlantTFBD (Jin et al. 2014), 2481 genes of TFs were identified in the flax genome and classified into 57 families. Altogether, 1624 genes were detected (FPKM > 1) in all of the stem portions analysed. Of the 629 expressed genes coding TFs (FPKM > 16) (Table S3), 27 genes were considerably up-regulated and 55 were down-regulated in FIB compared with the TOP using a cut-off log2FC > 2. Some of the down-regulated genes may be involved in intrusive elongation of fibres that is completed above the snap point (Gorshkova et al. 2003) and thus occurs only in the TOP sample. Almost half of the down-regulated genes in the FIB sample encode transcription factors with published activity in secondary cell wall formation (Kim et al. 2013; Zhong and Ye 2014; Taylor-Teeples et al. 2015); they are listed in Fig. 4 and Table S3. Among them, master switches triggering secondary cell wall biosynthesis that belong to NAC and MYB families of TFs, NST1 (Lus10002687 and Lus10017340), SND1 (Lus10001664), and MYB46 (Lus10002559, Lus10039610), were detected (q < 0.05). Moreover, the expression of several representatives of WRKY and ERF families was reduced in fibres compared with TOP. At the same time, the number of genes encoding TFs from G2-like, bZIP, bHLH, and MYB-related families is increased in fibres (Fig. 4). The function of most of the TFs with detected up-regulation in fibres depositing the tertiary cell wall is not yet characterized.
The up-regulated genes in fibres include those that are annotated as encoding various transporters (Table 1). Among them were transporters of manganese, potassium, iron, sulfate and nitrate, indicating the importance of specific ion balance for tertiary cell wall formation. Moreover, transcripts of Lus10029701 encoding MSL10 protein, which is involved in the formation of a plasma membrane-localized channel, were enriched in fibres. The MSL10-formed channel closes at lower tensions than it opens; together with homology of the bacterial channels responding to membrane stretching, it worth considering MSL10 as a plant mechanosensitive channel (Hamilton et al. 2015).
Within the hormone-related group, four out of five listed DEGs encode proteins involved in auxin signalling. Lus10015879 and Lus10005781 belong to the large auxin-responsive gene family of the small auxin-up RNA (SAUR) genes with largely unknown function. SAUR39 was suggested to act as a negative regulator of auxin synthesis and transport (Kant et al. 2009). IAA3 (Lus10039413) is a member of the auxin-induced Aux/IAA family and acts as a negative regulator in auxin signalling (Tian et al. 2002).
Out of ten detected transcripts of genes encoding isopentenyltransferase, the enzyme that catalyses the important step of cytokinin biosynthesis (Kakimoto 2001), one gene (IPT3, Lus10012372) had significantly increased transcript abundance specifically in FIB (Table 1). In Arabidopsis, IPT3 was shown to be expressed in phloem almost throughout the plant (Miyawaki et al. 2004). Two genes of ethylene-forming enzymes were considerably down-regulated in FIB: one (Lus10015153) compared to TOP and the other (Lus10031530) compared to MID; transcripts of two other members of the same gene family (Lus100353334 and Lus10029992) had a similar level of abundance in all analysed samples (Table S2).
The significant group of transcripts with differential abundance belonged to genes encoding proteins with unknown function (Table 2). All of the proteins encoded by the genes from Table 2 do not have any conserved domains. In some of them, transmembrane domains and signal peptides are present. One gene encoding a protein without an annotated domain (Lus10040842) belonged to the top 10 list of down-regulated genes (Table 1).
Peculiarities of the transcriptome in fibres at advanced stage of specialization included a number of down-regulated genes: the abundances of transcripts for 499 genes with FPKM > 16 in the TOP sample were decreased in fibres (log2FC > 2 and q < 0.05) (Table S2). The top 10 list of the most down-regulated genes in fibres depositing tertiary cell wall is given in Table 1. Several of these are genes for the proteins involved in secondary cell wall formation, such as tracheary element differentiation-related (Lus10018925, Lus10029760), laccase (Lus10034614), thaumatin family protein (Lus10008304), and zinc finger CCCH domain-containing protein (Lus10034624) (Endo et al. 2009; Berthet et al. 2011; Cassan-Wang et al. 2013; Wang et al. 2010). Moreover, there are genes encoding LTPs (Lus10032260, Lus10000397), germin-like protein 7 (Lus10029214), and gibberellin-regulated family protein GASA (Lus10024791) (Table 1).
Down-regulation of genes encoding enzymes involved in lignification and xylan biosynthesis
G-layers are known to contain little or no lignin, both in tension wood (Pilate et al. 2004; Kaku et al. 2009) and in phloem fibres (Love et al. 1994; Gorshkova et al. 2010). Xylan, the major matrix polysaccharide of lignified secondary cell wall, is also absent in G-layers (Bowling and Vaugn 2008; Gorshkova et al. 2010, 2015). Accordingly, the transcription of genes encoding proteins involved in lignin and xylan synthesis is down-regulated upon G-layer induction in tension wood (Andersson-Gunnerås et al. 2006; Mizrachi et al. 2015).
In the flax stem parts used in our experiments, secondary cell wall formation occurred in the xylem-containing TOP and MID samples but not in the fibres depositing the tertiary cell wall. RNA-Seq detected considerably lower levels of transcript abundance in fibres as compared to other samples for all known genes that are ascribed to xylan synthesis (Oikawa et al. 2010; Zhong and Ye 2014) (Fig. 3A and Table S2) and for most of those involved in phenylpropanoid metabolism leading to lignin formation (Fraser and Chapple 2011; Zhong and Ye 2014) (Fig. 3B and Table S2). These unravelled regularities serve as “physiological validation” of the performed whole transcriptome profiling.

Distribution of FPKM values for expressed genes involved in A xylan biosynthesis and B lignin biosynthesis in FIB (red), MID (yellow) and TOP (blue) samples. *P < 0.05; **q < 0.05
Cell wall formation as the major process occurring in fibres is obvious from label distribution after flax plant photosynthesis with 14CO2
For understanding the peculiarities of the transcriptome landscape in fibres, it is useful to characterize the major directions of fibre metabolism. For that, we let intact flax plants at the same age and developmental stage as for RNA-Seq experiments photosynthesize in the atmosphere with 14CO2, and 24 h later, we determined the label distribution in cell components in fibres isolated in a similar way as for the transcriptomic study. Such an approach permits distinguishing compounds synthesized at the time of the experiment from similar ones deposited earlier. This is especially important for cell wall polymers because they are rarely degraded and accumulate through the whole lifetime of the cell. For example, rhamnogalacturonans and polygalacturonic acid of the primary cell wall are present in fibres depositing the tertiary cell wall, but they would not be labelled by 14C because they were synthesized long in advance of exposure to 14CO2.
In the FIB sample, cell wall radioactivity constituted two-thirds of the total radioactivity of the alcohol-insoluble fraction (the alcohol-soluble fraction was removed during fibre isolation) (Fig. 5). The bulk (73 %) of the cell wall radioactivity accumulated in cellulose. The highest proportion of label among the monosaccharides of matrix polysaccharides was registered for galactose. In flax fibres, this monomer mainly belongs to the long β-(1→4)-galactans, which are the side-chains of the fibre- and stage-specific version of pectic polymer rhamnogalacturonan I (Gorshkova et al. 1996; Mikshina et al. 2012). Radioactivity of RG-I monomers (rhamnose, galacturonic acid, galactose and arabinose) constituted 74 % of the total radioactivity of all monosaccharides released from matrix polymers. Estimation of their specific radioactivity indicated higher values for RG-I monomers than for the rest of the monomers (xylose and glucose). The only exception, GalA, was likely due to the high content of polygalacturonic acid deposited in the primary cell wall at early stages of fibre development. The obtained data clearly indicate that among the major processes occurring in the FIB sample is the formation of the cell wall, mainly consisting of two polymers: cellulose and RG-I with side-chains of β-(1→4)-galactans.
Discussion
General picture of the transcriptome in flax fibres at advanced stage of specialization
The transcriptome profiling technique was applied for developing in planta individual type of plant cells - phloem fibres depositing the tertiary cell wall. The full flax genome assembly contains 43,486 protein-coding genes (Wang et al. 2012, Table S2), approximately two-thirds of which are expressed (FPKM > 1) in phloem fibres at advanced stage of specialization. Our results do not contradict most of the earlier data obtained on various fibre-containing tissue mixtures (Day et al. 2005; Andersson-Gunnerås et al. 2006; Roach and Deyholos 2007; Dash et al. 2014; Mizrachi et al. 2015; Gerttula et al. 2015) but rather expand them, giving a full transcriptome landscape specifically for fibres and revealing novel players.
Mature fibres are often considered as dead cells (Fahn 1990; Yilmaz 2015); according to our data, flax phloem fibres “lead a full life” during tertiary cell wall formation, having an active and diverse metabolism. The number of different transcripts that were revealed in the FIB sample was at the same level as in the TOP sample, which mainly consists of young tissues (Fig. 2B and Table S2). High expression levels of genes encoding ribosome constituents indicate intensive protein synthesis in fibres. Active cytoplasm in flax fibres at the stage of cell wall thickening is also evident from the electron microscopy study (Salnikov et al. 2008). Our data on label distribution after photosynthesis with 14CO2 (Fig. 5) and on transcriptome profiling, the same as data of flax fibre proteomic analysis (Hotte and Deyholos 2008), come together with the description of well-developed chloroplasts in flax fibres that may supply these symplastically isolated cells with self-assimilates (Ageeva et al. 2005; Salnikov et al. 2008). Active photosynthesis may distinguish phloem and xylem fibres depositing the tertiary cell wall because no expression of genes, the products of which are involved in the photosynthetic pathway, is revealed during transcriptome analysis of tension wood (Andersson-Gunnerås et al. 2006; Mizrachi et al. 2015).
The revealed DEGs often belong to large multi-gene families, which have members with tissue- and/or stage-specific characteristics of expression. In such cases, the general type of activity of the encoded proteins may be similar but with differences in regulation together with nuances of substrate and/or product specificity. This is especially true for the genes encoding cell wall proteins, like those most up-regulated in fibres XET/XTH and β-galactosidases. This is the way that a plant modifies the already deposited cell wall layers in a tissue- and stage-specific manner.
Important participants of cell metabolism regulation are transcription factors, the profile of which differs depending on cell physiological state. Among the up-regulated genes in the FIB sample are representatives of bZIP, C3H, CO-like, MYB-related, and TCP families. bZIP44 is involved in the network that controls carbohydrate partitioning via a mechanism that might include direct regulation of cell-wall invertase gene expression in Arabidopsis (Wobbes 2004). The set of transcription factors is quite specific in fibres forming the tertiary cell wall and does not include the TFs that were described for the secondary cell wall, including master switches NST1, SND1 and MYB46 (Kim et al. 2013; Zhong and Ye 2014; Taylor-Teeples et al. 2015). Genes for several MYB-related TFs are up-regulated in FIB (Fig. 4). Three of these genes, RSM3 (Lus10033212, Lus10010831, and Lus10028306), belong to the small RADIALIS-LIKE SANT/MYB (RSM1-4) subfamily. The RADIALIS gene has been identified as an important factor in the regulation of floral asymmetry; however, such TFs were also suggested to play roles in an as-yet unidentified developmental process that is not necessarily relevant to the floral architecture (Hamaguchi et al. 2008). Several members of the MYB-related family have binding sites in the promoters of COBL4 and IRX14 genes (Taylor-Teeples et al. 2015), differential expression of which was observed in fibres in our study (Table S2). One member of the CO-like family, CONSTANS (Lus10015619), may be involved in control of genes associated with proline or ethylene biosynthesis (Samach et al. 2000), whereas the representative of CCCH zinc finger proteins, AtC3H20/AtTZF2 (Lus10012922), may be involved in growth control and ABA and JA responses (Lee et al. 2012).

Hierarchical clustering and heatmap visualization of DEGs of transcription factors (FPKM > 16). FPKM values were used to cluster the DEGs using Pearson correlation and the average linkage method. Results were visualized using MeV v4.9.0. A Heatmap showing global visualization of 629 TF DEGs. Heatmap showing B down- and C up-regulated genes in isolated fibres using cut-off log2FC > 2. The bar with green-black-red gradation indicates high (red) to low (green) expression
Analysis of DEGs involved in hormonal regulation suggests that flax fibres at an advanced stage of specialization diminish the dependence on auxin and ethylene either by down-regulation of genes involved in their synthesis or by increasing transcript abundance for negative regulators of signalling. Importantly, the specific representatives of the multi-gene families were involved in tuning the response to hormones in fibres forming the tertiary cell wall. Though auxin redistribution was long considered as an inducer of G-layer deposition, no increase of its content in fibres was detected by sensitive analysis combined with cryosectioning of poplar tension wood (Hellgren et al. 2004). In yellow poplar (Liriodendron tulipifera), the expression of the majority of auxin-related genes was down-regulated in tension wood as compared to that in opposite wood (Jin et al. 2011). The effect of gibberellins on G-layer formation in tension wood of poplar was recently established (Gerttula et al. 2015); the only DEG involved in gibberellin metabolism or signalling detected in our experiments with the stringent criteria used was the GASA4 gene suppressed in the FIB sample (Table 1).
Upon transcriptome analysis, dramatic changes in expression level are detected for the large set of genes encoding various transporters (Table 1). Ion fluxes are known to be among the key regulators of cell metabolism (Hedrich 2012). Considering the origin and sequence of signals for tertiary cell wall induction and maintenance, the interplay between the changes in sets of transporters and intracellular regulatory proteins deserves further investigation, especially as the major participants are identified in the present study.
Cellulose deposition as the major process of tertiary cell wall formation
Tertiary cell wall, which is specific for fibres and is deposited at advanced stage of their specialization in many plant species, has very peculiar composition and structural design. These peculiarities may be reflected in the transcriptome, providing an opportunity for further insight into the mechanism of plant cell wall formation, which is still a stumbling block for molecular biologists.
One of the major processes ongoing in fibres at the analysed stage of development is cellulose synthesis (Fig. 5). It is performed by the multi-enzyme complex, which contains several types of highly homologous but distinct catalytic subunits—CESA glycosyltransferases (Richmond, Sommerville. 2000). Despite the very intensive cellulose synthesis in fibres, none of CESA genes was either significantly up-regulated in fibres relative to their levels in other samples or present among the most highly expressed genes. Recent studies reveal post-translational modifications, such as phosphorylation or s-acylation, as the factors that regulate cellulose biosynthesis (Jones et al. 2016; Kumar et al. 2016). They may be the reason for intensive cellulose biosynthesis during tertiary cell wall deposition despite the absence of an enriched abundance of corresponding transcripts. An additional factor could be the involvement of some auxiliary proteins in cellulose-synthesizing machinery (Jones et al. 2016). However, among the known ones, only COBL4 (Lus10035131) was up-regulated in FIB as compared to TOP. Transcription of genes for cellulose synthase-interactive protein 1 (CSI1) as well as KORRIGAN1 was not affected.

Distribution of radioactivity in the main cell components and in cell wall carbohydrates in particular in flax phloem fibres isolated 24 h after the intact plant photosynthesis with 14CO2
Two genes of metallothioneins 2, which are possible candidates for participation in cellulose synthesis, were at the very top of the genes with the most abundant transcripts. Some members of the metallothionein family were previously found to be enriched in phloem tissue in higher plants including flax (Guo et al. 2003; De Pauw et al. 2007; Roach and Deyholos 2007). In the study of cotton (Gossypium hirsutum L.) seed hairs, the most cellulose-enriched plant cell type, metallothionein, was proposed to be involved in the regulation of CESA complex formation by providing zinc ions to the Zn-binding domains of GhCESA proteins (Jacob-Wilk et al. 2006).
The next most highly expressed genes were those encoding lipid transfer proteins—small, soluble, cysteine-rich proteins characterized by a tunnel-like hydrophobic cavity—which makes them suitable for binding and transporting various lipids (Kader 1996; Salminen et al. 2016). Three of four LTP genes highly expressed in fibres (except Lus10015278) contain sequences encoding signal peptides for the secretory pathway. A number of LTPs are located in apoplast, making it unlikely these proteins play a role in intracellular lipid transfer (Kader 1996). Evidence has accumulated for the role of LTPs in the synthesis of lipid barrier polymers, such as cuticular waxes, suberin, cutin and sporopollenin (rev. Salminen et al. 2016). In flax fibres, contents of aromatics and waxes are extremely low (Morvan et al. 2003); therefore, high expression of LTPs in flax fibres during tertiary cell wall formation is unlikely to be associated with the synthesis of the lipid-containing polymers. Interestingly, tobacco NtLTP1.6 (TobLTP2) has in vitro cell wall loosening activity (Nieuwland et al. 2005) resembling that of expansins; the presence of the hydrophobic cavity is essential for such an effect (rev. Salminen et al. 2016). Moreover, it was shown that distribution of apoplastic GPI-anchored lipid transfer protein (LTPG) in Arabidopsis mature epidermal cells often showed a striated pattern resembling the pattern of cellulose microfibrils, and this pattern is dependent on cellulose microfibril formation (Ambrose et al. 2013). This may indicate that cell wall-localized LTPs are somehow involved in cellulose synthesis. High abundance of transcript encoding LTPs was previously described in other studies of cells with intensive cellulose deposition, including flax fibres (Roach and Deyholos 2007; Fenart et al. 2010), hemp fibres (De Pauw et al. 2007) and cotton seed hairs (Orford and Timmis 2000).
None of the annotated genes at least tentatively involved in cellulose synthesis and microfibril orientation are considerably up-regulated in fibres. This means that the described peculiarities of cellulose deposition in the tertiary cell wall, if dependent on newly synthesized proteins, are provided by the ones encoded by unknown (or not-fully-annotated) genes or proteins with relatively small changes in transcript abundance.
Genes encoding proteins potentially involved in synthesis and modification of rhamnogalacturonan I
The key matrix polysaccharide in tertiary cell walls belongs to the large and diverse family of RG-I pectic polymers, which are grouped together due to the presence of the backbone made of altering residues of rhamnose and galacturonic acid (Ridley et al. 2001; Yapo 2011; Mikshina et al. 2015a). The way of RG-I biosynthesis is understood quite poorly: several tens of glycosyltransferases (GT) are needed for its assembly (Ridley et al. 2001), among which only one involved in the formation of short side-chains has been reliably identified (Liwanag et al. 2012). As shown for flax and poplar fibres, G-layers contain tissue- and stage-specific version of RG-I (Gorshkova et al. 2010, 2015), which is the key agent of fibre function (Mikshina et al. 2015b). Because secondary and tertiary cell wall types have very distinct compositions of matrix polysaccharides with xylan dominating in the first one and absent in the other, and RG-I vice versa, the cells depositing secondary or tertiary cell walls may have corresponding differences in their transcriptomes. Indeed, genes involved in xylan synthesis are down-regulated in fibres forming the tertiary cell wall (Fig. 3A and Table S2) (Andersson-Gunnerås et al. 2006; Mizrachi et al. 2015). At the same time, RG-I synthesis is activated (Fig. 5), making it reasonable to search for the genes potentially involved in its formation and modification among those up-regulated in fibre genes.
Increased abundance of transcripts in fibres depositing the tertiary cell wall (Table 1) is evident for glycosyltransferases of GT2 (CSLG1, Lus10023057) and GT47 (Lus10013790) families of the CAZy database (http://www.cazy.org), same as for non-classified GT (FUT, Lus10035540). Lus10013790 is annotated as exostosin. In animals, such proteins are glycosyltransferases that take part in the synthesis of the extracellular matrix polymer heparin sulfate. This linear polysaccharide is composed of repeating disaccharide units (glucuronic acid linked to N-acetylglucosamine) (Esko and Selleck 2002) that to a certain extent resemble the RG-I structure. It is tempting to speculate that Lus10013790 is involved in the formation of the RG-I backbone of fibre-specific polysaccharide. The transcript levels of genes belonging to the GT47 family were increased upon induction of tension wood in poplar (Andersson-Gunnerås et al. 2006) and eucalypt (Mizrachi et al. 2015), but at the time of the cited papers, the presence of RG-I in the G-layer of tension wood fibres was not fully established, and the link between exostosin-like genes and RG-I could not be suggested. Though some enzymes of GT47, like MUR3 (Madson et al. 2003) and FRA8 (IRX7) (Zhong et al. 2005), were demonstrated to take part in quite different polysaccharide synthesis (MUR3 is required for the synthesis of xyloglucan, and FRA8 (IRX7) is required for the synthesis of glucuronoarabinoxylan), the homology of Lus10013790 to MUR3 and FRA8 (IRX7) genes is rather low (below 35 % of aa similarity). Thus, it is worth further effort to determine if members of GT47 in flax are indeed involved in RG-I backbone synthesis.
The other glycosyltransferase gene LusCSLG (Lus10023057), which is up-regulated in fibres, belongs to the GT2 family. The described CSLs synthesize polymeric chains made of β-(1→4)-linked monosaccharides: enzymes encoded by genes from CSLA clade were shown to synthesize β-(1→4)-mannans or glucomannans (Dhugga et al. 2004; Liepman et al. 2005) and CSLC—the backbone of xyloglucan made of β-(1→4)-glucose (Cocuron et al. 2007). CSLF, CSLH and probably CSLJ clades are involved in the formation of mixed-linkage glucan, which also contains β-(1→4)-glucose residues (Burton et al. 2006; Schwerdt et al. 2015). The functions of glycosyltransferases belonging to other CSL clades are not known. Putting together the high enrichment of flax fibre RG-I with long side-chains of β-(1→4)-galactans (Gorshkova et al. 1996; Mikshina et al. 2012) and the fibre- and stage-specific up-regulation of flax CSLG gene expression, it is reasonable to assume that the corresponding glycosyltransferase synthesizes β-(1→4)-galactans.
Previously, GALACTAN SYNTHASE 1 (GALS1) of GT92 was identified as a β-1,4-galactan: β-1,4-GalT in Arabidopsis (Liwanag et al. 2012). Such enzymes can also be involved in the addition of β-(1→4)-Gal side-chains to the RG-I backbone. Up-regulation of the GT92 gene was demonstrated during tension wood formation (Mizrachi et al. 2015). In our experiments, transcripts of five genes encoding enzymes of the GT92 family were detected: Lus10032118, Lus10014568, Lus10038387, Lus10024731 and Lus10008076 (Table S2). Among them, Lus10038387 showed the most evident pattern for specific transcript enrichment in the FIB sample.
RG-I deposited during G-layer formation is known to undergo substantial post-synthetic modification with the involvement of tissue-specific β-galactosidase (Roach et al. 2011; Mokshina et al. 2012; Gorshkova et al. 2015). Enrichment in transcripts encoding this enzyme was also revealed in fibres in our experiments (Table 1). Among the differentially expressed genes, increased transcript content for rhamnogalacturonate lyase family proteins (Lus10004281, Lus10019231) was also demonstrated. Rhamnogalacturonan lyase (RGL) specifically recognizes and cleaves α-(1→4)-glycosidic bonds between L-rhamnose and D-galacturonic acids in the backbone of RG-I (McDonough et al. 2004). Aspen tension wood (Andersson-Gunnerås et al. 2006) and fibre-enriched peels of flax stem appear to have increased levels of RGL transcripts (Roach and Deyholos 2007; Hobson et al. 2010). However, the presence of lyase activity in the plant cell wall is a debatable topic (Fry 2004). Most likely, enzymes annotated as RGLs do not act as lyases but rather modify RG-I in another way, such as binding to it and acting as an accessory protein for RG-I homo- or heterological interactions with cell wall polymers.
Concluding remarks
The RNA-Seq analysis of the flax phloem fibre whole transcriptome portrays plant cell at advanced stage of specialization and provides a solid basis for the in-depth analysis of fibre-specific processes and elucidation of the role of important players. In the case of cellulosic fibres, emphasis should be given, according to the results of RNA-Seq, to ion transporters, transcription factors, glycosyltransferases and accessory proteins involved in the formation of tertiary cell wall.
References
Ageeva MV, Petrovská B, Kieft H, Salnikov VV, Snegireva AV, van Dam JEG, Emons AMC, Gorshkova TA, van Lammeren AAM (2005) Intrusive growth of flax phloem fibers is of intercalary type. Planta 222:565–574
Ambrose C, DeBono A, Wasteneys G (2013) Cell geometry guides the dynamic targeting of apoplastic GPI-linked lipid transfer protein to cell wall elements and cell borders in Arabidopsis thaliana. PLoS ONE 8:e81215
Andersson-Gunnerås S, Mellerowicz EJ, Love J, Segerman B, Ohmiya Y, Coutinho PM, Nilsson P, Henrissat B, Moritz T, Sundberg B (2006) Biosynthesis of cellulose enriched tension wood in Populus: global analysis of transcripts and metabolites identifies biochemical and developmental regulators in secondary wall biosynthesis. Plant J 45:144–165
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist Soc Ser B 57:289–300
Berthet S, Demont-Caulet N, Pollet B, Bidzinski P, Cézard L, Le Bris P, Borrega N, Hervé J, Blondet E, Balzergue S et al (2011) Disruption of LACCASE4 and 17 results in tissue-specific alterations to lignification of Arabidopsis thaliana stems. Plant Cell 23:1124–1137
Bowling AJ, Vaughn KC (2008) Immunocytochemical characterization of tension wood: gelatinous fibers contain more than just cellulose. Am J Bot 95:655–663
Burton RA, Wilson SM, Hrmova M, Harvey AJ, Shirley NJ, Medhurst A, Stone BA., Newbigin EJ, Bacic A, Fincher GB (2006) Cellulose synthase-like CslF genes mediate the synthesis of cell wall (1,3;1,4)-β-D-glucans. Sci 311:1940–1942
Carpita N, McCann M (2000) The cell wall. In: Buchanan B, Gruissem W, Jones R (eds) Biochemistry and Molecular Biology of Plants. Rockwell, MD, American Society of Plant Physiologists, pp. 52–108
Cassan-Wang H, Goué N, Saidi MN et al (2013) Identification of novel transcription factors regulating secondary cell wall formation in Arabidopsis. Front Plant Sci 4:189. doi:10.3389/fpls.2013.00189
Cocuron J-C, Lerouxel O, Drakakaki G, Alonso AP, Liepman AH, Keegstra K, Raikhel NV, Wilkerson CG (2007) A gene from the cellulose synthase-like C family encodes a β-1,4 glucan synthase. Proc Natl Acad Sci USA 104:8550–8555
Dash PK, Cao Y, Jailani AK et al (2014) Genome-wide analysis of drought induced gene expression changes in flax (Linum usitatissimum). GM Crops Food, 5(2):106–119
Day A, Addi M, Kim W, David H, Bert F, Mesnage P, Rolando C, Chabbert B, Neutelings G, Hawkins S (2005) ESTs from the fibre-bearing stem tissues of flax (Linum usitatissimum L.): expression analyses of sequences related to cell wall development. Plant Biol 7:23–32
De Pauw MA, Vidmar JJ, Collins J, Bennett RA, Deyholos MK (2007) Microarray analysis of bast fibre producing tissues of Cannabis sativa identifies transcripts associated with conserved and specialised processes of secondary wall development. Funct Plant Biol 34:737–749
Devillard C, Walter C (2014) Formation of plant tracheary elements in vitro—a review. New Zeal J For Sci 44:22. doi:10.1186/s40490-014-0022-7
Dhugga KS, Barreiro R, Whitten B et al (2004) Guar seed beta-mannan synthase is a member of the cellulose synthase super gene family. Sci 303:363–366
Endo S, Pesquet E, Yamaguchi M, Tashiro G, Sato M, Toyooka K, Nishikubo N, Udagawa-Motose M, Kubo M, Fukuda H, Demura T (2009) Identifying new components participating in the secondary cell wall formation of vessel elements in Zinnia and Arabidopsis. Plant Cell 21(4):1155–1165
Esau K (1977) Anatomy of Seed Plants, 2nd edn. New York, John Wiley and Sons.
Esko JD, Selleck SB (2002) Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem 71:435–471
Fahn A (1990) Plant Anatomy. 4th Ed. Pergamon Press, Oxford, UK.
Fenart S, Ndong YP, Duarte J et al (2010) Development and validation of a flax (Linum usitatissimum L.) gene expression oligo microarray. BMC Genomics 11:592
Fraser CM, Chapple C (2011) The phenylpropanoid pathway in Arabidopsis. Arabidopsis Book 9:e0152. doi:10.1199/tab.0152
Fry SC (2004) Primary cell wall metabolism: tracking the careers of wall polymers in living plant cells. New Phytol 161:641–675
Fry SC, Smith RC, Renwick KF, Martin DJ, Hodge SK, Matthews KG (1992) Xyloglucan endotransglycosylase, a new wall-loosening enzyme activity from plants. Biochem J 282:821–828
Fukuda H (1992) Tracheary element formation as a model system of cell-differentiation. Int Rev Cytol 136:289–332
Gerttula S, Zinkgraf M, Muday G, Lewis D, Ibatullin FM, Brumer H, Hart F, Mansfield SD, Filkov V, Groover A (2015) Transcriptional and hormonal regulation of gravitropism of woody stems in Populus. Plant Cell 27:10.1105/tpc.15.00531
Goecks J, Nekrutenko A, Taylor J; The Galaxy Team (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11:R86
Goff L, Trapnell C, Kelley D (2013) cummeRbund: Analysis, exploration, manipulation, and visualization of Cufflinks high-throughput sequencing data. R package version 2.12.0.
Gorshkova TA, Wyatt SE, Salnikov VV, Gibeaut DM, Ibragimov MR, Lozovaya VV, Carpita NC (1996) Cell-wall polysaccharides of developing flax plants. Plant Physiol 110:721–729
Gorshkova TA, Chemikosova SB, Lozovaya VV, Carpita NC (1997) Turnover of galactans and other cell wall polysaccharides in developing flax plants. Plant Physiol 114:723–729
Gorshkova TA, Sal’nikov VV, Chemikosova SB, Ageeva MV, Pavlencheva NV, van Dam JEG (2003) The snap point: a transition point in Linum usitatissimum bast fiber development. Ind Crop Prod 18:213–221
Gorshkova TA, Gurjanov OP, Mikshina PV, Ibragimova NN, Mokshina NE, Salnikov VV, Ageeva MV, Amenitskii SI, Chernova TE, Chemikosova SB (2010) Specific type of secondary cell wall formed by plant fibers. Russ J Plant Physiol 57:328–341
Gorshkova T, Brutch N, Chabbert B, Deyholos M, Hayashi T, Lev-Yadun S, Mellerowicz EJ, Morvan C, Neutelings G, Pilate G (2012) Plant fiber formation: state of the art, recent and expected progress, and open questions. Cr Rev. Plant Sci 31(3):201–228
Gorshkova T, Mokshina N, Chernova T et al (2015) Aspen tension wood fibers contain β-(1→4)-galactans and acidic arabinogalactans retained by cellulose microfibrils in gelatinous walls. Plant Physiol 169:2048–2063
Goulao LF, Vieira-Silva S, Jackson PA (2011) Association of hemicelluloseand pectin-modifying gene expression with Eucalyptus globulus secondary growth. Plant Physiol Biochem 49:873–881
Guo WJ, Bundithya W, Goldsbrough PB (2003) Characterization of the Arabidopsis metallothionein gene family: tissue-specific expression and induction during senescence and in response to cupper. New Phytol 59:369–381
Hamaguchi A, Yamashino T, Koizumi N, Kiba T, Kojima M, Sakakibara H, Mizuno T (2008) A small subfamily of Arabidopsis RADIALIS-LIKE SANT/MYB genes: a link to HOOKLESS1-ediated signal transduction during early morphogenesis. Biosci Biotechnol Biochem 72(10):2687–2696
Hamilton ES, Schlegel AM, Haswell ES (2015) United in Diversity: Mechanosensitive Ion Channels in Plants. Annu Rev Plant Biol 66:113–137
Hayashi T, Kaida R, Kaku T, Baba K (2010) Loosening xyloglucanprevents tensile stress in tree stem bending but accelerates the enzymatic degradation of cellulose. Russ J Plant Physiol 57:316–320
Healey AL, Lee DJ, Furtado A, Simmons BA, Henry RJ (2015) Efficient Eucalypt cell wall deconstruction and conversion for sustainable lignocellulosic biofuels. Front Bioeng Biotechnol 3:190
Hedrich R (2012) Ion Channels in Plants. Physiol Rev 92(4):1777–1811
Hellgren JM, Olofsson K, Sundberg B (2004) Patterns of auxin distribution during gravitational induction of reaction wood in poplar and pine. Plant Physiol 135:212–220
Hobson N, Roach MJ, Deyholos MK (2010) Gene expression in tension wood and bast fibres. Russ J of Plant Physiol 5:321–327
Hotte NSC, Deyholos MK (2008) A flax fibre proteome: identification of proteins enriched in bast fibres. BMC Plant Biol 8:52
Huis R, Hawkins S, Neutelings G (2010) Selection of reference genes for quantitative gene expression normalization in flax (Linum usitatissimum L.). BMC Plant Biol 10:71. doi:10.1186/1471-2229-10-71
Jacob-Wilk D, Kurek I, Hogan P, Delmer DP (2006) The cotton fiber zinc-binding domain of cellulose synthase A1 from Gossypium hirsutum displays rapid turnover in vitro and in vivo. Proc Natl Acad Sci USA 103:12191–12196
Jin H, Do J, Moon D, Noh EW, Kim W, Kwon M (2011) EST analysis of functional genes associated with cell wall biosynthesis and modification in the secondary xylem of the yellow poplar (Liriodendron tulipifera) stem during early stage of tension wood formation. Planta 234:959–977
Jin JP, Zhang H, Kong L, Gao G, Luo JC (2014) PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucl Acids Res 42(D1):D1182–D1187
Jones DM, Murray CM, Ketelaar KDJ, Thomas JJ, Villalobos JA, Wallace IS (2016) The emerging role of protein phosphorylation as a critical regulatory mechanism controlling cellulose biosynthesis. Front Plant Sci 7:684
Kader JC (1996) Lipid transfer proteins in plants. Annu Rev Plant Physiol Plant Mol Biol 47:627–654
Kakimoto T (2001) Identification of plant cytokinin biosynthetic enzymes as dimethylallyl diphosphate:ATP/ADP isopentenyltransferases. Plant and Cell Physiol 42:677–685
Kaku T, Serada S, Baba K, Tanaka F, Hayashi T (2009) Proteomic analysis of the G-Layer in poplar tension wood. J Wood Sci 55:250–257
Kant S, Bi YM, Zhu T, Rothstein SJ (2009) SAUR39, a small auxin-up RNA gene, acts as a negative regulator of auxin synthesis and transport in rice. Plant Physiol 151:691–701
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi:10.1186/gb-2013-14-4-r36
Kumar M, Wightman R, Atanassov I, Gupta A, Hurst CH, Hemsley PA, Turner S (2016) S-Acylation of the cellulose synthase complex is essential for its plasma membrane localization. Science 353:166–169
Lee SJ, Jung HJ, Kang H, Kim SY (2012) Arabidopsis zinc finger proteins AtC3H49/AtTZF3 and AtC3H20/AtTZF2 are involved in ABA and JA responses. Plant Cell Physiol 53(4):673–686
Liepman AH, Wilkerson CG, Keegstra K (2005) Expression of cellulose synthase-like (Csl) genes in insect cells reveals that CslA family members encode mannan synthases. Proc Natl Acad Sci USA 102:2221–2226
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the \(2^{-\Delta\Delta{{\text{C}}}_{\text{T}}}\)method. Methods 25:402–408
Liwanag AJM, Ebert B, Verhertbruggen Y, Rennie EA, Rautengarten C, Oikawa A, Andersen MCF, Clausen MH, Scheller HV (2012) Pectin biosynthesis: GALS1 in Arabidopsis thaliana is a β-1,4-galactan β-1,4-galactosyltransferase. Plant Cell 24:5024–5036
Love GD, Snape CE, Jarvis MC, Morrison IM (1994) Determination of phenolic structures in flax fibre by solid state 13C NMR. Phytochem 35:489–492
Madson M, Dunand C, Li X, Verma R, Vanzin GF, Caplan J, Shoue DA, Carpita NC, Reiter W-D (2003) The MUR3 gene of Arabidopsis thaliana encodes a xyloglucan galactosyltransferase that is evolutionarily related to animal exostosins. Plant Cell 15:1662–1670
McDonough MA, Kadirvelraj R, Harris P, Poulsen JC, Larsen S (2004) Rhamnogalacturonan lyase reveals a unique three-domain modular structure for polysaccharide lyase family 4. FEBS Lett 565:188–194
Mellerowicz EJ, Gorshkova TA (2012) Tensional stress generation in gelatinous fibres: a review and possible mechanism based on cell-wall structure and composition. J Exp Bot 63:551–565
Mellerowicz EJ, Immerzeel P, Hayashi T (2008) Xyloglucan: the molecular muscle of trees. Ann Bot 102:659–665
Mikshina PV, Gurjanov OP, Mukhitova FK, Petrova AA, Shashkov AS, Gorshkova TA (2012) Structural details of pectic galactan from the secondary cell walls of flax (Linum usitatissimum L.) phloem fibres. Carbohydr Polym 87:853–861
Mikshina PV, Petrova AA, Faizullin DA, Zuev YF, Gorshkova TA (2015a) Tissue-specific rhamnogalacturonan I forms the gel with hyperelastic properties. Biochemistry-Moscow 80(7):915–924
Mikshina PV, Petrova AA, Gorshkova TA (2015b) Functional diversity of rhamnogalacturonans I. Russ. Chem. Bulletin 64(5):1014–1023
Miyawaki K, Matsumoto-Kitano M, Kakimoto T (2004) Expression of cytokinin biosynthetic isopentenyltransferase genes in Arabidopsis: tissue specificity and regulation by auxin, cytokinin, and nitrate. Plant J 37:128–138
Mizrachi E, Maloney VJ, Silberbauer J, Hefer CA, Berger DK, Mansfield SD, Myburg AA (2015) Investigating the molecular underpinnings underlying morphology and changes in carbon partitioning during tension wood formation in Eucalyptus. New Phytol 206:1351–1363
Mokshina NE, Ibragimova NN, Salnikov VV, Amenitskii SI, Gorshkova TA (2012) Galactosidase of plant fibers with gelatinous cell wall: Identification and localization. Russ J Plant Physiol 59(2):246–254
Mokshina N, Gorshkova T, Deyholos MK (2014) Chitinase-like (CTL) and cellulose synthase (CESA) gene expression in gelatinous-type cellulosic walls of flax (Linum usitatissimum L.) bast fibers. PLoS ONE 9(6):e97949
Morvan C, Andème-Onzighi C, Girault R, Himmelsbach DS, Driouich A, Akin DE (2003) Building flax fibres: more than brick in the walls. Plant Physiol Biochem 41:935–944
Muñoz C, Baeza J, Freer J, Mendonça RT (2011) Bioethanol production from tension and opposite wood of Eucalyptus globulus using organosolv pretreatment and simultaneous saccharification and fermentation. J Ind Microbiol Biotechnol 38:1861–1866
Nieuwland J, Feron R, Huisman BA, Fasolino A, Hilbers CW, Derksen J, Mariani C (2005) Lipid transfer proteins enhance cell wall extension in tobacco. Plant Cell 17:2009–2019
Oikawa A, Joshi HJ, Rennie EA, Ebert B, Manisseri C, Heazlewood JL, Scheller HV (2010) An integrative approach to the identification of Arabidopsis and rice genes involved in xylan and secondary wall development. PLoS ONE 5:e15481
Orford SJ, Timmis JN (2000) Expression of a lipid transfer protein gene family during cotton fibre development. Biochim Biophys Acta 1483(2):275–284
Paux E, Carocha V, Marques C, de Sousa AM, Borralho N, Sivadon P, Grima-Pettenati J (2005) Transcript profiling of Eucalyptus xylem genes during tension wood formation. New Phytol 167:89–100
Pilate G, Dejardin A, Laurans F, Leple J-C (2004) Tension wood as a model for functional genomics of wood formation. New Phytol 164:63–72
Richmond TA, Somerville CR (2000) The cellulose synthase superfamily. Plant Physiol 124:495–498
Ridley BL, O’Neill MA, Mohnen D (2001) Pectins: structure, biosynthesis, and oligogalacturonide-related signaling. Phytochem 57:929–967
Roach MJ, Deyholos MK (2007) Microarray analysis of flax (Linum usitatissimum L.) stems identifies transcripts enriched in fibre-bearing phloem tissues. Mol Genet and Genom 278:149–165
Roach MJ, Mokshina NY, Badhan A, Snegireva AV, Hobson N, Deyholos MK, Gorshkova TA (2011) Development of cellulosic secondary walls in flax fibers requires beta-galactosidase. Plant Physiol 156:1351–1363
Salminen TA, Blomqvist K, Edqvist J (2016) Lipid transfer proteins: classification, nomenclature, structure, and function. Planta 244(5):971–997
Salnikov VV, Ageeva MV, Gorshkova TA (2008) Homofusion of Golgi secretory vesicles in flax phloem fibers during formation of the gelatinous secondary cell wall. Protoplasma 233:269–273
Samach A, Onouchi H, Gold SE, Ditta GS, Schwarz-Sommer Z, Yanofsky MF, Coupland G (2000) Distinct roles of CONSTANS target genes in reproductive development of Arabidopsis. Science 288(5471):1613–1166
Schwerdt JG, MacKenzie K, Oehme D et al (2015) Evolutionary dynamics of the cellulose synthase gene superfamily in grasses. Plant Physiol 168:968–983
SEQC/MAQC-III Consortium (2014) A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat Biotechnol 32(9):903–914
Taylor-Teeples M, Lin L, De Lucas M, Turco G, Toal TW, Gaudinier A et al (2015) An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature 517:571–575
Thimm O, Blaesing OE, Gibon Y, Nagel A, Meyer S, Krueger P, Selbig J, Mueller LA, Rhee SY, Stitt M (2004) Mapman: a user-driven tool to display genomics datasets onto diagrams of metabolic pathways and other biological processes. Plant J 37:914–939
Tian Q, Uhlir NJ, Reed JW (2002) Arabidopsis SHY2/IAA3 inhibits auxin-regulated gene expression. Plant Cell 14:301–319
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7(3):562–578
Updegraff DM (1969) Semi-micro determination of cellulose in biological materials. Anal Biochem 32:420–424
Wang H, Avci U, Nakashima J, Hahn MG, Chen F, Dixon RA (2010) Mutation of WRKY transcription factors initiates pith secondary wall formation and increases stem biomass in dicotyledonous plants. Proc Natl Acad Sci USA 107:22338–22343
Wang ZW, Hobson N, Galindo L et al (2012) The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J 72:461–473
Wendel JF, Cronn RC, Alvarez I, Liu B, Small RL, Senchina DS (2002) Intron size and genome size in plants. Mol Biol Evol 19:2346–2352
Wobbes B (2004) Control of plant carbohydrate partitioning by the Arabidopsis thaliana ATB2 bZIP transcription factor gene. Dissertation, University of Utrecht, Niederlande
Yapo BM (2011) Rhamnogalacturonan-I: A structurally puzzling and functionally versatile polysaccharide from plant cell walls and mucilages. Polym Rev 51:391–413
Yilmaz ND (2015) Agro-residual fibers as potential reinforcement elements for biocomposites. In: Thakur VJ (ed) Lignocellulosic Polymer Composites: Processing, Characterization, and Properties. Scrivener Publishing, MA, pp 233–270
Zhong R, Ye Z-H (2014) Complexity of the transcriptional network controlling secondary wall biosynthesis. Plant Sci 229:193–207
Zhong R, Peña MJ, Zhou GK, Nairn CJ, Wood-Jones A, Richardson EA, Morrison WH 3rd, Darvill AG, York WS, Ye ZH (2005) Arabidopsis fragile fiber8, which encodes a putative glucuronyltransferase, is essential for normal secondary wall synthesis. Plant Cell 17:3390–3408
Acknowledgements
This work was partially supported by grants of the Russian Foundation for Basic Research (#14-04-01778; RNA-Seq of isolated fibres, qRT-PCR experiments), the Russian Science Foundation (#16-14-10256; RNA-Seq of top and middle parts of flax stem, 14C-distribution experiments, bioinformatics).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material

Rights and permissions
About this article

{kind=link}
Cite this article
Gorshkov, O., Mokshina, N., Gorshkov, V. et al. Transcriptome portrait of cellulose-enriched flax fibres at advanced stage of specialization. Plant Mol Biol 93, 431–449 (2017). https://doi.org/10.1007/s11103-016-0571-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-016-0571-7