
The new conjugate compound pyrrolylcarnosine was synthesized from the natural antioxidant carnosine and the aromatic five-membered nitrogen heterocycle pyrrole. The synthesis of pyrrolylcarnosine was described. Its physicochemical properties and biological activity in various oxidative stress models were evaluated. The results showed that pyrrolylcarnosine was characterized by resistance to hydrolysis by serum carnosinase and exhibited high antioxidant activity in model experiments. It had a neuroprotective effect under oxidative stress induced by the neurotoxin AAPH, increasing the viability of a differentiated culture of human neuroblastoma SH-SY5Y and protecting it from death. In general, the results indicated creation of a new drug based on pyrrolylcarnosine was promising.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Oxidative stress (OS) is the main molecular mechanism of neuron death in neurodegenerative and cerebrovascular diseases [1]. Various cytoprotectors and antioxidants are used to prevent oxidative damage of neuronal cells in experimental neuropharmacology, although drugs with efficacies confirmed in clinical trials do not currently exist [2]. In this respect, the natural histidine-containing peptide carnosine, the protector efficacy of which was demonstrated in various CNS disease models including Parkinson’s disease and stroke, can be considered a potential antioxidant with neuroprotector activity [3, 4]. New data on its mechanisms of neuroprotector activity under OS conditions induced by mitochondrial neurotoxins, heavy metals, and NMDA excitotoxicity were obtained. The ability of carnosine to penetrate neurons using PEPT2 carrier peptides and to suppress the accumulation of reactive oxygen species (ROS) within cells, leading to a reduction of their death and an increase of their viability, was shown in neuronal cultures [3, 5, 6].
Use of carnosine as an adjuvant (at a daily dose of 1.5 – 2 g) in addition to the main pharmacotherapy in test clinical trials could improve the efficacy of the standard treatment of patients with various types of Parkinsonism and ischemic CNS injuries [7, 8]. However, carnosine has not been incorporated into routine clinical practice as a dosage form because it is rapidly destroyed in the blood pool by the specific enzyme carnosinase, which necessitates the use of extremely high doses of it. A review of randomized controlled trials by Menon, et al. [9] in 2018 proposed carnosine and other histidine-containing dipeptides (anserine and N-acetylcarnosine) and their components (β-alanine) as potential strategies for prevention of several chronic diseases [9].
Pyrrole is a five-membered nitrogen-containing aromatic compound that is used to synthesize a broad spectrum of drugs such as antimicrobial and anticancer agents and many other compounds [2, 10, 11]. Several medicinal compounds containing a pyrrole component have already reached the pharmaceutical market. Nonsteroidal anti-inflammatory drugs are particularly interesting among them [12, 13].
Pyrrolidine is a pyrrole derivative and starting material for synthesizing piracetam, a drug used as a nootropic agent with neuroprotector efficacy that is included in the list of vitally important drugs in the Russian Federation. A broad spectrum of new pyrrole-containing compounds based on pyrrole-2,5-dione, many of which exhibit significant experimental antiradical efficacy [14], including in cell culture experiments with modeled OS [15], have been designed in the last decade.
The conjugate of pyrrole and carnosine, pyrrolylcarnosine, was synthesized and patented [16] based on existing data for their biological effects. However, an assessment of its physicochemical properties, resistance to the action of serum carnosinase, antioxidant activity in in vitro systems, cytotoxicity for neuronal culture, and ability to pass through intracellular membranes and an evaluation of its neuroprotector efficacy in a general OS model are unresolved issues.
The aim of the present study was to assess the physicochemical properties and biological activity of pyrrolylcarnosine in various OS models.
Experimental Chemical Part
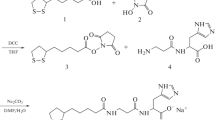
Synthesis of pyrrolylcarnosine (Fig. 1).

Synthetic scheme for 3-(N-pyrrolyl)propanoyl-1-histidine (pyrrolylcarnosine, PC).
The synthesis of pyrrolylcarnosine (PC) used L-carnosine (98% purity; Swedlight AB, Sweden); 2,5-dimethoxytetrahydrofuran (99% purity, mixture of cis- and trans-isomers; Acros Organics); MeOH, EtOH, and HOAc (Khimmed, Russia); and deionized H2O.
L-Carnosine (10 g, 44.2 mmol), 2,5-dimethoxytetrahydrofuran (1.1 eq, 6.4 g, 48.7 mmol), and HOAc (2.5 eq, 6.6 g, 110.5 mmol) were dissolved in deionized H2O (60 mL), placed in a tightly closed flask into a bath heated to 102 – 103°C, and stirred for 3 h at that temperature. The homogeneous dark-brown solution was evaporated to a brown caramel that was successively dissolved in H2O (2 × 120 mL) and evaporated after each dissolution. The hard solid was dissolved in hot MeOH (60 mL). The resulting solution was immediately placed evenly onto silica gel for flash chromatography using MeOH (Rf 0.62). Fractions containing product were combined and evaporated. The resulting light-yellow powder (10 g) was refluxed for 2 h in EtOH (25 mL). The suspension was cooled to room temperature (12 h). The while crystalline precipitate was rinsed on the filter with cold EtOH. Yield 9.5 g (78%), mp 242 – 246°C. NMR spectroscopy was consistent with 99% of the target compound in the precipitate. The only trace impurity was MeOH.
The molecular structure of the pyrrole-containing carnosine derivative was confirmed using NMR spectroscopy and liquid chromatography in combination with high-resolution mass spectrometry (HRMS).
PMR and 13C NMR spectra were taken in D2O on a Bruker Avance 500 NMR spectrometer (Bruker Daltonics Inc., USA) at 500 MHz for 1H and 125 MHz for 13C.
PMR (500 MHz, D2O, δ, ppm): 2.58 – 2,68 (m, 2H), 2.86 (dd, 1H, J 15.5, 8.9 Hz), 3.06 (dd, 1H, J 15.5, 4.6 Hz), 4.01 – 4.12 (m, 2H), 4.33 (dd, 1H, J 8.9, 4.6 Hz), 6,00 (t, 2H, J 1.9 Hz), 6.59 (t, 2H, J 1.9 Hz), 6.85 (s, 1H), 8.42 (d, 1H, J 1.0 Hz).
13C NMR (125 MHz, D2O, δ, ppm): 27.3, 37.7, 45.1, 54.0, 107.9, 116.6, 121.3, 129.5, 133.1, 173.2, 176.5.
High-resolution mass spectra were taken on a Bruker micrOTOF II instrument using electrospray ionization (ESI). HRMS in positive-ion mode of the target compound found m/z [M + H] = 277.1287 g/mol, which corresponded to 3-(N-pyrrolyl)propanoyl-L-histidine (pyrrolylcarnosine, C13H16N4O3) in the protonated form with [M + H] = 277.1295 g/mol. Thus, the molecular mass of PC determined by HRMS was 277.1295 g/mol.
A patented in-house LC-MS method (HPLC-MS) [17] was developed by us for quantitative determination of PC in biological materials. PC was extracted from biological samples using deproteinization. A sample of biological material (plasma, tissue homogenate, cell culture lysate) (100 μL) was treated with an internal standard solution (400 μL, L-alanylcarnosine, 10 μg/mL) in trichloroacetic acid (TCA, 10%) to denature the proteins. The resulting suspension of denatured proteins was precipitated in a chilled centrifuge at 16,000 g. The supernatant was transferred to a chromatography vial that was placed into the chromatograph autosampler for LC-MS analysis. The supernatant liquid was injected into a 10-μL chromatography loop. The PC concentration was determined by LC-MS on a Finnigan Surveyor LC Pump Plus chromatograph in combination with an LCQ Fleet MS mass-spectrometric detector (ion trap). Compounds were analytically separated using an Ultrasphere 5 ODS chromatography column (Hichrom, Ltd., Great Britain) (250 × 4.6 mm, 5 μm). The eluent was a mixture of two solutions, i.e., ammonium acetate (10 mM) acidified with glacial HOAc to pH 3.7 (solution A) and MeCN containing a solution of acidified ammonium acetate (90:10; solution B), taken in a 90:10 (A:B) ratio. The chromatography was performed in isocratic mode at eluent flow rate 0.7 mL/min. The injected volume was 10 μL. The separation temperature was 35°C. The chromatographic analysis took 10 min. The retention time of PC was 8.2 ± 0.1 min; of the internal standard (L-alanylcarnosine, Hamari Chemicals, Ltd.), 5.8 ± 0.1 min. Mass spectrometric detection used fragment ions with m/z 156.02, 231.10, and 258.98 that formed via decomposition of the quasi-molecular peak of PC with m/z 277.29 at normalized impact energy 35 eV (Fig. 2 shows the second-order mass spectrum for PC). The internal standard (L-alanylcarnosine) was identified using the total ion current of daughter ions with m/z from 75 to 300 that formed via fragmentation of the quasimolecular L-alanylcarnosine ion with m/z 298.3. The mass spectrometer recorded positive ions charged by ESI. The electrospray was generated at a voltage of 5 kV. The nebulizer gas (N2) flow was 5 L/min; spraying pressure, 100 psi. The capillary temperature was 350°C; heater temperature, 300°C. The excitation amplitude on the trap end electrodes was 0.1 V. The damping gas was high-purity He (99.9999%). Data were analyzed using the Xcalibur 2.1 w/Foundation 1.0.1 computer program. The quantitative concentration of PC was determined using the internal standard method. The mathematical relationship of the chromatographic peak areas of the analyte and internal standard as a function of the nominal concentration of the target compound was measured for calibration. Linear regression by a least-squares method was used for the calculations. The calibration curve obeyed a linear function in the concentration range from 0.5 to 10,000 μg/mL. The PC content was determined using the formula
where C is the PC concentration (ng/mL); S, PC chromatographic peak area normalized to the internal standard peak area. The relative uncertainty of the method for PC determination was <10%.

Comparison of resistance of PC (1) and carnosine (2) to hydrolysis by serum carnosinase.
Preparative purification was not required. The compound obtained from flash chromatography followed by refluxing in EtOH was 99% pure according to NMR data. Standard laboratory silica gel (0.060 – 0.200 mm, 60 Å; Acros Organics) was used for the chromatography. The purity of the obtained compound was also determined by HPLC using a Prontosil 120C18aq column. The purity of the obtained PC was >99% according to the HPLC data.
Experimental Biological Part
Determination of PC resistance to hydrolysis by blood serum carnosinase
The method proposed by Pegova, et al. [18] was used to determine the resistance of PC to serum carnosinase. It was based on evaluation of the degree of hydrolysis in the presence of donor blood serum carnosinase. A mixture of serum samples obtained from several donors was used in the research. Serum carnosinase was activated by incubation with CaCl2 (5 mM) at 37°C for 30 min. Then, PC solution at a final concentration of 2 mM was added to the incubation medium. The samples were incubated for 0, 15, 30, 45, 60, 90, 120, 150, and 180 min and mixed with cold TCA (20%) in a 1:1 ratio. Denatured proteins were precipitated by centrifugation at 16,000 g for 30 min at 4°C. The supernatant was used for the analysis. The controls were samples incubated without added serum. The content of the new compound in the samples was determined by the in-house HPLC method. The instrument response in arbitrary integration units from starting samples (0 min) containing PC at a concentration of 2 mM was taken as 100%. Other samples were normalized to the peak area of the starting sample. Data were processed using the Xcalibur 2.1 Foundation 1.0.1 program.
Iron-induced chemiluminescence of biological samples
The in vitro antioxidant activity of PC was evaluated using a lipid peroxidation (LPO) model initiated by Fe2+ ions in low-density and very low-density lipoproteins isolated from blood serum of patients with chronic cerebrovascular pathology that was proposed by Vladimirov, et al. [19] and adapted to a Luminometer-1251 instrument (LKB, Sweden) [20].
Preparation of analytical samples. Serum (200 μL) was treated with CaCl2 solution (2,000 μL, 0.28%) and heparin solution (40 μL, 1%), left at room temperature for 5 min, and centrifuged (3,000 rpm, 15 min). The supernatant was discarded. The precipitated drug was treated with phosphate buffer (800 μL, pH 7.45, 60 mM KH2PO4 containing 105 mM KCl), and carefully stirred. The resulting drug suspension was transferred to an analytical cuvette (800 μL) and treated with the tested compound (100 μL) at the required concentration.
Recording of parameters. The cuvette with the resulting suspension was placed into the instrument sample chamber (heated to 37°C with constant stirring). Background readings were recorded. Chemiluminescence (CL) was initiated using a pipette to add Fe2+ solution (0.1 mL, FeSO4·7H2O) to a final concentration of 2.5 mM. The luminescence curve was recorded.
Parameters for the CL rapid flash (h, mV), the intensity of which characterized the level of preformed LPO products (primarily lipid hydroperoxides) and the latent period (τ, s), indicating the resistance of the substrate to further oxidation and reflecting the endogenous antioxidant potential, were analyzed.
Cell culture
The inherent toxicity and neuroprotector properties of PC were studied in vitro using a fast-growing passaged cell culture of SH-SY5Y human neuroblastoma (ATCC®, USA). The cells were cultivated in Petri dishes in a mixture of Eagle’s MEM with Earle’s salts and glutamine and F-12 without glutamine in a 1:1 ratio with added penicillin-streptomycin solution (1%, PanEco, Russia) and fetal bovine serum (10%, Biosera, South America). The culture was kept in a cell incubator (Shel Lab) at 37°C, 90% humidity, and 5% CO2. The experimental procedures used cells that were seeded onto plates and differentiated for a week by adding retinoic acid (10 μM) to the culture medium and reducing the amount of serum in the culture medium to 1%.
Measurement of PC content in cell culture lysates by LC-MS
The efficiency of PC transport into neuronal cells was determined by seeding SH-SY5Y neuroblastoma culture onto 6-well plates and differentiating according to the protocol. The wells were treated with PC (0.5 mM) on the day of the experiment and incubated for 1, 3, 6, or 24 h. Then, the cells were rinsed (5×) with Hank’s solution and lysed by two freeze(thaw cycles. The resulting lysates were treated with cold TCA (10% with added alanylcarnosine internal standard, 10 μg/sample) and centrifuged at 16,000 g for 30 min at 4°C. The supernatant was used for HPLC-MS analysis as described above. The precipitate was dissolved in NaOH solution (0.1 N). The amount of protein in it was determined using a DC Protein Assay Kit (Bio-Rad) according to the manufacturer’s protocol. The amount of PC was calculated using the internal standard and the PC peak area normalized to the peak area of alanylcarnosine. In turn, the resulting concentration was normalized to the amount of protein in the sample.
Determination of cell viability (MTT assay)
The viability of differentiated SH-SY5Y human neuroblastoma cells was assessed using the MTT assay [21]. Cells were seeded in 96-well plates calculated for 40,000 cells per well. The culture medium was replaced by fresh medium one day before performing the experimental procedures. The cells were treated with PC to final concentrations from 0.5 to 10.0 mM to assess the inherent cytotoxicity of PC. PC was not added to control wells. The cytoprotective activity of PC was assessed by treating the cells with an OS inducer [2 mM AAPH, 2, 2_-azobis(2-methylpropionamidine) dihydrochloride] and/or PC at the corresponding concentrations (0.5, 0.75, 1, and 2 mM). Then, the cells were incubated for 24 h. The medium was collected for the lactate dehydrogenase (LDH) test. Next, wells with cells were treated with MTT [3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Diaem, Russia] solution prepared in culture medium to a final concentration of 0.5 mg/mL. Transformation of the yellow tetrazolium salt into its violet formazan product was indicative of mitochondrial activity of viable cells. This method made it possible quickly and accurately to determine the quantitative survival of cells using spectrometric measurements. The cells were incubated with the MTT reagent for 3 h. The medium was carefully removed from the wells without disturbing the formazan crystals. The dry formazan residue in each cell was dissolved in DMSO (100 μL; Dia-M) to produce a violet solution. The plates were placed into a Synergy H4 microplate spectrophotometer (BioTek), in which the solutions in the wells were stirred for 6 min and the optical densities at λ = 570 nm and λ = 660 nm were measured. The optical densities at λ = 660 nm were subtracted from those at λ = 570 nm [21, 22]. The data were given as percent of the optical density of the solution in control wells, the viability of the cells in which was taken as 100%. Results were processed using the Microsoft Excel program.
Assessment of cell death (LDH test)
LDH in medium collected before the MTT assay was determined according to the manufacturer’s protocol for the Lactate Dehydrogenase Activity Assay Kit (MAK066, Sigma, USA). The principle of the method consisted of LDH released into the culture medium from dead cells reducing NAD to NADH to form a color, the intensity of which was determined by colorimetric analysis from the optical density of the solution at 450 nm. Measurements were made on a Synergy H4 microplate spectrophotometer (BioTek) every 5 min as long as the sample activity was less than that of the standard with the highest concentration (12.5 nmol/well). The values were expressed as the mean for groups in percent of the mean for control wells and were compared to the results of the MTT assay for the same plate.
Determination of amount of ROS
The amount of ROS was determined using the fluorescent dye 2′,7′-dichlorodihydrofluorescein diacetate (DCFH2-DA; Invitrogen) at a concentration of 10 μM. Neuroblastoma cells were seeded in 24-well plates and differentiated according to the protocol. The wells were treated with an OS inducer (AAPH, 2 mM) and/or PC at concentrations selected based on the MTT and LDH assays and incubated for 1 h. DCFH2-DA (10 μM final concentration in the sample) was added to the wells 30 min before the end of incubation. The plates were left in the cell incubator. Then, the cells were rinsed (3×) with Hank’s solution. The DCF fluorescence intensity was detected at λem = 520 nm in a Synergy H1 plate reader (BioTek) using Gen5 software.
Data were given as mean ± standard error of the mean (M ± SEM). Results were statistically processed using GraphPad Prism 8 software and the Shapiro(Wilk test for determining normal distributions and one-factor dispersion analysis (one-way ANOVA) with the Bonferroni multiple-comparison adjustment for a set with a normal distribution. The Dunn correction for multiple comparisons was used for a set with a nonnormal distribution. Differences between groups were considered statistically significant for p < 0.05.
Results and Discussion
Determination of PC resistance to hydrolysis by serum carnosinase
Figure 2 shows the hydrolysis kinetics of molecular carnosine and PC by serum carnosinase. Carnosine was 86% hydrolyzed by serum carnosinase in 1 h and 100%, in 3 h. PC was practically not hydrolyzed in 3 h by serum carnosinase. The data were given in percent of the initial concentration (2 mM) of the tested compounds.
Determination of PC antioxidant activity by Fe-induced chemiluminescence
The antioxidant activity of PC as compared to carnosine was evaluated. Tables 1 and 2 present these changes of the Fe-induced CL parameters under the influence of the tested compounds. PC began to act at a minimum concentration of 0.05 mM, reducing the level of lipid hydroperoxides by 25.8% (relative to the control taken as 100%); at 0.25 mM, by 40.1%; at 0.5 mM, by 45.2%; and at 1 mM, by 50.1%. These values were proportional to its concentration in the sample. Carnosine less effectively reduced lipid hydroperoxides at all tested concentrations; i.e., at 0.05 mM, by 13.7%; 0.25 mM, by 29.3%; 0.5 mM, by 37.5%; and 1 mM, by 48.2% (Table 1).
PC and carnosine increased the length of the CL latent period (t, s), which characterized the endogenous antioxidant potential, in proportion to their concentrations in the samples (relative to the control taken as 100%); e.g., at a concentration of 0.05 mM, by 20.1 and 9.2%, respectively; 0.25 mM, by 29.1 and 35.1%; 0.5 mM, by 48.1 and 47.3%; 1 mM, by 63.6 and 61.7%, respectively (Table 2).
In general, the antioxidant activity of PC, which was characterized by the ability to reduce the lipid hydroperoxide level and increase the endogenous antioxidant potential, was comparable to that of carnosine and, in several instances, greater than it. The results were consistent with high antioxidant activity of the new compound (PC).
Uptake of PC into SH-SY5Y human neuroblastoma cells
The contents of PC at an initial concentration of 0.5 mM in lysates of cells incubated for 1, 3, 6, or 24 h were 0.48 ± 0.06, 1.17 ± 0.07, 1.15 ± 0.18, and 1.81 ± 0.16 μg/mg of protein, respectively; and did not differ at all time points (Fig. 3). Thus, PC effectively passed through the membranes into the cells.

Uptake of PC (0.5 mM) into neuronal cells (human neuroblastoma SH-SY5Y).
Effect of PC on viability of SH-SY5Y human neuroblastoma cells, inherent cytotoxicity
The inherent cytotoxicity of PC was assessed over a broad concentration range (0.5, 1, 2.5, 5.0, and 10.0 mM) from its effect on the viability of SH-SY5Y human neuroblastoma cells using the MTT assay. Cultures with PC were incubated for 24 h (Fig. 4). PC was shown not to exhibit inherent toxicity upon incubation of the cultures over a broad concentration range (from 0.5 t0 10.0 mM).

Effect of pyrrolylcarnosine on viability of SH-SY5Y human neuroblastoma cells, inherent cytotoxicity.
Assessment of PC neuroprotector activity in a general AAPH-induced OS model
MTT assay. The effect of PC on the viability of cells under OS conditions was assessed using the MTT assay. General OS was induced by adding AAPH (2 mM) to the culture medium. The results indicated that incubation of the cell culture with AAPH (2 mM) for 24 h reduced the cell viability up to 59.8 ± 1.1% relative to intact cells, the viability of which was taken as 100% (Fig. 5).

Effect of PC at concentrations of 0.5, 0.75, and 1 mm with 24-h incubation on survival of neuroblastoma cells with AAPH (2 mM) toxicity; in % of mean level in intact cells. * p < 0.05 vs. intact cells, # vs. cells with AAPH.
The cell viability increased by an average of up to 74% if PC at concentrations of 0.5, 0.75, 1.0, 1.5, and 2.0 mM was added to the incubation medium simultaneously with AAPH, regardless of the PC concentration.
Thus, PC increased the cell viability by an average of 24% (relative to cells subjected to the action of AAPH) under OS conditions induced by AAPH (2 mM), although the control level was not reached.
LDH test. Incubation of cells with AAPH (2 mM) for 24 h led to an increase in the LDH content in the medium by 27.0 ± 5.6% relative to intact cells taken as 100% (Fig. 6). Addition of PC to the incubation medium containing AAPH (2 mM) prevented an increase in the LDH content, e.g., at a concentration 0f 0.5 mM, up to 105.8 ± 11.0%; 0.75 mM, up to 106.6 ± 9.6%; and 1 mM, up to 108.5 ± 2.3%. PC, regardless of its concentration, reduced the LDH content by an average of 20% relative to cells incubated with AAPH.

Effect of PC at concentrations of 0.5, 0.7, and 1 mM on LDH content 24 h after its simultaneous addition to culture medium with added neurotoxin AAPH (2 mM); * p < 0.05 vs. intact cells, # vs. cells with AAPH.
Effect of PC on ROS level under AAPH-induced OS conditions
The effect of PC on the increase of ROS under AAPH-induced OS conditions was evaluated from DCF fluorescence intensity. The DCF fluorescence intensity tripled relative to that of intact cells after incubation for 1 h if AAPH (2 mM) was added to an attached culture (in a microplate reader). This was indicative of a significant increase in ROS (Fig. 7).

Effect of PC at concentrations of 0.5, 0.75, and 1 mM on DCF fluorescence intensity 1 h after its simultaneous addition to culture medium with AAPH (2 mM); in % of fluorescence intensity of intact cells. * p < 0.05 vs. intact cells; # p < 0.05 vs. cells with AAPH.
Addition of PC at concentrations of 0.5, 0.75, and 1 mM simultaneously with AAPH (2 mM) followed by incubation for 1 h reduced the fluorescence intensity by an average of 19% relative to that of cells incubated with AAPH, regardless of the PC concentration in the sample. However, the fluorescence intensity remained elevated relative to that of intact cells.
The search for effective neuroprotectors with high pharmacological activity and the minimum number of adverse effects is an important problem of modern neuropharmacology. The new compound PC was synthesized by us to solve this problem. Its structure contained a pyrrole heterocycle in combination with a fragment of the natural peptide antioxidant carnosine.
Carnosine in the human body is known to undergo enzymatic peptidolysis by serum carnosinase to the amino acids β-alanine and L-histidine [23]. In contrast to carnosine, PC displayed high resistance to hydrolysis by carnosinase because of the modification of carnosine at the β-alanine amino-acid fragment. New chemical derivatives of carnosine with compounds such as a-lipoic acid, Trolox (water-soluble analog of tocopherol), and salicylic acid that also demonstrated high resistance to enzymatic hydrolysis were synthesized by us in previous studies [24,25,26].
The method of Fe-induced chemiluminescence of lipoproteins in blood serum of healthy volunteers, the specificity of which was demonstrated by us in previous studies [25, 26], was used to assess the antioxidant potential of PC. The results indicated that PC statistically significantly reduced the lipid hydroperoxide level and substantially increased the antiradical activity, which were comparable to those of the natural antioxidant carnosine, and in several instances exceeded its activity. The significant antioxidant activity of the new compound was due to the action of the carnosine fragment itself [3, 4] and of pyrrole, derivatives of which also exhibit pronounced antioxidant efficiency [14, 15].
An important aspect of evaluating the mechanisms of PC neuroprotector activity was its ability to permeate into neuronal cells. An in-house HPLC-MS method was developed by us for quantitative determination of PC.
The effectiveness of the protector activity of PC was assessed by us under OS conditions induced by AAPH, which directly generated ROS followed by development of OS in the aqueous phase within cells and in the extracellular milieu [27]. PC under these conditions was shown to increase the viability of SH-SY5Y human neuroblastoma cells as evaluated by the MTT assay. Simultaneously, PC prevented the death of neurons, which was determined from the content of LDH, which catalyzed the transformation of pyruvate into lactate. Release of LDH into the culture medium was indicative of damage to cell membranes. Because LDH is a rather stable enzyme, it is often used to determine cell damage and the degree of action of various cytotoxic compounds in various tissues and cell cultures [28]. In general, it was concluded by us based on a comparison of the LDH and MTT assays that PC was an efficient neuroprotector under OS conditions induced by AAPH.
Also, a fluorimetric method was used to show that PC efficiently reduced the intracellular level of ROS generated under AAPH-induced OS conditions after incubation for 1 h. Therefore, the neuroprotector efficacy of PC was due to its ability to permeate into neuronal cells and prevent an increase of ROS.
It could be concluded based on the above that the new conjugate of the antioxidant carnosine and the heterocycle pyrrole that was synthesized by us was characterized by resistance to the hydrolase activity of human serum carnosinase and pronounced antiradical activity in experimental models of neurotoxicity. It also exhibited cytoprotective activity under OS conditions induced by the neurotoxin AAPH, statistically significantly increased the viability of SY-SY5Y human neuroblastoma cell culture, and prevented the death of the cells. Thus, these results indicated that the creation of a new pharmaceutical based on PC was promising.
Conflict of interest
We declare no conflict of interest.
Financing
The work was supported by the Ministry of Education and Science of the RF in the framework of a State Task for the Research Center of Neurology (Topic No. AAAA-A20-120052790037-4).
Contributions of authors
TNF, VAM, and SLS developed the concept; OIK, OAM, and DAA performed the research; TNF, OIK, and DAA described the results, formulated conclusions of the research, and prepared and edited the text; TNF, VAM, and SLS approved the final version of the article.
References
A. M. Pisoschi and A. Pop, Eur. J. Med. Chem., 97, 55 – 74 (2015).
M. A. Piradov, M. M. Tanashyan, M. Domashenko, et al., Ann. Klin. Eksp. Nevrol., 9(1), 41 – 50 (2015).
D. Berezhnoy, S. Stvolinsky, A. Lopachev, et al., Amino Acids, 51, 139 – 150 (2019).
A. A. Boldyrev, G. Aldini, and W. Derave, Physiol. Rev., 93(4), 1803 – 1845 (2013); https://doi.org/10.1152/physrev.00039.2012.
A. Lopachev, O. Lopacheva, D. Abaimov, et al., Biokhimiya, 81(5), 678 – 689 (2016).
A. Lopachev, O. Lopacheva, E. Akkuratov, et al., Neirokhimiya, 34(1), 49 – 53 (2017).
A. Boldyrev, T. Fedorova, M. Stepanova, et al., Rejuvenation Res., 11(4), 821 – 827 (2008).
T. Fedorova, M. Belyaev, O. Trunova, et al., Biochemistry (Moscow), Suppl. Ser. A: Membr. Cell Biol., 3, 62 – 65 (2009).
K. Menon, A. Mousa, and B. de Courten, BMJ Open, 8(3), e020623 (2018).
S. P. Adams, M. Tsang, and J. M. Wright, Cochrane Database Syst. Rev., 12, (2012).
V. Bhardwaj, D. Gumber, V. Abbot, et al., RSC Adv., 5(20), 15233 – 15266 (2015).
A. E. Karateev, Sovrem. Revmatol., 2, 72 – 78 (2014).
A. Matveev, A. Krasheninnikov, and E. Egorova, Russ. Med. Zh. Med. Obozr., 2(4), 34 – 39 (2018).
G. M. Reddy, A. Camilo, Jr, and J. R. Garcia, Bioorg. Chem., 106, 104465 (2021).
D. Tzankova, S. Vladimirova, D. Aluani, et al., Acta Pharm., 70(3), 303 – 324 (2020).
T. N. Fedorova, S. L. Stvolinskii, V. A. Migulin, et al., RU Pat. No. 2,777,391 (13) C1, Aug. 3, 2022.
R. D. Seifulla, D. A. Abaimov, A. K. Sariev, et al., RU Pat. No. 2,585,115 C1, May 27, 2016.
A. Pegova, H. Abe, and A. Boldyrev, Comp. Biochem. Physiol. Part B: Biochem. Mol. Biol., 127(4), 443 – 446 (2000).
Y. Vladimirov, L. Packer, M. Traber, and W. Xin, Natural Antioxidants: Molecular Mechanism and Health Effects, L. Packer, M. G. Traber, and W. Xin (eds.), AOCS Press, Champaign, Illinois (1996), p. 125.
T. Fedorova, A. Logvinenko, V. Poleshchuk, and S. Illarioshkin, Neirokhimiya, 34(4), 344 – 349 (2017).
E. E. Akkuratov, O. M. Lopacheva, M. Kruusmagi, et al., Mol. Neurobiol., 52, 1726 – 1734 (2015).
F. Gunz, Br. J. Cancer, 2(1), 41 (1948).
J. F. Lenney, Biochim. Biophys. Acta, Enzymol., 429(1), 214 – 219 (1976).
S. Stvolinskii, N. Antonova, O. Kulikova, et al., Biomed. Khim., 64(3), 268 – 275 (2018).
S. Stvolinsky, E. Bulygina, T. Fedorova, et al., Cell. Mol. Neurobiol., 30, 395 – 404 (2010).
O. I. Kulikova, S. L. Stvolinsky, V. A. Migulin, et al., DARU, J. Pharm. Sci., 28, 119 – 130 (2020).
L. Rangaswamy, G. Aruna, U. Sathisha, et al., Chem.-Biol. Interact., 203, 448 – 455 (2013).
G. Holmgren, J. Synnergren, Y. Bogestal, et al., Toxicology, 328, 102 – 111 (2015).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiko-Farmatsevticheskii Zhurnal, Vol. 58, No. 4, pp. 17 – 25, April, 2024.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fedorova, T.N., Kulikova, O.I., Migulin, V.A. et al. New Derivative of Pyrrole and Carnosine: Synthesis, Physicochemical Properties, and Biological Acivity. Pharm Chem J 58, 617–624 (2024). https://doi.org/10.1007/s11094-024-03185-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-024-03185-z