
Abstract
Aim
The pharmacokinetic (PK) properties of cyclosporine (CsA) in renal transplant recipients are patient- and time-dependent. Knowledge of this time-related variability is necessary to maintain or achieve CsA target exposure. Here, we aimed to identify factors explaining variabilities in CsA PK properties and characterize time-varying clearance (CL/F) by performing a comprehensive analysis of CsA PK factors using population PK (popPK) modeling of long-term follow-up data from our institution.
Methods
In total, 3674 whole-blood CsA concentrations from 183 patients who underwent initial renal transplantation were analyzed using nonlinear mixed-effects modeling. The effects of potential covariates were selected according to a previous study and well-accepted theoretical mechanisms. Model-informed individualized therapeutic regimens were also evaluated.
Results
A two-compartment model adequately described the data and the estimated mean CsA CL/F was 32.6 L h−1 (relative standard error: 5%). Allometrically scaled body size, hematocrit (HCT) level, CGC haplotype carrier status, and postoperative time may contribute to CsA PK variability. The CsA bioavailability in patients receiving a prednisolone dose (PD) of 80 mg was 20.6% lower than that in patients receiving 20 mg. A significant decrease (52.6%) in CL/F was observed as the HCT increased from 10.5% to 60.5%. The CL/F of the non-CGC haplotype carrier was 14.4% lower than that of the CGC haplotype carrier at 3 months post operation.
Conclusions
By monitoring body size, HCT, PD, and CGC haplotype, changes in CsA CL/F over time could be predicted. Such information could be used to optimize CsA therapy. CsA dose adjustments should be considered in different postoperative periods.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cyclosporine (CsA), a potent calcineurin inhibitor, is commonly used to prevent allograft rejection after renal transplantation [1, 2]. With the introduction of CsA combination therapy, the survival rates of transplant patients, particularly short-term outcomes, have greatly improved [3]. However, prolonged use of CsA leads to substantial toxicity, which may reduce long-term renal allograft survival as well as increase the risk of cardiovascular morbidity and mortality [4,5,6]. Most adverse events and rejection rates may correlate with the concentration of CsA administered.
CsA exhibits unique pharmacokinetics (PK), including low bioavailability (approximately 25%; range 10–89%) owing to its poor aqueous solubility and low transmembrane permeability in the intestine mediated by P-glycoprotein (P-gp) [1, 7]. In addition, CsA binds extensively to erythrocytes [8], is predominantly metabolized by cytochrome P450 (CYP) 3A isoenzymes [9], and is subsequently eliminated in the bile [10]. Meanwhile, CsA has a narrow therapeutic index and large inter- and intra-individual PK variability, making it essential to conduct routine therapeutic drug monitoring (TDM) to optimize the CsA dosage regimen and minimize adverse effects [11].
Population PK (popPK), which is a superior approach to classical PK analysis, can be used to obtain population standard values and identify covariates with sparse sampling during TDM [12,13,14]. Currently, numerous popPK models have been developed to quantitatively describe the PK characteristics of CsA [15]. Several clinical factors, including body weight, postoperative time, and hematocrit (HCT), have also been identified to explain the PK variability of CsA [16].
The PK parameters of CsA in renal transplant recipients are patient- and time-dependent. The dose required to achieve targeted whole-blood concentrations of CsA varies considerably among patients and according to the time after transplantation. Two distinct challenges exist for CsA dosage individualization in transplant recipients, i.e., predicting the initial dose of CsA without any known concentration–time data in a particular patient and adjusting the dose over time after transplantation. To maintain or achieve CsA target exposure, knowledge of time-varying PK characteristics is necessary.
The postoperative day (POD) is a combined reflection of various time-related factors, such as the recovery of gastrointestinal function [17] and tapering of co-administered steroid doses [18]. During the early stages after transplantation, gastrointestinal function is abnormal, which decreases the bioavailability of CsA. With the recovery of gastrointestinal function during therapy, CsA absorption improves, thereby decreasing the apparent clearance (CL/F) [19]. However, tapering the steroid dosage during immunosuppressive therapy may reduce the expression of CYP3A and P-gp, leading to increased absorption and accelerated metabolism of CsA [20]. Moreover, an increase in the HCT level leads to increased binding of CsA to erythrocytes, resulting in elevated concentrations and decreased CL/F [21,22,23]. In addition to the abovementioned considerations, other unidentified confounding factors may contribute to the complexity of the impact of POD. Therefore, a clearer understanding of POD as a covariate is required for dose optimization.
Although various studies have been conducted to describe the tendency of CsA CL/F over POD [21, 24], most studies have had a short follow-up (less than 1 year), resulting in limited information regarding long-term time-related variability in CsA PK [15, 16]. Only one study conducted by Fanta et al. reported time-varying CsA PK properties in a long-term follow-up (> 16 years) of pediatric patients [25]. They reported that the bioavailability of CsA increases rapidly in the immediate postoperative period and then decreases gradually to reach the initial value approximately 1–1.5 years after transplantation. Saturable presystemic metabolism and the effects of NR1I2 polymorphism were found to contribute to this tendency. However, no such research has been reported in adults.
In this study, we aimed to identify factors affecting variability in CsA PK and characterize the time-varying CL/F of CsA in a comprehensive analysis of the effects of demographic, clinical, and genetic factors on CsA PK using popPK modeling of long-term follow-up data available from our institution. Model-informed individualized therapeutic regimens were also evaluated.
Materials and Methods
Patients and Data Collection
Data were collected from 183 adults (122 men and 61 women) who underwent renal transplantation at Huashan Hospital. The inclusion criteria were as follows: (1) allograft renal transplantation for the first time, (2) age greater than or equal to 18 years, and (3) CsA-based triple immunosuppressive regimen used. The exclusion criteria were as follows: (1) administration of the conventional oral formulation of CsA, (2) undergoing dialysis treatment, and (3) required covariate data missing. Demographic and pathophysiological data were obtained during routine clinical visits from July 2003 to December 2016. Missing data were determined according to adjacent available data. The methods used to handle missing covariate data have been described in Table S1.
In total, 3674 whole-blood CsA concentrations were available for model development. Of these, 3326 sparse-PK samples of CsA predose concentrations (C0) and 2-h postdose concentrations (C2) were retrospectively collected from follow-up TDM. Additionally, 24 concentration–time profiles (259 full-PK samples) were evaluated during the 12 h after the morning CsA dose within the first 90 days after transplantation. Whole-blood samples were primarily obtained before dosing and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, and 12 h after dosing. In addition, 89 other samples were collected during the absorption phase (1, 2, 3, 4, and 6 h after dosing). All samples were stored at − 20 °C until analysis.
The study protocols were approved by the Ethics Committee of Huashan Hospital and conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent before enrolment in the study.
Immunosuppressive Therapy
All patients were administered combined immunosuppressive therapy, including a CsA microemulsion (Neoral; Novartis Pharma Schweiz AG, Emberbach, Germany), mycophenolate mofetil (MMF; CellCept; Roche Pharma Ltd., Shanghai, China) and corticosteroids. The initial dose of CsA was 5 mg kg−1 day−1, administered as two doses under fasting conditions immediately after surgery. Subsequent doses were empirically adjusted to achieve target concentrations based on local guidelines (Text S1) [26]. MMF (0.5–3 g day−1) was administered according to body size and POD. This schedule was followed with oral prednisolone (80 mg day−1), and the dosage was gradually decreased by 10 mg day−1 until reaching 20 mg day−1 after 10 d. The dosage was further tapered to 15, 10, and 5 mg day−1 by months 1, 3, and 6, respectively.
Determination of CsA Concentration
Whole-blood samples were collected from July 2003 to April 2011 and analyzed using a well-validated fluorescence polarization immunoassay (FPIA) on an AxSYM Abbott diagnostic system (Abbott Diagnostics, Chicago, IL, USA). Samples collected from May 2011 to December 2016 were analyzed using a chemiluminescent microparticle immunoassay (CMIA) on an Architect I2000 system (Abbott Diagnostics).
The following formula (Eq. 1) [27] was used to convert the CMIA measured C0 before modeling, owing to the systematic biases and cross-reactivity of metabolites between the methods.
AxSYM represents the FPIA performed using an AxSYM Analyzer, whereas CMIA was performed on an Architect system, as described above.
For AxSYM, the limit of detection (LOD) was 21.8 ng mL−1, and the calibration range was 40–800 ng mL−1; for CMIA, the LOD was 25 ng mL−1, and the calibration range was 30–1500 ng mL−1.
Genotyping and Haplotype Analysis
Five single-nucleotide polymorphisms (SNPs), i.e., CYP3A4*1G, CYP3A5*3, and ABCB1 C1236T, G2677T/A, and C3435T, were genotyped by an independent external contractor (GeneCore BioTechnologies Co., Ltd., Shanghai, China) using a DNA sequencing apparatus (Applied Biosystems 3730; Thermo Fisher Scientific, Waltham, MA, USA) [28]. Deviations from the Hardy–Weinberg equilibrium were examined using Pearson’s χ2-test. Linkage disequilibrium (LD) between different pairs of ABCB1 SNPs was determined using the absolute standardized LD coefficient. Further details are available in Text S2.
PopPK Model Development
PopPK model was developed using nonlinear mixed-effects modeling software (NONMEM version 7.4; ICON Development Solutions, Ellicott City, MD, USA), with Pirana 2.9 as an interface for Perl Speaks NONMEM (PsN; version 4.9.0) [29]. Graphical analyses were processed using R software (version 3.5.0; http://www.r-project.org/). The first-order conditional estimation method, including η-ε interactions (FOCE-I), was employed throughout the method-building procedure [30].
Base Model
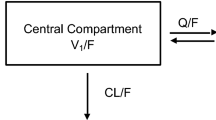
Based on visual inspection of the data and a review of the literature, a two-compartment model with first-order absorption and lag time was used to describe CsA PK [16]. The estimated parameters included CL/F, central volume of distribution (Vc/F), inter-compartmental clearance (Q/F), apparent peripheral volume of distribution (Vp/F), absorption rate constant (Ka), absorption lag time (Tlag), and bioavailability (F) relative to a population standard value defined as 1.
Between-subject variability (BSV) was estimated for all parameters, except Tlag, and was assumed to be log-normally distributed. Proportional and combined proportional as well as additive structures were tested to describe the residual unexplained variability.
Covariate Model
Demographic and pathophysiological data, as well as concomitant medications (Table 1), were evaluated as covariates. Data extracted from medical records, including body size, HCT, PODs, and prednisolone dose (PD), were evaluated as possible covariates of CsA PK. These covariates were selected according to a previous study and their clinical relevance [16]. The correlations between preselected covariates and the changes of covariates with POD were investigated graphically.
As the most frequently identified covariate in the final models, the change in CL/F and volume of distribution as a function of body weight was described allometrically [31, 32]. Body size was based on fat-free mass (FFM) predicted from total body weight, height, and sex (Text S3) [33].
Considering the hypothesis that tapering PD may significantly influence bioavailability [34, 35], the relationship between PD and F was described using linear, Emax or sigmoidal Emax model (Eq. 2–4), as follows:
F20 is the bioavailability of CsA for a PD of 20 mg, assumed to be 1. Slope is a dose-dependent linear change in F. Parameters Fmax and ED50 are the maximal increase in F and the dose above 20 mg, which corresponds to half the Fmax, respectively. The parameter γ is a shape parameter.
As for substrates of CYP3A and P-gp, we also comprehensively evaluated the influence of genotype, including single gene sites, combined genotypes, and haplotypes of the drug-metabolizing enzymes CYP3A5*3 and CYP3A4*1G and the multidrug resistance transporter ABCB1 with C1236T, G2677T/A, and C3435T polymorphisms.
Potential covariates were screened using a stepwise approach [30]. The influence of other continuous covariates was explored using a linear, exponential, and power function model. Categorical variables, such as SNPs and concomitant medications, were investigated by estimating the fractional change in one group compared with that in the other groups.
Model Selection Criteria
Visual model fit was evaluated using standard goodness-of-fit (GOF) criteria, reductions in the objective function value (OFV), and acceptable precision on estimates [30]. A covariate was considered significant if its inclusion decreased the OFV by more than 3.84 (χ2-test, P < 0.05, df = 1) and if backward elimination of the covariate increased the OFV by more than 10.83 (χ2-test, P < 0.001, df = 1). Moreover, a clear pharmacological or biological basis was also considered as covariates were added. During the model development process, the condition numbers were calculated, and no more than 1,000 were kept to avoid over-parameterization [36].
Model Evaluation
Fifty-six patients from the evaluation group were included in the analysis to examine the predictability of the final model. The adequacy of the model was externally evaluated using GOF plots, prediction-corrected visual predictive checks (pcVPCs), and normalized prediction distribution errors (NPDEs) [37, 38]. A nonparametric bootstrap was employed to assess the robustness of the model parameter estimates [39].
The dataset was simulated 2000 times for the pcVPCs and NPDEs. The 95% confidence intervals for the median, and the 5th and 95th percentiles of the simulations at different time points were calculated and graphically compared with the observations in pcVPCs, using automatic binning. The NPDE was determined using an add-on R package [38]. A histogram of the NPDE distribution as well as plots of NPDE versus PRED and NPDE versus POD were used to evaluate the final model.
For the nonparametric bootstrap procedure, 500 bootstrap datasets were generated by random sampling with replacement using Perl modules [40]. The median and the 2.5th to 97.5th percentiles of the parameters after bootstrap runs with successful convergence were compared with the final model parameter estimates.
Dosing Regimen Optimization
Monte Carlo simulations were conducted using parameter estimates from the final model to determine the optimal starting dosing regimen and achieve the target concentration during different postoperative periods (Text S1) [26]. The CsA dose was simulated from 50 mg q12h to 300 mg q12h for a standard-sized subject (FFM 50 kg) with different covariate levels (Table S2). Time-concentration profiles were simulated based on 1000 hypothetical individuals, and the steady-state C0/C2 value for each simulated subject was calculated. The median and the 25th to 75th percentiles of a steady-state C0/C2 value were calculated to select the optimal dosing regimen.
Results
Patients
Demographic characteristics and clinical data of the study population are presented in Table 1. In total, 3674 CsA whole-blood measurements were available from 183 renal transplant recipients. A median of 15 CsA observations was obtained for each patient (range 2–50). A description of the sampling points is provided in Table S3. Sampling occasions varied from day 1 to day 5998 (> 16 years) after transplantation (Table S4) and CsA doses ranged from 25 to 300 mg twice daily.
All allele frequencies of CYP3A4*1G, CYP3A5*3, and ABCB1 genetic polymorphisms were in Hardy–Weinberg equilibrium (Table 2). D’ values between ABCB1 G2677T/A or C1236T and C3435T were 0.69 and 0.75, respectively, whereas that between ABCB1 G2677T/A and C1236T was 0.60. These results indicate that ABCB1 C1236T and G2677T/A were in LD with C3435T. Haplotype frequencies of ABCB1 C1236T-G2677T/A-C3435T were calculated using SHEsis [41]. Only haplotypes with frequencies and patient proportions greater than 8% were analyzed (Table S5).
Patients were divided into two groups: data from 127 patients were used for model development, and data from 56 patients were used for model evaluation. Sixteen patients without genetic information were included in the evaluation dataset. No concentrations below the lower quantification limit were included in the analysis.
PopPK Model Development
Base Model
A two-compartment model with first-order absorption and lag time was selected as the base model to describe CsA PK. The data was not sufficiently informative to support estimation of complex absorption models, such as zero-order model, Erlang model, and Weibull model [42,43,44]. The exponential model provided the best results for the residual variability. The BSV of the mean CL/F in the base model was 24.2%, with a relative standard error of 9.6%. The parameter estimates and associated precisions are listed in Table S6.
Covariate Model
Before the stepwise process, the correlation between PK parameters and potential covariates was investigated graphically. The CsA CL/F tended to first decrease with POD after renal transplantation, and then increase, eventually reaching stability (Fig. 1), consistent with the results from Fanta et al. [25]. This was further demonstrated by the boxplots of CsA daily dose normalized C0 over different postoperative periods (Figure S1). Therefore, POD was incorporated into the base model exponentially; the GOF improved, and the OFV decreased by − 54.1 (Table S6). The GOF plots are presented in Figure S5.

Empirical Bayes estimates of cyclosporine clearance (CL/F) during the post-transplantation follow-up. The model-predicted typical CL/F (red dashed line) and the individual CL/F (blue circles) are shown
Although the base model with POD could partly describe the changes in CsA CL/F over time, a model in which covariates describe this time dependency is more useful for TDM. The correlations between preselected covariates and the changes of covariates with POD were investigated graphically. First, the changes in covariates and POD were examined by graphical inspection to identify potential covariates (Figure S2 and Figure S3). Then, the identified covariates were incorporated into the base model to test the effects on CsA PK properties.
The influence of patient body size on CsA disposition was best described by allometric scaling based on FFM rather than total body weight for all PK disposition parameters, with the OFV reduced by − 11.2 (P < 0.001). Considering that the HCT level was low during the early postoperative period and increased during the first months after transplantation, the effect of the HCT on CsA clearance was investigated. The OFV substantially decreased by − 41.4 (P < 0.001) when the HCT was included, indicating a significant model improvement. A decrease (52.6%) in CL/F was observed as the HCT increased from 10.5% to 60.5%.
As the PD was tapered dramatically during the initial stage of transplantation, the effects of prednisolone on F were added to the model because a high PD may influence CsA absorption nonlinearly. Therefore, nonlinear Emax model describing the effect of PD on F (Eq. 3) reduced the OFV by − 24.4 (P < 0.001) and was superior to a linear model (ΔOFV 261.9, Eq. 2). Fmax and ED50 were estimated as − 0.288 and 23.8 mg, respectively. The relative standard errors estimated for Fmax and ED50 were 24.2% and 35.4%, respectively. Moreover, CsA bioavailability in patients receiving a PD of 80 mg was 20.6% lower than that in patients receiving a PD of 20 mg. The parameter γ in the sigmoidal Emax model (Eq. 4) was not estimated successfully and the function was simplified to the Emax model.
Multiple genetic variants in genes encoding the CsA-metabolizing enzymes CYP3A4/5 and the multidrug resistance transporter ABCB1 were analyzed [45]. However, no significant effects of the selected SNPs on CsA PK were found. Previous genotyping analysis results indicated that ABCB1 C1236T and G2677T/A are in strong LD with C3435T. Thus, the influence of the ABCB1 C1236T-G2677T/A-C3435T haplotypes on CsA PK was considered. The CL/F of the non-CGC haplotype carrier was 14.4% less than that of the CGC haplotype carrier 3 months following the operation (ΔOFV − 15.0, P < 0.001).
In addition to pathophysiological and genetic factors, concomitant medications were also investigated, but none displayed significant relationships with PK parameters. After other covariates were tested, the incorporation of POD reduced the OFV by − 79.1 (P < 0.001), indicating that other sources of time-related variability should be considered in further analyses. The CL/F-POD relationship improved in different postoperative periods after including POD-related factors (Figure S4).
In the final model, all retained covariates caused a significant increase in OFV upon removal. For these reasons, this model was accepted as the definitive final model. Equation 5 shows the results of the covariate analysis for CsA CL/F:
In this equation, influence scopes were adjusted according to their respective median values as determined from the dataset. The parameter estimates and associated precisions are shown in Table 3. The data was not sufficiently informative to support estimation of BSV on F. Additionally, shrinkage values of BSVs and RUVs, except for Vc/F and Q/F, were < 35%, which may be due to the relatively small intensive sample size. The condition number of the final model was 240.1.
Model evaluation
The GOF plots externally evaluated for the final models are presented in Fig. 2. Compared with the base model and the model incorporating only POD, the final model was greatly improved and showed no obvious bias. Over 99.6% (1141/1146) of the observations were within ± 4 conditional weighted residuals.

Externally evaluated diagnostic goodness-of-fit plots for the final model. A Observations versus population predictions; B observations versus individual predictions; C conditional weighted residuals (CWRES) versus population predictions; D CWRES versus PODs. (A–D) The locally weighted regression line (red dashed lines). (A, B) the line of unity (black solid lines), and (C, D) y = 0 (solid lines) are shown
The pcVPC results demonstrated good predictability of drug concentrations. The pcVPCs of the final model are depicted in Fig. 3. The simulated data corresponded well with the observed data, indicating a lack of significant model misspecifications. In the NPDE analysis, no trend in the scatterplots was observed (Fig. 4). The P values obtained using the Wilcoxon signed rank test were > 0.05, while those obtained via the Fisher’s variance test were < 0.05, indicating an inconsistency between the evaluation and simulation data for variance. Bootstrap parameter estimates closely matched the mean estimates from the population model, confirming model stability (Table 3).

Prediction-corrected visual predictive checks (pcVPCs) stratified on postoperative days (PODs) for the final model, based on 2000 simulations. The median observed values per bin (red solid line), the 5th and 95th percentiles (red dashed lines) of the observations (blue circles), as well as the 95% confidence interval of the 5th and 95th percentiles (blue areas) and the confidence interval of the median (green area) are shown

Normalized prediction distribution error (NPDE) plots for the final model. A Q-Q plot of the NPDE; B histogram of the NPDE; C NPDE versus postoperative days; D NPDE versus population predicted concentration (PRED)
Dosing Regimen Optimization
The results of the Monte Carlo simulation are shown in Table S7. The predicted time course of CsA concentrations in a ‘typical’ patient (i.e., patients with median covariates), which was simulated based on 1000 hypothetical individuals in different postoperative periods, is shown in Fig. 5. A stable recommended dosing regimen could maintain median concentrations within a desired concentration range, indicating that the final model could be applied to design the dosing regimen.

The concentration–time profiles of cyclosporine (CsA) from Monte Carlo simulation of 1000 hypothetical individuals with median covariates following different postoperative periods. The 25th and 75th percentiles of the simulation data (light blue), the median of the simulated data (red solid line), and both the therapeutic range of CsA predose concentrations (black solid lines) and 2-h postdose concentrations (black dashed lines) are shown. HCT, hematocrit; PD, prednisolone daily dose; POD, postoperative days. The simulation scheme with an asterisk (*) indicates the ABCB1 CGC haplotype carrier
Discussion
Although nearly 20 CsA popPK studies have been reported in adult renal transplant recipients, no large long-term follow-up cohort study has been conducted to elucidate the time-related variability in CsA PK [16]. Moreover, CsA PK characteristics may change with POD, thereby requiring dose adjustment to achieve target exposure. Therefore, it is essential to identify factors that can explain the variability in CsA PK and characterize the time-varying CL/F of CsA.
To the best of our knowledge, this is the first popPK study of long-term changes in CsA CL/F with POD in adult renal transplant patients. A two-compartment model with first-order absorption and lag time adequately described the CsA PK properties. In the final model, FFM, HCT level, POD, and the presence of the ABCB1 CGC haplotype were the most influential covariates with regard to CsA clearance. Tapering of the PD following the initial stage of transplantation may contribute to changes in bioavailability.
In a study conducted by Fanta et al. as PK data on intravenously and orally administered CsA were available, potential factors affecting the absorption process, which may be related to long-term PK properties, were analyzed [25]. Dose-dependent absorption or saturable presystemic metabolism contributed to this tendency. In this study, the factors that may influence CsA distribution were also considered, and we found that time-varying clearance could be largely attributed to changes in erythrocyte binding owing to increased HCT levels with time after transplantation.
Although available models have identified covariates primarily through empirical investigation, in our previous study, we found that theory-based modeling is helpful to improve model predictability [46]. Unlike empirical covariate selection, theory-based covariate selection allows the incorporation of relationships linking parameters and covariates based on a fundamental understanding of PK processes rather than on the available data alone, and may improve model predictability [47]. Therefore, in this study, our modeling was based on theoretical mechanisms and the data properties. First, the correlation between the changes in potential covariates and POD were investigated graphically. We then included identified covariates in the model to exam the statistical significance. After all POD-related covariates were analyzed, we incorporated the POD in final model.
In the final model, the oral bioavailability of CsA increased by approximately 35.6% in the first month after transplantation, consistent with a previous report [25]. CsA exposure in the early phases after transplantation was 15.4% lower than that in the stable period when the same CsA dose was administered. The increased CsA bioavailability in the initial period may correlate with improved absorption in the intestine [48] and reduction in dose of co-administered steroids [46]. Steroids can activate the pregnane X receptor (PXR) to upregulate CYP3A and P-gp activity [35, 49]. Thus, high doses can lead to reduced bioavailability. Specifically, the maximum reduction, estimated in the final model, was induced by steroids in F above 20 mg was 0.288. Moreover, differences in steroid doses from 20 mg, at which half the Fmax is reached, was 23.8 mg, corresponding to a dose of 43.8 mg. The model further predicts CsA bioavailability in patients receiving a PD of 80 mg to be 20.6% lower than that in patients receiving a PD of 20 mg. However, there is a lack of intravenous vs. oral data to verify these results.
Approximately 58% of circulating CsA is bound to red blood cells [50]; therefore, changes in the HCT level with POD may influence CsA PK. HCT level was low in the early postoperative period but increased during the first months after the transplantation, which is consistent with previous findings [51, 52]. The effect of the HCT on CsA clearance was retained in the final model, and a reduction in the HCT from 60.5% to 10.5% led to a 1.1-fold increase in CsA exposure. This relationship is consistent with known physiological properties [53]. Moreover, increases in the HCT levels led to the elevation of CsA binding to erythrocytes, which could partly prevent CsA extraction via the liver and distribution into peripheral tissues, resulting in elevated concentrations and decreased CL/F [21,22,23]. However, the potential clinical significance requires further validation.
During the modeling process, we also attempted to estimate the effect of HCT on CsA clearance based on theoretical relationships, as reported in our previous study [46]. However, compared to the empirical method (ΔOFV − 31.3, P < 0.001), the theory-based method (ΔOFV − 28.7, P < 0.001) did not significantly reduce the OFV. Moreover, a potential challenge was noted regarding the clinical application of this model. That is, the parameters used to predict CsA plasma concentrations (Cp) were assumed to be constant, resulting in the Cp exhibiting a linear correlation with CsA whole-blood concentration (Text S4), which does not accurately reflect clinical samples [54]. Besides, CsA Cp is not determined in routine practice, which may limit the use of this theory-based model. Therefore, we included the effect of HCT on CL/F empirically.
Multiple intrinsic/extrinsic factors, including demographic factors, genetic polymorphisms in drug-metabolizing enzymes and transporters, disease progression, concomitant medications, and their combined effects, may influence the in-vivo behaviors of drugs in the clinic [55]. Most of these covariates contribute to inter- and intra-individual variability [16]. POD is a surrogate for many time-varying factors [56]. Changes in covariates with POD were visually inspected and tested based on theoretical mechanisms in this study. In the final model, the tendency of CsA CL/F over POD was described by the POD-related covariates (known factors, such as PD and HCT) and POD (other unknown factors). More valuable information may be provided as these time-varying covariates can partly explain the effects of POD as a covariate on inter-individual variability [57]. Other currently unknown POD-related factors represent additional sources of inconsistencies, as the model describes variability, and should be evaluated in future studies. Meanwhile, a combination of these factors, the minimum CL/F value would be reached within one to six months, the required time for which would depend on the prednisolone daily dose.
In addition to time-varying factors, ABCB1 genetic polymorphisms were also included in the final model. In this study, we found that ABCB1 C1236T-G2677T/A-C3435T haplotypes may be an effective index for the characterization of CsA PK. The CL/F of the non-CGC haplotype carrier was 14.4% lower than that of the CGC haplotype carrier 3 months post operation. P-gp is distributed widely on the brush border surface of the intestine and mainly influences the CsA absorption process [58]. Mutations at the above three sites may decrease the amount or activity of P-gp, resulting in the reduced excretion of CsA into the intestinal lumen and increased bioavailability [19, 59]. However, the clinical significance must be further confirmed in future studies. Fanta et al. reported that the NR1I2 genotype, which encodes PXR, may influence CsA bioavailability [25]. However, no such information was available for this study, and additional studies are needed to evaluate this mechanism.
In this study, the recommended dosing regimens in different simulation scenarios were given based on the prior distributions of the final model. Clinicians can design an optimal regimen for each patient based on the individual’s status. According to the simulation results in this study, different dose regimens were needed to achieve different target exposures during different postoperative periods. However, all of these regimens were obtained based on popPK parameter distributions in the final model. Enhanced individualized dose predictions can be designed via Bayesian forecasting using available concentrations as prior information [16, 60].
One potential limitation of this study was its retrospective observational design. And the information pertaining to CsA exposure and drug-induced response relationship was not available. Moreover, there was no way to confirm whether patients adhered to their prescribed dosage regimen. Additionally, this was a single-center study. Multicenter validation is necessary to improve model predictability and assess the impact of CsA minimization and precision-dosing strategies.
In conclusion, a popPK model for adult renal transplant recipients was developed based on a large long-term follow-up cohort study, and the time-varying CL/F of CsA was comprehensively analyzed. Allometrically scaled body size, HCT level, CGC haplotype carrier, and postoperative time may mediate CsA PK variability. Additionally, tapering of PD during the initial stage of transplantation may contribute to changes in bioavailability. Increases in HCT resulted in the enhancement of CsA binding to erythrocytes, leading to increased CsA concentrations and decreased CL/F, which may affect the time-varying CL/F of CsA. Therefore, CsA dose adjustments should be considered during different postoperative periods.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Fahr A. Cyclosporin clinical pharmacokinetics. Clin Pharmacokinet. 1993;24:472–95.
Meier-Kriesche HU, Li S, Gruessner RW, Fung JJ, Bustami RT, Barr ML, Leichtman AB. Immunosuppression: evolution in practice and trends, 1994–2004. Am J Transplant. 2006;6:1111–31.
Sam R, Leehey DJ. Improved graft survival after renal transplantation in the United States, 1988 to 1996. N Engl J Med. 2000;342:1837–8.
Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. CJASN. 2009;4:481–508.
Hesselink DA, Hoorn EJ. Improving long-term outcomes of kidney transplantation: the pressure is on. Neth J Med. 2014;72:248–50.
Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: a critical reappraisal. Am J Transplant. 2011;11:450–62.
Akhlaghi F, Trull AK. Distribution of cyclosporin in organ transplant recipients. Clin Pharmacokinet. 2002;41:615–37.
Legg B, Gupta SK, Rowland M, Johnson RW, Solomon LR. Cyclosporin: pharmacokinetics and detailed studies of plasma and erythrocyte binding during intravenous and oral administration. Eur J Clin Pharmacol. 1988;34:451–60.
Dai Y, Iwanaga K, Lin YS, Hebert MF, Davis CL, Huang W, Kharasch ED, Thummel KE. In vitro metabolism of cyclosporine A by human kidney CYP3A5. Biochem Pharmacol. 2004;68:1889–902.
Venkataramanan R, Starzl TE, Yang S, Burckart GJ, Ptachcinski RJ, Shaw BW, Iwatsuki S, Van Thiel DH, Sanghvi A, Seltman H. Biliary excretion of cyclosporine in liver transplant patients. Transplant Proc. 1985;17:286–9.
Shaw LM, Bowers L, Demers L. Critical issues in cyclosporine monitoring: report of the task force on cyclosporine monitoring. Clin Chem. 1987;33:1269–88.
Ette EI, Williams PJ. Population pharmacokinetics I: background, concepts, and models. Ann Pharmacother. 2004;38:1702–6.
Sun H, Fadiran EO, Jones CD, Lesko L, Huang SM, Higgins K, Hu C, Machado S, Maldonado S, Williams R, Hossain M, Ette EI. Population pharmacokinetics. A regulatory perspective. Clin Pharmacokinet. 1999;37:41–58.
Sheiner LB, Rosenberg B, Marathe VV. Estimation of population characteristics of pharmacokinetic parameters from routine clinical data. J Pharmacokinet Biopharm. 1977;5:445–79.
Han K, Pillai VC, Venkataramanan R. Population pharmacokinetics of cyclosporine in transplant recipients. AAPS J. 2013;15:901–12.
Mao JJ, Jiao Z, Yun HY, Zhao CY, Chen HC, Qiu XY, Zhong MK. External evaluation of population pharmacokinetic models for ciclosporin in adult renal transplant recipients. Br J Clin Pharmacol. 2018;84:153–71.
Drewe J, Beglinger C, Kissel T. The absorption site of cyclosporin in the human gastrointestinal tract. Br J Clin Pharmacol. 1992;33:39–43.
Kelly P, Kahan BD. Review: metabolism of immunosuppressant drugs. Curr Drug Metab. 2002;3:275–87.
Chen B, Zhang W, Gu Z, Li J, Zhang Y, Cai W. Population pharmacokinetic study of cyclosporine in Chinese renal transplant recipients. Eur J Clin Pharmacol. 2011;67:601–12.
Klintmalm G, Sawe J. High dose methylprednisolone increases plasma cyclosporin levels in renal transplant recipients. Lancet. 1984;1:731.
Wu KH, Cui YM, Guo JF, Zhou Y, Zhai SD, Cui FD, Lu W. Population pharmacokinetics of cyclosporine in clinical renal transplant patients. Drug Metab Dispos. 2005;33:1268–75.
Yin OQ, Lau SK, Chow MS. Population pharmacokinetics of cyclosporine in Chinese cardiac transplant recipients. Pharmacotherapy. 2006;26:790–7.
Fanta S, Jonsson S, Backman JT, Karlsson MO, Hoppu K. Developmental pharmacokinetics of ciclosporin–a population pharmacokinetic study in paediatric renal transplant candidates. Br J Clin Pharmacol. 2007;64:772–84.
Porta B, Perez-Ruixo JJ, Gorriz JL, Crespo JF, Sancho A, Pallardo LM, Jimenez NV. Population pharmacokinetics of cyclosporine in kidney transplant patients. Transplant Proc. 1999;31:2246–7.
Fanta S, Jonsson S, Karlsson MO, Niemi M, Holmberg C, Hoppu K, Backman JT. Long-term changes in cyclosporine pharmacokinetics after renal transplantation in children: evidence for saturable presystemic metabolism and effect of NR1I2 polymorphism. J Clin Pharmacol. 2010;50:581–97.
Shi BY, Yuan M. Guidelines for immunosuppressive therapy in Chinese renal transplant recipients. Organ Transplant. 2016;7:327–31.
Serdarevic N, Zunic L. Comparison of architect I 2000 for determination of cyclosporine with axsym. AIM. 2012;20:214–7.
Geng F, Jiao Z, Dao YJ, Qiu XY, Ding JJ, Shi XJ, Li ZD, Zhong MK. The association of the UGT1A8, SLCO1B3 and ABCC2/ABCG2 genetic polymorphisms with the pharmacokinetics of mycophenolic acid and its phenolic glucuronide metabolite in Chinese individuals. Clin Chim Acta. 2012;413:683–90.
Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT: Pharmacomet Syst Pharmacol. 2013;2:e50.
Beal SL, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM user’s guides. Ellicott City: Icon Development Solutions; 1989.
West GB, Brown JH, Enquist BJ. A general model for the origin of allometric scaling laws in biology. Science. 1997;276:122–6.
Anderson BJ, Holford NH. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet. 2009;24:25–36.
Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet. 2005;44:1051–65.
Pascussi JM, Drocourt L, Fabre JM, Maurel P, Vilarem MJ. Dexamethasone induces pregnane X receptor and retinoid X receptor-alpha expression in human hepatocytes: synergistic increase of CYP3A4 induction by pregnane X receptor activators. Mol Pharmacol. 2000;58:361–72.
Pascussi JM, Drocourt L, Gerbal-Chaloin S, Fabre JM, Maurel P, Vilarem MJ. Dual effect of dexamethasone on CYP3A4 gene expression in human hepatocytes. Sequential role of glucocorticoid receptor and pregnane X receptor. Eur J Biochem. 2001;268:6346–58.
Owen JS, Fiedler-Kelly J. Introduction to population pharmacokinetic/pharmacodynamic analysis with nonlinear mixed effects models. New Jersy: Wiley; 2014.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13:143–51.
Comets E, Brendel K, Mentre F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed. 2008;90:154–66.
Ette EI, Williams PJ, Kim YH, Lane JR, Liu MJ, Capparelli EV. Model appropriateness and population pharmacokinetic modeling. J Clin Pharmacol. 2003;43:610–23.
Ette EI. Stability and performance of a population pharmacokinetic model. J Clin Pharmacol. 1997;37:486–95.
Shi YY, He L. SHEsis, a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell Res. 2005;15:97–8.
Bourgoin H, Paintaud G, Buchler M, Lebranchu Y, Autret-Leca E, Mentre F, Le Guellec C. Bayesian estimation of cyclosporin exposure for routine therapeutic drug monitoring in kidney transplant patients. Br J Clin Pharmacol. 2005;59:18–27.
Saint-Marcoux F, Marquet P, Jacqz-Aigrain E, Bernard N, Thiry P, Le Meur Y, Rousseau A. Patient characteristics influencing ciclosporin pharmacokinetics and accurate Bayesian estimation of ciclosporin exposure in heart, lung and kidney transplant patients. Clin Pharmacokinet. 2006;45:905–22.
Rietbrock S, Merz PG, Fuhr U, Harder S, Marschner JP, Loew D, Biehl J. Absorption behavior of sulpiride described using Weibull functions. Int J Clin Pharmacol Ther. 1995;33:299–303.
Haufroid V, Mourad M, Van Kerckhove V, Wawrzyniak J, De Meyer M, Eddour DC, Malaise J, Lison D, Squifflet JP, Wallemacq P. The effect of CYP3A5 and MDR1 (ABCB1) polymorphisms on cyclosporine and tacrolimus dose requirements and trough blood levels in stable renal transplant patients. Pharmacogenetics. 2004;14:147–54.
Mao J, Jiao Z, Qiu X, Zhang M, Zhong M. Incorporating nonlinear kinetics to improve predictive performance of population pharmacokinetic models for ciclosporin in adult renal transplant recipients: a comparison of modelling strategies. Eur J Pharm Sci. 2020;153:105471.
Danhof M, de Lange EC, Della Pasqua OE, Ploeger BA, Voskuyl RA. Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol Sci. 2008;29:186–91.
Wilms HW, Straeten V, Lison AE. Different pharmacokinetics of cyclosporine A early and late after renal transplantation. Transplant Proc. 1988;20:481–4.
Lamba J, Lamba V, Schuetz E. Genetic variants of PXR (NR1I2) and CAR (NR1I3) and their implications in drug metabolism and pharmacogenetics. Curr Drug Metab. 2005;6:369–83.
Legg B, Rowland M. Cyclosporin: erythrocyte binding and an examination of its use to estimate unbound concentration. Ther Drug Monit. 1988;10:16–9.
Awni WM, Kasiske BL, Heim-Duthoy K, Rao KV. Long-term cyclosporine pharmacokinetic changes in renal transplant recipients: effects of binding and metabolism. Clin Pharmacol Ther. 1989;45:41–8.
Malyszko J, Oberbauer R, Watschinger B. Anemia and erythrocytosis in patients after kidney transplantation. Transplant Int. 2012;25:1013–23.
Sikma MA, Hunault CC, Huitema ADR, De Lange DW, Van Maarseveen EM. Clinical pharmacokinetics and impact of hematocrit on monitoring and dosing of tacrolimus early after heart and lung transplantation. Clin Pharmacokinet. 2019;59:403–8.
Legg B, Rowland M. Saturable binding of cyclosporin A to erythrocytes: estimation of binding parameters in renal transplant patients and implications for bioavailability assessment. Pharm Res. 1988;5:80–5.
Grillo JA, Zhao P, Bullock J, Booth BP, Lu M, Robie-Suh K, Berglund EG, Pang KS, Rahman A, Zhang L, Lesko LJ, Huang SM. Utility of a physiologically-based pharmacokinetic (PBPK) modeling approach to quantitatively predict a complex drug-drug-disease interaction scenario for rivaroxaban during the drug review process: implications for clinical practice. Biopharm Drug Dispos. 2012;33:99–110.
Ette EI, Ludden TM. Population pharmacokinetic modeling: the importance of informative graphics. Pharm Res. 1995;12:1845–55.
Wahlby U, Thomson AH, Milligan PA, Karlsson MO. Models for time-varying covariates in population pharmacokinetic-pharmacodynamic analysis. Br J Clin Pharmacol. 2004;58:367–77.
Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29.
Johne A, Kopke K, Gerloff T, Mai I, Rietbrock S, Meisel C, Hoffmeyer S, Kerb R, Fromm MF, Brinkmann U, Eichelbaum M, Brockmoller J, Cascorbi I, Roots I. Modulation of steady-state kinetics of digoxin by haplotypes of the P-glycoprotein MDR1 gene. Clin Pharmacol Ther. 2002;72:584–94.
Zhao CY, Jiao Z, Mao JJ, Qiu XY. External evaluation of published population pharmacokinetic models of tacrolimus in adult renal transplant recipients. Br J Clin Pharmacol. 2016;81:891–907.
Funding
This work was supported in part by grants from the ‘2019 Key Clinical Program of Clinical Pharmacy’ (No. shslczdzk06502), and ‘Weak Discipline Construction Project’ (No. 2016ZB0301-01) of Shanghai Municipal Health and Family Planning Commission. We would like to thank Editage (www.editage.cn) for English language editing.
Author information
Authors and Affiliations
Contributions
JJM, XYQ, and WWQ participated in the research design; JJM, XYQ, WWQ, LYX, MZ, and MKZ implemented and conducted the study; JJM, XYQ, and WWQ performed the research and analyzed the data. JJM and WWQ drafted the manuscript, which was revised and approved by all the authors. There are no other relationships or activities that could appear to have influenced the submitted work.
Corresponding authors
Ethics declarations
Conflict of interest
There are no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years, and no other relationships or activities that could appear to have influenced the submitted work. All authors have no conflicts of interest to declare.
Ethical Approval
All procedures involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mao, J., Qiu, X., Qin, W. et al. Factors Affecting Time-Varying Clearance of Cyclosporine in Adult Renal Transplant Recipients: A Population Pharmacokinetic Perspective. Pharm Res 38, 1873–1887 (2021). https://doi.org/10.1007/s11095-021-03114-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-021-03114-9