
Abstract
Alloys and coatings used in high temperature applications are often subject to surface degradation influenced by the presence of deposits. Typical examples are fireside corrosion in coal-fired boilers and hot corrosion of blades and vanes in the hot sections of gas turbines. Depending on the source, deposit compositions may occur in a wide range from primarily sulfate to primarily oxide and various combinations of the two. There does not seem to be evidence of severe corrosion caused by gaseous Na2SO4. Generally, severe corrosion occurs when the deposits are liquid. However, significant corrosion has been observed in some cases with deposits, which are nominally solid. Corrosion is often caused by liquid deposits, in which negative oxide solubility gradients for alloy components are established across the deposits by rapid interface reactions. Hot corrosion can occur at temperatures near 700 °C by a variety of mechanisms if a phase which allows rapid transport is formed. This includes compounds such as Na2MoO4 (even in atmospheres without SO3), MSO4–Na2SO4 solutions or metastable nanostructured phases. Calling all corrosion in this temperature regime “Type II” can be misleading with regard to mechanism. There are similarities in the underlying mechanisms of some forms of hot corrosion in the 700 °C range and fireside corrosion in that they involve synergistic fluxing. The corrosive species responsible for fireside corrosion of ferrous alloys is a liquid (Na,K)2SO4–Fe2(SO4)3 solution and not alkali iron trisulfate. The propagation mechanism involves a synergistic dissolution process of protective Cr2O3 and Fe2O3. Terms such as “Type I,” “Type II” and “gas-phase-induced acidic fluxing” should be used with care in describing mechanisms of deposit-induced corrosion.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Alloys and coatings used in high temperature applications are subject to surface degradation by oxidation. However, in most practical applications the surface degradation is also influenced by the presence of deposits on the component surfaces. Typical examples are corrosion in coal-fired boilers resulting from ash deposits (generally termed fireside corrosion) and corrosion of blades and vanes in the hot sections of gas turbines (generally termed hot corrosion when the deposit consists primarily of sulfate). Early summaries of fireside corrosion [1] and hot corrosion [2] provide a wealth of practical information on the subject. In subsequent decades, significant research was conducted on the corrosion mechanisms. Detailed descriptions of this work have been provided in relatively recent reviews of fireside corrosion [3] and hot corrosion [4]. The purpose of this article is to compare various forms of deposit-induced corrosion and incorporate the results of recent studies to amplify aspects of the mechanisms, which have heretofore not been adequately explained. Thus, the nature of the article is primarily “review,” but in a few instances new data have been added to illustrate particular mechanisms. The sequence will be to first describe various mechanisms of hot corrosion and then fireside corrosion since the former provides a basis for understanding the latter.
Origin of Deposits and Deposit Composition
The composition of the deposits, which is determined by their origin, is a primary factor in determining the nature and extent of corrosion. Based on the concepts of slag chemistry, Ndamka et al. [5] characterized various oxides which are commonly found in deposits as follows:
-
Basic Oxides—CaO and MgO
-
Acidic Oxide—SiO2
-
Amphoteric Oxide—Al2O3
and introduced a “Basicity Index” (B.I.) as
The composition of the ash deposits from coal combustors depends on the composition of the coal. Table 1 shows typical ash compositions derived from coal from two regions of the USA (after [6]). The ash from the low-sulfur coal contains higher amounts of SiO2 and lower amounts of CaO and MgO (low B.I.) and lower amounts of SO3, i.e., smaller amounts of sulfates. The ash from the high-sulfur coal contains lower amounts of SiO2 and higher amounts of CaO and MgO (high B.I.) and higher amounts of SO3, i.e., larger amounts of sulfates. The high-sulfur coal ash contains larger amounts of iron oxides, which are significant in the production of liquid ash deposits. The effects of coal ash composition will be discussed more fully in the section on Fireside Corrosion.
Deposits on components in the hot section of gas turbines can be caused by impurities in the fuel and/or intake air. Conventional wisdom from early studies on corrosion in marine gas turbines was that the deposits consisted mainly of sodium sulfate (Na2SO4), which formed from NaCl in the intake air and SO3 formed by oxidation of sulfur in the fuel. However, it is now clear that deposits can arise from several sources. One of the first evaluations of turbine deposits was carried out by Tschinkel [7] who analyzed deposits on blades and vanes in turbines flown for times of 1000–5000 h by several airlines. Table 2 is based on Table 4 of the original paper—blades with insignificant amounts of deposit have not been included and small amounts of Mg and Si have been ignored. The key points of this analysis are:
-
Sources 9 and 10 are listed as being from “coastal airfields.” Unfortunately, this is not expanded upon.
-
Aviation fuel specifications at the time allowed 0.3 wt%S.
-
Tschinkel notes the excess of Ca and suggests the possibility of concrete dust from runways—this was apparently the first time it was recognized that not all the deposits came from reaction with the fuel.
-
Tschinkel analyzed the thermodynamics of the condensation of Na2SO4 and found it could only occur during idling and low power.
-
Calculations indicated Na2SO4 should evaporate and dissociate at high temperatures—Tschinkel comments it is not clear if deposits have accumulated over time or formed during the last idling cycle.
Deposit compositions on industrial gas turbines, which were operating in the vicinity of seacoasts, were carefully analyzed by Bornstein and Allen [8, 9]. Table 3 presents the compositions formed on the surfaces exposed to the hot gas path and only to the cooling air. The compositions in both regions were quite similar and had contents of the metallic elements, which were similar to deposits found in the compressor. Deposit compositions on external blade surfaces and in cooling passages were similar—indicating they formed by impaction of ingested particles, not condensation. Additionally the deposits contained significant amounts of K, Ca and Mg in addition to Na. The earlier Tschinkel paper was not cited.
Smeggil [10] summarized several studies of the composition of deposits formed on components in marine gas turbines. The emphasis was on water-soluble species, which were mainly Na2SO4 and CaSO4 with some amounts of MgSO4 and, in some cases, chlorides. Species, which are not soluble in water, e.g., oxides, were apparently not examined.
Useful information has been gained from analysis of deposits formed on insulating coatings, thermal barrier coatings (TBCs), which typically consist of ZrO2-based ceramics [11]. Fragments of deposits obtained from air plasma-sprayed TBC-coated first-stage vanes of an electric power generating gas turbine fueled by syngas produced from petroleum coke were examined after approximately 16,000 h of operation [12]. There were two groups of particle sizes. X-ray diffraction and EDS analyses showed that the larger particles were zirconia from the TBC. The smaller particles contained Fe, Ni and oxygen as well as some calcium and sulfur. The Fe and Ni presumably came from hardware in the gas turbines while the Ca and S suggested the presence of CaSO4. Characterization of the deposit from a turbine blade also indicated the presence of Fe2O3 and SiO2.
Braue et al. [13,14,15] conducted extensive analyses of deposits formed on electron beam physically vapor deposited (EBPVD) TBCs from components in an aero turbine from a commercial airline. Figure 1 shows a simple schematic diagram, which summarizes a number of their results. The outer deposit is an oxide mixture consisting primarily of CaO, MgO, Al2O3 and SiO2 known as CMAS [11]. Such a deposit can form from ingestion of runway dust, ash from volcanic eruptions or sand. The TBC was found to have CaSO4 present in all the columnar boundaries of the coating. This result is interesting in that the melting point of CaSO4 is 1460 °C (Table 4), which is well above the operating temperature of the TBC. A reaction zone was observed between interdiffused CaSO4/CMAS components (e.g., Na, Mg, Ca, S) and the upper TGO layer just above a columnar alumina zone, which had formed on the bond coat. This observation has two significant implications. First, it indicates that metallic components can be exposed to sulfate deposits, even if they are covered by a TBC. Second, it suggests that Na2SO4, which is liquid at the TBC operating temperature (Table 4), may have facilitated the penetration of Ca through the TBC columnar boundaries. Bohna [16] recently demonstrated one way this can occur. The top two images of Fig. 2 show a micrograph and Ca X-ray map for a vertically cracked air plasma-sprayed TBC, which was coated with a CMAS-type deposit containing 65 at.% CaO and exposed in dry air at 1100 °C for 72 h. It is clear that no Ca has penetrated into the TBC. The bottom two images show a micrograph and Ca X-ray map for the same coating which was exposed to the same mixture plus 2 at.% K2SO4, which melts at 1071 °C (Table 4). In this case, Ca has penetrated completely through the TBC, presumably by being carried with the sulfate as it wet the crack surfaces. Tolpygo [17] proposed that penetration of CMAS components through EBPVD TBCs can occur by vapor transport of their hydroxides and showed interaction with the TGO in burner rig experiments. However, the reaction products were found to contain Na, which had originated in the silica used. The sulfur content of the fuel used in the burner rig was not specified. Lutz et al. [18] showed that hydroxides could transport from CMAS and react with exposed TGOs in furnace tests but did not produce reaction with the TGO below an EBPVD TBC. Further analysis of TBC penetration is beyond the scope of this article.

Schematic diagram of a CMAS deposit on the surface of a TBC from a commercial airline gas turbine described in [13,14,15]. A middle section of the YSZ top coat displayed CaSO4 infiltration of open porosity. A reaction zone was observed between interdiffused CaSO4/CMAS components (e.g., Na, Mg, Ca, S) and the upper TGO layer just above a columnar alumina zone (CAZ), which had formed on the bond coat

Micrograph and Ca X-ray map for a vertically cracked TBC which was coated with a CMAS-type deposit containing 65 at.% CaO and exposed in air at 1100 °C for 72 h (top). Micrograph and Ca X-ray map for the same coating, which was exposed to the same mixture plus 2 at.% K2SO4 (bottom) [16]
Shifler [19] has pointed out that current US Navy fuel specifications require the sulfur content to be below 15 ppm and that sulfate deposits in gas turbines now result from atmospheric pollution (SO2 and particulate matter), particularly in some parts of the world. However, it should be pointed out that there are many examples in the early literature [2] in which vanadium contamination in the fuel resulted in severe corrosion. The effects of vanadium have been reviewed by Hancock [20]. The current significance of those early results is that deposits can indeed form by condensation and that maintaining low impurity levels in the fuel should still be beneficial.
In summary, depending on the source, deposit compositions may occur in a wide range from primarily sulfate to primarily oxide:
However, rarely will they be pure oxide or pure sulfate. Figure 3 summarizes the thermodynamic stability of various relevant compounds. The form of deposit species will be:
-
Na and K will always be present in sulfates (with the exception of high-SiO2 deposits where they may be in silicates).
-
Mg and Ca will be present in sulfates or oxides (depending on T and pSO3).
-
Si and Al (data not shown) will always be present in binary or ternary oxides

Equilibrium SO3 partial pressures for converting various oxides to sulfates
The significance of mixed deposits includes:
Alkali sulfates (and possibly water vapor) may result in penetration of components of CMAS through TBCs at temperatures below the melting point of CMAS.
The presence of oxides may alter the fluxing behavior of sulfate deposits (this will be discussed in several subsequent sections).
Finally, with regard to NaCl which may be ingested in the combustion air, Pahlavanyali [21] has shown that small amounts of NaCl degrade the cyclic oxidation resistance of the superalloy CMSX-4 at 1000 and 1100 °C and the cyclic oxidation resistance of Pt–Al-coated CMSX-4 at 1100 °C with modest effect at 1000 °C. These results are not believed to be significant for blades and vanes in the hot gas path because NaCl would be reacted in the combustor, i.e., the very high exposure temperatures are not relevant. Pettit [4] has pointed out that there are characteristic microstructures associated with NaCl-induced corrosion, which are rarely seen on hot section components. However, NaCl in intake air could survive in cooling passages for significant times and produce corrosion as component temperatures are increased. Smeggil [10] has shown that small amounts of chlorides can significantly lower the melting temperatures of deposits. Therefore, cyclic oxidation experiments on superalloys and “marginally resistant coatings” with small amounts of NaCl and exposed at realistic T (≈ 700–900 °C) would be worthwhile.
Physical State of Deposits
Vapor Results
DeCrescente and Bornstein [22] showed significant hot corrosion of a susceptible superalloy, B-1900, when Na2SO4 could condense but none when it remained in the vapor phase. Knutsson et al. [23] published results nominally on corrosion by Na2SO4 in the vapor phase at 900 °C. However, the conditions of the experiments were such that condensation occurred—thus producing small amounts of liquid salt and mild hot corrosion conditions. It should be pointed out that condensation may occur by reaction with Cr-oxides and/or wetting of alumina (Na2SO4 has been observed to wet alumina, particularly under basic fluxing conditions [24]). Thus, there does not seem to be evidence of severe corrosion caused by Na2SO4 when it remains in the vapor phase.
Liquid Results
It is generally presumed that deposits must be molten to cause severe corrosion. Table 4 presents the melting temperatures of various compounds and solutions, which are important in deposit-induced corrosion. The melting point of pure Na2SO4 is 884 °C. However, interaction with various transient oxides can produce liquid at much lower temperatures. The Fe2(SO4)3–K2SO4 phase diagram is presented in Fig. 4 [25]. A liquid K2SO4–Fe2(SO4)3 solution forms with small additions of Fe2(SO4)3 and a eutectic temperature of 627 °C. Luthra [26] has commented upon similar low melting phases in the CoSO4–Na2SO4 and NiSO4–Na2SO4 systems. The mechanisms of corrosion in liquid deposits will be discussed in the sections on Fluxing Reactions in Liquid Deposits and Temperature Effects.

K2SO4–Fe2(SO4)3 phase diagram after Ref. [25]
Solid Results
Significant corrosion has been observed in some cases with deposits, which are nominally solid. Chiang et al. [27] reported on the degradation of chromia- and alumina-forming alloys with deposits of MgO and CaO.
-
Both CaO and MgO deposits increased the degradation of Cr2O3-forming alloys by accelerated CrO3 reactive evaporation at temperatures as low as 850 °C. The mechanism was found to involve “gettering” of the CrO3 vapor by the CaO or MgO, which led to more rapid evaporation from the Cr2O3 scale and more extensive depletion of Cr from the alloys. This mechanism may be relevant to chromide coatings used in cooling passages of gas turbine components.
-
Reaction between CaO and Cr2O3 scales on chromia-forming alloys or transient Cr2O3 on MCrAlY alloys at temperatures above 1000 °C resulted in catastrophic corrosion by liquid formation.
-
MgO did not produce the catastrophic attack because of the absence of low melting phases in the MgO–Cr2O3 system.
The effects of solid deposits will be discussed more fully in a section on Oxide Deposits.
Fluxing Reactions in Liquid Deposits
When a deposit covers the alloy surface, illustrated for Na2SO4 in Fig. 5, it separates the alloy from the gaseous environment [28]. While the reaction–product barrier, which forms beneath the deposit on the alloy surface, often exhibits features similar to those for the gas–alloy reaction in the absence of the deposit, there are differences because sulfur also enters the alloy from the deposit. The hot corrosion process occurs in two stages: initiation stage and propagation stage. During the initiation stage, the alloy undergoes a process similar to simple oxidation with the difference that in the hot corrosion process the oxidizing species comes from the salt deposit. The interaction between the oxides formed on the alloy and the salt can lead to extremely corrosive conditions as the salt becomes basic or acidic. Na2SO4 exhibits an acid–base character with SO3(g) being the acidic component and Na2O(s) the basic component. The composition of the Na2SO4 melt at a fixed temperature can therefore be described by the oxygen partial pressure and either the activity of Na2O in the melt, \({\text{a}}_{{{\text{Na}}_{ 2} {\text{O}}}}\), or the SO3 partial pressure, through the equilibrium constant for Eq. 2 [4].
The salt may become more basic (higher Na2O activity) or more acidic (lower Na2O activity) when interacting with the base alloy and its oxides, i.e., species dissolving into the salt can alter its basicity. A phase stability diagram for the Na–S–O system (Fig. 6) shows the compositional changes, which occur in the salt deposit during the initiation stage [28]. Once the salt deposit becomes sufficiently basic or acidic, rapid degradation can occur, i.e., the propagation stage begins. In some cases, the end of the initiation stage occurs when the deposit becomes liquid, penetrates the oxide scale and spreads along the alloy/scale interface.

Schematic diagram illustrating oxygen consumption by the alloy during the initiation stage of hot corrosion [28]

A thermodynamic stability diagram for the Na–O–S system showing possible composition shifts of the Na2SO4 phase [28]
Our understanding of the processes involved in fluxing of oxides has been greatly enhanced by the excellent fundamental studies of Rapp and coworkers [29,30,31,32,33]. The oxide solubility plots [29] presented in Fig. 7 show that a protective oxide scale can be dissolved due to melts with low aNa2O (acidic melts). The Na2SO4 deposit can become acidic due to SO3 in the gas atmosphere, or by the dissolution of transient oxides of Mo or W, which are added to superalloys for solid solution strengthening or V oxides, which arise from fuel contamination. Once a deposit has been formed on the surface of combustion hardware, the amount of corrosion depends significantly on whether or not the deposit melts. If the temperature of the combustion environment is above the melting point of Na2SO4 (Tm = 884 °C), the corrosion is called Type I hot corrosion or high temperature hot corrosion. Below the melting point of Na2SO4, the salt deposit can become molten because of a reaction between the combustion gas and the oxide scale grown on the alloy. This type of corrosion is called Type II hot corrosion or low temperature hot corrosion. It will be pointed out subsequently that the designations “Type I” and “Type II” are somewhat unfortunate in that they sometimes lead to incorrect assumptions regarding mechanisms.

Adapted from [29]
Measured oxide solubilities in Na2SO4 at 927 °C and 1 atm O2.
Rapp and Goto [30] proposed a criterion by which the continued self-sustaining hot corrosion attack can occur. This is known as the Rapp–Goto Criterion and is given by Eq. 3, where Coxide is the solubility of the protective oxide and x is the distance into the molten salt deposit from the oxide/salt interface.
When the solubility gradient is positive, the salt can become saturated with oxide and a protective scale is able to form over the metal surface. A negative solubility gradient in the oxide solubility at the oxide/salt interface results in dissolution of the protective oxide scale and reprecipitation of the oxide as discontinuous non-protective particles in the outer regions of the molten salt deposit where the solubility is lower. This is shown schematically in Fig. 8. It is important to note that, in situations where an acidifying species is coming from the vapor phase (e.g., high pSO3 or V coming from the fuel), a positive solubility gradient will be established and self-sustaining fluxing does not occur. This is because the oxide solubility will be greater at the salt/gas interface than it is at the scale/salt interface, i.e., the solubility gradient will be positive. Thus, significant hot corrosion requires large amounts of the deposited salt to be present.

Schematic diagram illustrating the Rapp–Goto criterion for the dissolution and reprecipitation of a hypothetical oxide “MO”
Type I hot corrosion typically occurs at high temperatures of approximately 900–1000 °C. There are two propagation modes for this type of hot corrosion. These occur when there is dissolution of the protective oxide from a highly basic molten salt deposit where aNa2O is high, and when there is dissolution of the protective oxide from a highly acidic molten salt deposit where aNa2O is low as the result of reaction with acidic oxides formed from the alloy [4]. These are called basic fluxing and alloy-induced acidic fluxing, respectively. These two forms of Type I hot corrosion occur due to interactions between the salt deposit and the underlying surface.
The hot corrosion of Ni provides a significant example of basic fluxing. For exposures in air, as oxidation begins the oxygen potential in the Na2SO4 decreases since NiO begins to form on the nickel and oxygen cannot move rapidly enough through the Na2SO4 from the gas. As the oxygen potential in the Na2SO4 decreases, the sulfur potential concomitantly increases. Eventually sulfur potentials can be achieved which are sufficient to form nickel sulfide. The net effect of the oxygen and sulfur removal from the Na2SO4 deposit on nickel oxidized in air is to increase the activity of Na2O in the Na2SO4 at the salt/metal interface, which increases the local solubility of NiO, and the Rapp–Goto criterion is satisfied [4].
An important extension of the study of Ni was provided by Otsuka and Rapp [31] for the hot corrosion of Ni by basic fluxing in a gas with an SO3 partial pressure for which dissolution of NiO should have occurred by acidic fluxing. They performed experiments with preoxidized nickel with a Na2SO4 deposit in an oxygen–SO2 mixture and measured the Na2O activity and oxygen partial pressure at the NiO/Na2SO4 interface as a function of time. A schematic diagram of the results is presented in Fig. 9, where a stability diagram is used to show the phases of nickel which are stable as a function of the Na2O activity and oxygen partial pressure. The basic solubility of NiO is dependent on the oxygen partial pressure according to reaction 4:
so the solubility minimum shifts with pO2 as indicated by the dashed line in Fig. 9.

A stability diagram is used to show the phases of nickel, which are stable as a function of the SO3 partial pressure (Na2O activity) and oxygen partial pressure and the reaction path when Ni is coated with Na2SO4 and exposed in an O2–SO2 atmosphere. (Schematic diagram based on results in Ref. [31].)
The sequence in which these measurements were made is also qualitatively indicated by successively higher numbers on this stability diagram. The salt, which stands in equilibrium with the bulk gas composition, is indicated by the final point, #5. It can be seen that as the oxygen partial pressure is decreased substantially due to reaction with nickel and the region of sulfide stability is entered (point #2), the activity of Na2O increases due to oxygen and sulfur removal from the Na2SO4 (point #3). The attack occurs by basic fluxing even though the bulk gas composition is such that, absent reaction, NiO should dissolve by acidic fluxing. This result is highly significant in that it shows rapid interface reactions can control the hot corrosion mechanism with little or no influence from the gas phase.
The hot corrosion of Ni–Cr–Al alloys also occurs by basic fluxing [4]. In simple terms, Al removes oxygen from the sulfate and Cr removes sulfur so that the local SO3 partial pressure becomes low and the salt at the salt/alloy interface becomes highly basic. This creates negative solubility gradients for the various oxides and rapid fluxing occurs.
If Mo is added to Ni–Cr–Al alloys, the propagation mechanism changes. A description of the corrosion mechanism for alloy-induced acidic fluxing Type I hot corrosion will now be discussed in more detail for Mo-containing alumina-forming nickel-based alloys [4]. During the first stages of oxidation, transient MoO3 and NiO oxides as well as Al2O3 form at the alloy surface. MoO3 reacts with the oxide ions in the Na2SO4 salt deposit to decrease the Na2O activity of the melt, which prevents the basic fluxing mechanism from occurring. This reaction is shown in Eq. 5.
MoO3 will decrease the Na2O activity in Na2SO4 to levels where acidic fluxing of the protective Al2O3 scale can occur. The attack initiates near Mo-rich particles because the activity of MoO3 in the Na2SO4 is highest in these areas. The acidic fluxing of the Al2O3 scale causes Al3+ and MoO42− ions to diffuse through the salt where Al2O3 reprecipitates as a porous non-protective scale, and the MoO3 evaporates. This form of attack is self-sustaining, because MoO3 is able to continue to form at the alloy/salt interface and evaporate at the salt/gas interface to maintain a negative solubility gradient. A typical microstructure for this form of attack is presented in the right-hand micrographs of Fig. 10.

Comparison of corrosion of NiCrAlMo model alloy (Ni–8Cr–6Al–6Mo wt%) with a Na2SO4 deposit exposed in air for 100 h at 700 °C (left) and for 100 h at 900 °C (right). The latter exposure is considered typical of Type I hot corrosion conditions
Another important example of limited effect of the gas phase is the work of Hwang and Rapp [32] who proposed a model to explain an increase in Fe2O3 powder solubility in fused Na2SO4 at 927 °C (1200 K) under an O2 + 1% SO2 gas mixture in the presence of Cr2O3 powder and vice versa. The model was called synergistic dissolution, which can be defined as an acid–base coupling reaction process, wherein the simultaneous dissolution of two or more oxides in contact with a given molten salt (e.g., Na2SO4) can support each other by the dissolution of one oxide shifting the local salt composition in the basic direction while that of the second shifts it in the acidic direction. The conditions of acidity or basicity in a molten salt (i.e., activity of Na2O or \({\text{p}}_{{{\text{SO}}_{ 3} }}\)) containing two or more oxides which allows synergistic fluxing must be such that its value lies between the minimum solubility values of the individual oxides. Under these conditions, the more acidic oxide(s) will be dissolved via basic fluxing releasing SO3 into the liquid salt, and thus, the acidity of the melt will be increased locally. This increase in melt acidity will establish conditions for acidic dissolution of the more basic oxide(s). Therefore, as the more basic oxide(s) dissolve via acidic fluxing releasing oxide ions (e.g., Na2O in the form of O2− ions) into the liquid salt, the basicity of the melt will be increased again. Under this cyclic acid–base coupling reaction, the negative solubility gradients (Rapp and Goto criterion) are simultaneously satisfied for both oxides, at least locally during different stages of the synergistic process. It is important to point out that the cyclic process described above must be very localized occurring at the interfaces of two different oxide particles very close to each other (probably some tens of nanometers). In other words, the global acidity (or basicity) measured by an external probe will give an average value of the melt around the probe. This kind of analysis allowed first Hwang and Rapp [32] and later Longa-Nava et al. [33] to study the synergistic dissolution kinetics of Fe2O3–Cr2O3 and NiO–Cr2O3, respectively. Based on these studies, the synergistic dissolution can be considered a self-sustaining process. This process is shown schematically in Fig. 11 for the synergistic fluxing of the oxides of Co and Al where Co3O4 dissolves by acidic fluxing and Al2O3 dissolves by basic fluxing.

Solubility plots illustrating the synergistic dissolution of Co3O4 and Al2O3 in Na2SO4
It is important to note that in the case of synergistic fluxing the salt basicity is determined by rapid local reactions and not that which would be dictated by the SO3 partial pressure in the gas phase.
Temperature Effects
The previous section described alloy-induced acidic fluxing at high temperatures of around (900–1000 °C). It is also possible to have hot corrosion at lower temperatures between 650 and 750 °C, i.e., below the melting point of Na2SO4 (884 °C), see Table 4. Corrosion in this temperature regime is generally called Type II hot corrosion. In Type II hot corrosion, the accelerated corrosion is generally believed to be caused by the formation of a (Na,K)2SO4–MSO4 eutectic melt that has a melting point well below that of (Na,K)2SO4 [25, 26]. It is also known that a partial pressure of SO3 of about 10−5 atm is required for this melt to form. This amount of SO3 in the gas atmosphere is not uncommon in industrial gas turbines. However, deposit melting in some cases occurs without the influence of SO3.
Disk alloys used in advanced gas turbine engines often contain significant amounts of Mo (2 wt% or greater), which is known to cause corrosion under Type I hot corrosion conditions (at temperatures around 900 °C) due to alloy-induced acidic fluxing. However, it has been shown that several model and commercial Ni-based disk alloys with different amounts of Mo can severely corrode at 700 °C in air in the presence of a Na2SO4 deposit [34]. Alloys with 2 wt% or more Mo showed degradation products similar to those observed previously in Mo-containing alloys, which undergo alloy-induced acidic fluxing Type I hot corrosion even though the temperatures were in the Type II hot corrosion range. A typical alloy-induced acidic fluxing Type I hot corrosion microstructure from a 900 °C exposure is shown in Fig. 10 for a NiCrAlMo alloy compared to the results from the same alloy tested at 700 °C in air. The degradation is much greater for the 900 °C hot corrosion image. The appearance of Mo-rich oxides at the alloy/oxide interface is a key characteristic of alloy-induced acidic fluxing. The Mo-rich oxide is more pronounced on the 900 °C image, but Mo-rich oxide was detected not only on the NiCrAlMo model alloy but on all model and commercial alloys with a Mo content higher than 2 wt% tested in air at 700 °C. The reason for the degradation observed at 700 °C is the formation of sodium molybdate with a melting point (687 °C), which is below the temperature tested, see Table 4. The reaction forming sodium molybdate reduces the Na2O activity and causes the melt to become highly acidic, producing a negative solubility gradient at the oxide/salt interface which results in dissolution and fluxing of the NiO or Al2O3 oxide scales. The fluxing of both the transient NiO and protective Al2O3 scale causes Ni2+, Al3+ and MoO42− ions to diffuse through the salt where Al3+ and/or Ni2+ are reprecipitated where the pO2 is higher as a non-protective scale, and the MoO3 evaporates. The mechanism which is occurring for the Mo-containing disk alloys at 700 °C with a Na2SO4 deposit in air is very similar to the mechanism for alloy-induced acidic fluxing Type I hot corrosion. This is important, as alloy-induced acidic fluxing is believed to only occur at high temperatures (above 900 °C). A low temperature (700 °C) alloy-induced acidic fluxing mechanism is able to occur for Mo-containing Ni-based alloys in air atmospheres.
The presence of Ni-rich oxide was not only observed as an outside layer but also at what appeared to be the deposit particles edges (Fig. 12). EDS and WDS measurements indicate that there are significant areas of pure Na2SO4 present in the deposits. These presumably remained solid during the exposure. Apparently Mo has only interacted with the Na2SO4 at the salt/alloy interface and boundaries between the Na2SO4 particles, and the Na2MoO4 is able to wet the interparticle boundaries. The interparticle veins are the Ni transport paths through the deposit to form the NiO observed at the salt/gas interface. Degradation is less than that observed at 900 °C where the entire deposit is liquid and provides a much larger transport cross section for Ni.

SEM images of Mo-containing alloys (2, 4, and 6 wt% Mo) with Na2SO4 deposit after exposure in air at 700 °C for 100 h
Tests performed on alloys with greater than 2 wt%Mo at 650 °C, below the melting point of sodium molybdate (Table 4), produced no severe corrosion and tend to confirm that Na2MoO4 is the cause of corrosion. Figure 13 shows a schematic diagram of the corrosion rate versus temperature for NiCrAl and NiCrAlMo alloys. The “double peak” generally observed for NiCrAl alloys [4] is absent for NiCrAlMo alloys. Since the salt can become molten at any temperature above the melting point of Na2MoO4 (687 °C) and the dissolution of Mo into the salt creates acidic conditions at the salt/alloy interface, alloy-induced acidic fluxing can occur over the entire temperature range with the corrosion rate increasing monotonically with temperature.

Schematic diagram comparing the temperature dependence of the hot corrosion of NiCrAl alloys (two peaks) and NiCrAlMo alloys (single peak)
Type II HC is often described as gas-phase-induced acidic fluxing given that gas conditions, which tend to make the salt acidic are required to establish liquid salt formation for alloys without Mo (or W). However, as mentioned previously, since SO3 is entering the salt from the gas phase a positive solubility gradient would be established for the various oxides. The most widely accepted model for Type II HC attack is the one proposed by Luthra [35,36,37] for Co-based alloys, which is based on the rapid dissolution of the more noble metal (Co) or metal oxide (CoO) via acidic fluxing which prevents the formation of a protective M2O3 scale. It was also postulated that the driving forces for the dissolution at the scale/liquid salt interface, transport through the liquid salt, and subsequent precipitation at the liquid salt/gas interface are a negative activity gradient of Co2+ cations and positive gradient of oxygen partial pressure (\({\text{p}}_{{{\text{O}}_{ 2} }}\)) and that the Rapp–Goto solubility criterion is not necessary [35, 37]. This mechanism does not seem to explain all experimental observations and an alternative mechanism for corrosion of Co-based alloys has been recently developed [38] and will now be described.
Figure 14 shows the development of the corrosion morphology at the hot corrosion front on a Co–20Cr–26Al–0.4Y (at.%) specimen coated with 2.5 mg/cm2 Na2SO4 and exposed to O2 + 1000 ppm SO2 at 700 °C, which combined with EDS results (not shown) indicated a clear Al and Cr depletion region at the alloy/salt interface (seen as a disappearance of the Al-rich β-phase). This observation suggests that the rapid dissolution of Co is not the controlling factor in the degradation, since an enrichment of γ-phase, which is richer in Co, is observed at the alloy/salt interface.

Temporal evolution of the hot corrosion morphology for a Co–20Cr–26Al–0.4Y (at.%) alloy coated with 2.5 mg/cm2 Na2SO4 and exposed to O2 + 1000 ppm SO2 (\({\text{p}}_{{{\text{SO}}_{ 3} }}\) ~ 7.2 × 10−4 atm.): a, b 1 h, c, d 5 h, e, f 10 h and h, i 20 h
The following mechanism based on synergistic fluxing has been proposed [38] as an alternative to the Luthra model for Co-based alloys. First, assume that the conditions to stabilize the liquid CoSO4–Na2SO4 solution by the sulfation of transient Co3O4 and/or CoO have been established due to the reaction with the SO3 in the gas.
Once a liquid salt film is formed, different chemical gradients will be established through the liquid as a result of the O2 and SO3 consumption due to the alloy oxidation and oxide sulfation, respectively (see Fig. 15a-1, a-2). Based on this, a significant decrease in the melt acidity with depth will occur due to the positive gradient in \({\text{p}}_{{{\text{SO}}_{ 3} }}\) established through the liquid salt. Under these conditions, at least locally, the basicity of the melt will be such that the melt satisfies the conditions for basic fluxing of the Al2O3 and/or Cr2O3 (see Fig. 15a-3) at the thermally grown oxide (TGO)/salt interface via
As a result of the basic fluxing of Al2O3 and/or Cr2O3, the acidity of the melt must increase at the TGO/salt interface due to the release of SO3 during NaAlO2 and Na2CrO4 formation (which are dissociated in the melt as AlO2− and CrO42− ions). Similarly, the Cr2O3 may also be dissolved by basic fluxing to form chromite ions (CrO2−).

Schematic representation of: a basic fluxing of either Al2O3 or Cr2O3 and b acidic fluxing of Co3O4 or CoO
Figure 15a-1 to a-3 summarizes the basic dissolution process in the synergistic region. At the same time, the continuous increase in the melt acidity results in the continuous acidic fluxing of the cobalt oxides via Eqs. 8 and 9
Similarly, Fig. 15b-1 to b-3 illustrates the acidic dissolution process in the synergistic region. Conversely, this process will decrease the acidity of the melt again (at least locally) as a result of the release of Na2O to the melt during the formation of CoSO4 in solution as Co2+ and SO42−ions. This provides the conditions for the basic dissolution of the Al2O3 and/or Cr2O3 to take place again. It is important to point out at this stage that the thickness and extent of the synergistic region, as well as the local potentials (e.g., O2 and \({\text{p}}_{{{\text{SO}}_{ 3} }}\)), are unknown and these have only been drawn to illustrate qualitatively what is happening during the dissolution process. However, it is postulated that within the synergistic region, particularly at the sites where the dissolution process takes place, the real gradients can oscillate from site to site (from acidic to basic) at the scale/salt interface in order to satisfy the mutual dissolution process and thus the Rapp–Goto criterion for a negative solubility gradient (see Fig. 16), even when a positive \({\text{p}}_{{{\text{SO}}_{ 3} }}\) gradient is observed outside the synergistic region.

Schematic representation of the synergistic dissolution process: (1) initial gas condition; (2) a locally decrease in the melt acidity is expected once a liquid salt film is established at the scale/salt interface 1 as a result of oxide sulfation, giving the conditions for the Al2O3 and/or Cr2O3 basic fluxing; (3) the acidity of the melt increases due to basic fluxing of the Al2O3 and/or Cr2O3; (4) the local increase in the melt acidity gives the conditions for the non-protective Al2O3 and/or Cr2O3 reprecipitation in the melt; (5) at the same time, the increase in the melt acidity gives the conditions for a localized acidic fluxing of cobalt oxides at the scale/salt interface 2; (6) the basicity of the melt increases due to acidic fluxing of the CoO and/or Co3O4; (7) the local increase in the melt basicity gives the conditions for the non-protective cobalt oxides reprecipitation and the Al2O3 and/or Cr2O3 basic fluxing again (stage 2)
Once the synergistic region has been formed with the Co2+, AlO2−, CrO2− and CrO42− species in solution in the melt, the conditions required for diffusion and their subsequent reprecipitation as Co-, Al- and Cr-compounds must be established through the liquid salt. These conditions must be satisfied above the synergistic region, since within it, the continuous increase and decrease in acidity would tend to keep different species in solution. Outside of the synergistic region, it is expected that the various chemical gradients follow chemical equilibria.
Summary of Newly Proposed Mechanism for Type II Hot Corrosion for Co Alloys
Liquid Formation
Na2SO4 melting results from a partial pressure of SO3 which is large enough to stabilize a Na2SO4–CoSO4 solution which is liquid at the exposure temperature [37].
Initial Attack
The Na2SO4–CoSO4 solution dissolves γ-Al2O3 and Cr2O3 by basic fluxing. This results in depletion of the β-phase from γ to β type alloys and a shift in the local salt composition in the acidic direction as Na2O is consumed.
Synergistic Fluxing
The basic fluxing of the Al2O3 and Cr2O3 shifts the salt composition back in the acidic direction and causes acidic fluxing of Co-oxides. This in turn creates local conditions for further basic fluxing of Al2O3 and Cr2O3.
Transport and Reprecipitation
The Al and Cr transport occurs outwardly by diffusion of aluminate and chromate ions with reprecipitation of the oxides occurring where the local salt composition is more acidic. This apparently occurs at rather short distances from the dissolution sites as the alumina and chromia are found below the original specimen surface. Finally the cobalt transport through the melt is conducted via the 3Co2+/2Co3+ exchange reaction as proposed by Luthra [37] following
If the proposed synergistic fluxing mechanism is correct, it should provide a description for how the corrosion rates are affected by significant variables, such as alloy composition, SO3 partial pressure and the presence of oxide contaminants in the Na2SO4.
Effect of SO3 Partial Pressure
The rate of hot corrosion at 700 °C increases with increased pSO3. This is the result of:
-
1.
More extensive and rapid formation of liquid in the deposit.
-
2.
More rapid outward transport of dissolved Co2+ via the 3Co2+/2Co3+ exchange reaction [37].
-
3.
Reprecipitation of Al2O3 and of Cr2O3 at more shallow depths, which shortens the transport paths for these species.
Effect of Cr Content
It is generally accepted that the rate of Type II HC of Co alloys decreases with increased alloy chromium concentration. This is the result of:
-
1.
The volume fraction of the selectively attacked β-phase is decreased.
-
2.
The concentration of acidic species in the melt is increased.
Effect of Salt Composition
The sodium sulfate in the deposits in operating gas turbines is often contaminated by oxides, which are ingested in the intake air. The proposed mechanism indicates that those contaminants, which shift the basicity of the salt, could alter the synergistic fluxing.
Figure 17 presents micrographs showing the effect of alloy composition and CaO additions to the deposit on the extent of degradation of MCrAlY alloys exposed at 700 °C with a Na2SO4 deposit in O2 +1000 ppm SO2 [39]. The rate increases with Co content in the alloy. This is primarily the result of more rapid melting of the deposit and a larger volume fraction of liquid. The CaO results in a considerable decrease in the extent of corrosion for the CoCrAl alloy (Alloy 2) and a slight decrease for Ni-based alloys. These rate decreases caused by CaO in the deposit occur because it shifts the melt to higher basicity, which is outside the range for efficient synergistic fluxing.

Cross sections, which show the effects of alloy composition and CaO additions to the deposit on the extent of degradation of several MCrAlY alloys exposed for 100 h at 700 °C with a Na2SO4 deposit in O2 +1000 ppm SO2. The corrosion rate is slightly decreased by CaO in the deposit (higher basicity)
Figure 18 presents micrographs showing the effect of alloy composition and SiO2 additions to the deposit on the extent of degradation of the same MCrAlY alloys exposed at 700 °C with a Na2SO4 deposit in O2 +1000 ppm SO2 [39]. The rate is decreased for all three alloys by SiO2 in the deposit because it shifts the melt to higher acidity, which is again outside the range for efficient synergistic fluxing.

Cross sections which show the effects of alloy composition and SiO2 additions to the deposit on the extent of degradation of several MCrAlY alloys exposed for 100 h at 700 °C with a Na2SO4 deposit in O2 +1000 ppm SO2. Corrosion rate increases with Co content. Corrosion rate decreased by SiO2 in deposit (higher acidity)
The mechanism just described was developed specifically for CoCrAl-type alloys. However, it should apply in modified form to alumina-forming NiCrAl-type alloys. Finally, with regard to the latter, it should be mentioned that it is often stated incorrectly in the literature that sulfides are absent from the alloy for Type II corrosion. This is generally the case for Co-based alloys but, as pointed out by Pettit [4], not so for alloys, which contain significant amounts of Ni. Figure 19 shows a cross section of a cast Ni–20Co–18Cr–12.5Al–0.6Y–0.4Si–0.25Hf (wt%) after 10 h exposure at 700 °C with a Na2SO4 deposit in O2 + 1000 ppm SO2. There are clearly copious amounts of Cr-sulfide in the underlying alloy, which probably result from SO3 release in reactions such as 6 and 7 above.

Cross section of Ni–20Co–18Cr–12.5Al–0.6Y–0.4Si–0.25Hf(wt%) after 10 h exposure at 700 °C with a Na2SO4 deposit in O2 + 1000 ppm SO2
A recent transmission electron microscopy (TEM) investigation of the corrosion of a coated superalloy obtained from service [40] has indicated the presence of nanostructured oxides and sulfides at the base of a corrosion pit. The authors speculate that this observation precludes the possibility of Type II corrosion occurring by mixed sulfate attack. Unfortunately, other than the exposure temperature being in the range 600–750 °C, no details are given about the exposure conditions, e.g., deposit composition or loading, SO3 content in the exposure environment, exposure time, etc. Furthermore, there is no discussion regarding whether the component was exposed to liquid water during service, e.g., washing, or whether water-based solutions were used anywhere in the specimen preparation. Exposure to water would explain the absence of Na or K in the corrosion products. However, the appearance of nanostructured phases is consistent with the result of another recent study of an uncoated 2nd generation single-crystal superalloy [41], which was carried out under well-controlled conditions.
It should be pointed out that this type of superalloy does not form a continuous alumina or chromia scale in this temperature range, even in the absence of deposits, as has been mentioned by Smeggil [10]. This is illustrated for PWA 1484 (nominal composition in Table 5) in Fig. 20. At 1100 °C, the alloy forms a continuous alumina scale. However, at 700 °C it undergoes some internal oxidation in dry air and profuse internal oxidation in air + water vapor.

Oxidation products formed on PWA 1484: a at 1100 °C in dry air, b at 700 °C in dry air and c at 700 °C in air + 10% H2O
In the presence of a Na2SO4 deposit, hot corrosion was found to occur on this alloy at 550–650 °C, where liquid formation does not occur (see Table 4) [41]. The corrosion morphology consisted of porous (Ni,Co)O at the gas interface, with entrapped Na2SO4 and an internal oxide/sulfide zone. Channels of (Ni,Co) O appeared to form within the sulfate deposit, as shown in Fig. 21. A time study showed that the reaction progresses by consuming the sulfate deposit, starting from the sulfate/gas interface. Dry air exposures did not cause accelerated corrosion, indicating that SO2/SO3 in the environment is essential for the observed low temperature corrosion to occur.

Cross-sectional SEM images of the corrosion products formed on PWA 1484 after 20 h hot corrosion exposure in a air and O2-(2.5, 10, and 100 ppm) SO2 and with 2.5 mg/cm2 Na2SO4 deposit; b phase identification for 600 °C in O2-2.5 ppm SO2 [41]
Pure nickel corrosion exposures were also conducted at 500–650 °C with the aim to more clearly elucidate a mechanism for the inferred solid-state attack [41]. The nickel gave similar corrosion morphologies to those obtained for the alloy. Experiments conducted on pure nickel with crystals of Na2SO4 on the surface revealed a rapid “wetting” of the sulfate. The effect of pre-oxidation temperature was also studied and the results revealed that low temperature pre-oxidation, which produced a more defective oxide, was necessary to sustain the observed sulfate wetting and associated rapid attack.
TEM analysis of exposed nickel-based superalloy and pure nickel specimens identified a nanocrystalline Na2Ni2SO5 phase present next to NiO filaments, which penetrated into and along the Na2SO4. The Na2Ni2SO5 was assessed to be metastable, with eventual decomposition resulting in porous NiO plus particles of Na2SO4. The nanocrystalline structure of the Na2Ni2SO5 allowed for the requisite rapid nickel transport for the accelerated attack. Note the similarity between Figs. 12 and 21b where, in the latter, liquid Na2MoO4 is the phase, which wets the Na2SO4 grains and provides the rapid transport path for Ni. The following three criteria are deemed necessary for the proposed solid-state mechanism to operate: a significantly high nickel flux; SO3 in the atmosphere to stabilize Na2Ni2SO5 formation; and sufficiently slow (Na2SO4) deposit sintering to allow for adequate wetting by NiO to occur.
Summary of Temperature Effects
Hot corrosion can occur at temperatures near 700 °C by a variety of mechanisms if a phase which allows rapid transport is formed. This includes compounds such as Na2MoO4 (even in atmospheres without SO3), MSO4–Na2SO4 solutions or metastable nanostructured phases. The term gas-phase-induced acidic fluxing is somewhat of a misnomer in that, in some cases, the gas is unimportant and, in others, serves only to produce the rapid transport phase.
Oxide Deposits
As pointed out in the section on Physical State of Deposits, nominally solid deposits can cause significant corrosion, e.g., CaO and MgO deposits increased the degradation of Cr2O3-forming alloys by accelerated CrO3 evaporation at temperatures as low as 850 °C [27]. Also reaction between CaO and Cr2O3 scales at temperatures above 1000 °C resulted in catastrophic corrosion by liquid formation.
Jung [12] and Jung et al. [42] studied the effects of Na2SO4, CaO and CaSO4 deposits on alumina-forming alloys, particularly Pt-modified aluminide coatings at 950 and 1150 °C in dry air under thermal cycling conditions (1 h cycles). Figure 22 shows cross sections of platinum aluminide coatings on Rene′ N5 (nominal composition in Table 5) exposed at 950 °C for 200 h in dry air with various deposits. The degradation of the specimen with a CaO deposit was much more severe than any of the other specimens (note different magnifications) and the otherwise protective alumina scale had reacted with the CaO to form Ca-aluminates. The CaSO4 resulted in less attack because of the lower CaO activity in the sulfate. Figure 23 shows cross sections of René N5 without the aluminide coating after 140 h of cyclic oxidation at 950 °C in dry air with no deposit and a CaO deposit. The no-deposit specimen is covered by a thin alumina scale, while the specimen with the CaO deposit is severely degraded with a thick NiO outer scale over an inner zone of Ca–Al mixed oxides. Figure 24 shows cross sections of the aluminide coated Rene′ N5 exposed at 1150 °C for 40 h in dry air with various deposits. The corrosion is significant on all specimens because of the high temperature, but the degradation caused by the CaO is more extensive than that caused by the other deposits.

Scanning electron micrographs of cross sections of platinum aluminide coatings on Rene′ N5 exposed at 950 °C for 200 h in dry air with various deposits

Cross sections of René N5 after 140 h of cyclic oxidation at 950 °C in dry air with no deposit (top) and a CaO deposit (bottom)

Cross sections of platinum aluminide coatings on Rene′ N5 exposed at 1150 °C for 40 h in dry air with various deposits
The attack of the Pt-aluminide coating by CaO and CaSO4 was the result of interaction between the alumina scales and CaO. Several experiments were conducted to investigate the interactions between bulk alumina and CaO. Specimens of polycrystalline alumina and sapphire were exposed for 60 h at 950 °C while embedded in CaO powder. EDS indicated some Ca on the surface of the exposed specimen and CaAl4O7 was identified from the XRD spectra. Figure 25 shows the pseudo-binary phase diagram for the CaO-Al2O3 system, which indicates that CaO and Al2O3 should react to form a series of ternary Ca–Al oxides. Ca aluminates, other than CaAl4O7, did not form probably because of the kinetics under the exposure conditions. Nevertheless, this indicates there is reactivity between CaO and alumina. Thus, it was proposed that a major effect of the CaO in damaging a growing alumina scale is that depicted in the schematic diagram of Fig. 26 as follows:
-
Step #1: CaO deposits accumulate on an alumina layer.
-
Step #2: CaO reacts with a normally protective Al2O3 layer to form Ca aluminates, which are less protective against oxidation.
-
Step #3: Cracks are initiated and propagate along the interfaces between Ca aluminates and an Al2O3 layer during thermal cycling, which causes spallation of the Ca aluminate layer.
-
Step #4: An Al2O3 protective layer is recovered when the coating is exposed to high oxygen partial pressure after the spallation. At the same time, CaO is continuously deposited on the Al2O3 layer.
Step #1–#4 repeat until the coating material is completely degraded.

Phase diagram for the Al2O3–CaO system [43]

Schematic diagram showing the proposed effect of CaO on a growing alumina scale
Subsequently, Gheno et al. [43] studied two-phase β–γ MCrAlY alloys exposed to CaO at 1100 °C. The alloys were found to suffer significantly more degradation than in the absence of a deposit. The reaction with CaO proceeded according to two distinct mechanisms. During the initial stage, formation of a liquid calcium chromate led to the rapid consumption of the Cr-rich γ-phase. The extent of degradation was particularly important for a single-phase γ composition and was significantly reduced by increasing the alloy β fraction. In a subsequent stage, a continuous Al2O3 layer established at the base of the scale, which led to a much lower oxidation rate. However, the protective oxide reacted with CaO to form calcium aluminates, and as the Al2O3-scale thickness was reduced, the instantaneous oxidation rate was higher than in the absence of a deposit.
A temperature decrease to 900 °C was found to be detrimental to the kinetic stability of the protective oxide, mainly because of the longer persistence of the metastable and faster growing θ-Al2O3. Failure to maintain an external Al2O3 scale resulted in internal oxidation of Al for an Al-lean alloy, and formation of a Cr-rich oxide scale. This did not have as severe consequences as at 1100 °C since 900 °C is below the calcium chromate eutectic.
A recent study [44] has shown that bi-thermal exposures to CaO/CaSO4 deposits produce degradation which is similar to that observed in some field-exposed components fabricated from the superalloy N5, which involved extensive internal oxidation. CaSO4-induced degradation occurs during high temperature (≈ 1150 °C) exposure and causes the subsurface depletion of Al and Cr and enrichment in S through reaction (11)
The subsurface-depleted alloys were susceptible to internal oxidation when exposed at lower temperatures (≈ 900 °C).
Summary of Oxide Deposits
Deposits, which are primarily oxides, produce degradation in several ways: accelerating evaporation from chromia scales and producing non-protective phases such as liquids or friable aluminates.
Fireside Corrosion Mechanisms
The deposits formed in coal-fired boilers are complex. Alkali iron trisulfates, which are stabilized by SO3 in the gas atmosphere, have been cited to be the major corrosive species for many years [1]. Recently, the propagation mechanism for fireside corrosion was investigated using T92, a typical ferritic boiler steel (Fe–9Cr–2W–0.3Ni–0.3Mo wt%) and a model austenitic Fe–Ni–Cr alloy (Fe–18Cr–12Ni wt%) in contact with synthetic coal ash deposits at 700 °C [45]. (Results would be expected to be the same for Grade P92 as those for T92.) The deposits were:
-
1.
Na2SO4:K2SO4 in a 1:1 molar ratio (designated as M1). This deposit will remain solid until it reacts with sufficient thermally grown iron oxide and SO3 in the gas atmosphere to form a liquid (Na,K)2SO4-Fe2(SO4)3 solution, see Fig. 4.
-
2.
Na2SO4:K2SO4:Fe2O3 in a 1.5:1.5:1.0 molar ratio. This was designated the Standard Corrosion Mix (SCM). The ratios for this mix are the stoichiometric coefficients of alkali iron trisulfates. This deposit is designed to form alkali iron trisulfates even without the presence of thermally grown iron oxides on the specimen surface.
The specimens were placed into alumina crucibles so that half of the specimen was covered by powder and half was not so the effect of deposit thickness on the corrosion of the alloy could be determined. This provides three deposit zones for each specimen shown schematically in Fig. 27: a thick deposit zone, a thin deposit zone and a no-deposit zone.

Schematic diagram of a partially embedded fireside corrosion specimen
Figure 28 shows a macroscopic photograph and cross sections at two locations for T92, which was exposed in SCM for 160 h in O2 +1000 ppm SO2. The pits consist of thick iron oxide scales and internal corrosion products rich in Fe, Cr, S, and O with layers rich in alkali sulfates. The sulfur content increases with depth into the pits until a thick layer of iron and chromium sulfide (identified with EDS) forms at the base.

Macrophotograph and cross section of a T92 specimen partially embedded in an SCM deposit and exposed for 160 h in O2 +1000 ppm SO2
Figure 29 presents typical results for the FeNiCr alloy, which was exposed in M1, as surface micrographs and cross sections. The attack begins in the thin deposit zone and, as time increases (80–160 h), the corrosion spreads into the no-deposit zone, and the scale consists of thicker iron oxide scales as the liquid wet the upper part of the specimen. The thick and thin deposit zones were severely degraded after each test duration. The corrosion started out covering large areas in the deposit zone, and these areas and the amount of corrosion increased with time until the entire deposit zone was completely degraded. The corrosion products consisted of external thick porous iron oxide scales, which grew around remaining alkali sulfates from the deposit. Below the thick iron oxide scales are internal corrosion pits rich in Fe, Cr, S, and O with layers rich in alkali sulfates. The sulfur content increases with depth into the pits until a thick layer of iron and chromium sulfide forms at the base. The results were similar for FeNiCr and T92 for the thick and thin deposit zones, even though the chromium content is double that for T92 alloy.

Cross-sectional micrographs of the thin deposit zone of FeNiCr tested at 700 °C with the M1 deposit in a crucible in O2 + 1000 ppm SO2 for 20, 40, 80, and 160 h
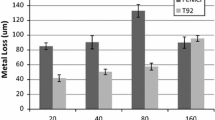
Figure 30 compares the metal loss for the FeNiCr alloy in the M1 and SCM deposits. The metal losses are significantly higher for the M1 deposit than the SCM. The same trend held for T92.

Comparison of thick deposit zone metal loss for FeNiCr exposed in the M1 and SCM deposits at 700 °C in O2 + 1000 ppm SO2 for 160 h
Based on the results, the following fireside corrosion initiation and propagation mechanism was proposed. During the initiation stage, transient iron oxides and chromium oxide grow on the surface of the alloy. The alloy is covered by the deposit, which contains alkali sulfates K2SO4 and Na2SO4. The SO3 from the catalyzed O2 + SO2 mixture migrates to the oxide/deposit interface, and when the sulfur potential becomes sufficient, the transient Fe2O3 reacts to form Fe2(SO4)3. When sufficient Fe2(SO4)3 is dissolved in the alkali sulfates the deposit melts, as seen on the phase diagram in Fig. 4. The formation of the liquid salt allows dissolution and fluxing of the protective oxide scales and base metal to take place, which is responsible for the lack of protective Cr2O3 and the observed metal loss. It is at this point that the propagation stage occurs.
Based on these results, the cause of the increased corrosion of the M1 deposit and the likely controlling fireside corrosion propagation mechanism was proposed to be synergistic dissolution of oxides in the molten salt deposit similar to that proposed by Rapp and Hwang for hot corrosion [32] and described above for Type II corrosion of CoCrAl alloys [38]. Once a liquid eutectic melt has formed, it reacts with the protective oxide scales and base metal becoming highly basic or acidic and causing dissolution and fluxing, creating the observed corrosion products. If the oxide dissolution reactions are considered individually, Fe2O3 would be expected to undergo acidic dissolution by the reaction given in Eq. 12. This reaction would consume SO3 in the form of pyrosulfate:
The Cr2O3 will undergo basic dissolution according to reaction (13).
When the two oxides are both in contact with the salt deposit and the melt basicity is in a position between the solubility minima for Fe2O3 and Cr2O3, synergistic fluxing and the simultaneous occurrence of the two dissolution reactions for Cr2O3 and Fe2O3 support each other by creating negative solubility gradients for both species and accelerating corrosion by short circuiting the rate-limiting diffusion process. The basic dissolution of Cr2O3 disrupts protective scale formation and makes the melt more acidic so that the melt is then under favorable conditions for further acidic dissolution of the Fe2O3. This can repeat indefinitely, so that an acidic/basic cycle is created in the melt to produce continued accelerated corrosion. The repeating of the basic and acidic dissolution reactions is shown in the schematic diagrams in Fig. 31a, b and solubility curves in Fig. 31c. The arrows in Fig. 31c show the negative solubility gradients and display the local shift in acidity and basicity of the melt. There is a negative solubility gradient set up where the iron oxide has a higher solubility at the oxide/salt interface than at the gas/salt interface. The iron oxide precipitates where there is a locally low solubility and a higher pO2. The chromium solute also experiences a negative solubility gradient. However, the results imply that the solubility of Cr2O3 in the melt must be less than that for Fe2O3, because the chromium precipitates nearer to the alloy surface, creating the observed internal corrosion pits.

Negative solubility gradients on the Fe2O3 and Cr2O3 solubility curves due to synergistic fluxing and schematic of precipitation process
The addition of Fe2O3 to the base M1 deposit (SCM), significantly decreases the amount of metal loss of these alloys. The SCM is less corrosive than the M1 deposit because the iron oxide in the deposit changes the local basicity of the melt so that it is no longer in a region where the maximum corrosion rate can occur. Synergistic dissolution has been slowed, but not stopped completely.
Supporting Experiments
To further assess if synergistic dissolution was occurring, the FeNiCr and T92 alloys were exposed to a deposit containing M1 + 30%Cr2O3 (M1–Cr) at 700 °C in O2 + 1000 ppm SO2 for 160 h. Figure 32 shows mass change and thick deposit metal loss values for T92 compared with the results under the same conditions when using the M1 and SCM deposits. The M1-Cr deposit is even less corrosive than the SCM since the Cr2O3 increased the local acidity of the melt so much that it essentially stopped synergistic dissolution.

Mass change and thick deposit zone metal loss for T92 exposed at 700 °C with the M1, SCM, and M1 + 30%Cr2O3 powder deposits in a crucible in O2 + 1000 ppm SO2 for 160 h
Additional experiments were performed with other additions to M1: (Al2O3: M1-A, CaO: M1-C, and SiO2: M1-S). Thick deposit metal loss plots are shown in Fig. 33 compared with the results for the M1 deposit. (The M1-S bar for the FeNiCr alloy cannot be seen because the metal loss was minimal). Alumina in the deposit reduced the amount of corrosion. Alumina is a neutral oxide and may undergo acidic or basic fluxing when reacting with alkali sulfates and can shift melt basicity away from the maximum synergistic zone.

Thick deposit zone metal loss of FeNiCr and T92 exposed in various deposits containing M1 with oxide additions at 700 °C in O2 + 1000 ppm SO2 for 160 h
Calcia is a highly basic oxide and its addition to the alkali sulfates makes the melt highly basic and therefore much less corrosive. Synergistic fluxing is significantly slowed down due to the localized increase in basicity of the melt. Calcia is more basic than alumina, so the effects are even greater.
Silica is a highly acidic oxide, which undergoes basic dissolution when reacting with alkali sulfates according to Eq. (14).
The formed Na2Si2O5 has a melting point of 874 °C, and the basicity shift prevents the basic fluxing portion of the sequence.
The modified corrosion mechanism provides the following practical advantages:
-
1.
Allows evaluation of different protection schemes.
-
2.
Provides basis for better semi-empirical correlations.
With regard to the second point, Wright and Shingledecker [46] established a “Relative Corrosion Potential” (RCP): assuming alkali-Fe-trisulfates were the corrosive species and performing a mass balance to determine the relative amounts of these species which could form based on the composition of the coal. An low-sulfur coal, which would form ash similar to that in Table 1, was taken as a reference and given an RCP of “10.” It was assumed that Cl affects the alkali release and that some of the CaO and MgO would react with Na2O and K2O to form high melting point compounds. Thus, RCP = f(Na, K, Ca, Mg, Cl). Wright and Shingledecker achieved a reasonable correlation between the corrosion rate of several austenitic alloys and the RCP of the coal being used. It is likely that, recognition that mixed sulfates are the underlying corrodent, incorporation of “synergistic fluxing” and considering the effect of SiO2 will provide better correlation with coal compositions. It is likely there are competing effects between the acidic and basic oxides in real coal ash.
Fireside Corrosion Summary
The corrosive species responsible for degradation was a (Na,K)2SO4–Fe2(SO4)3 solution and not alkali iron trisulfates. The formation of the liquid deposit is similar to Type II hot corrosion. The propagation mechanism involves a synergistic dissolution process, whereby simultaneous basic and acidic dissolution of protective Cr2O3 and Fe2O3 disrupts protective oxide formation and locally produces negative solubility gradients at the oxide/salt interface. The dissolved Fe2O3 and Cr2O3 reprecipitate where there is lower solubility, creating the observed corrosion products. Thus, fireside corrosion in coal ash involves the fundamental process of degradation by liquid alkali sulfate solutions, which is modified by the presence of Fe2O3 and other oxides, which are present in the ash.
Summary
Alloys and coatings used in high temperature applications are often subject to degradation influenced by the presence of surface deposits on the alloy surfaces, e.g., fireside corrosion, hot corrosion etc. Depending on the source, deposit compositions may occur in a wide range from primarily sulfate to primarily oxide:
However, rarely will they be pure oxide or pure sulfate.
There does not seem to be evidence of severe corrosion caused by gaseous Na2SO4.
Generally, severe corrosion occurs when the deposits are liquid. However, significant corrosion has been observed in some cases with deposits, which are nominally solid. Deposits, which are primarily oxides, produce degradation in several ways: accelerating evaporation from chromia scales and producing non-protective phases such as liquids or friable compounds in place of the normally protective scale.
In the case of liquid sulfate deposits, Rapp and Goto [30] have proposed a criterion by which the continued self-sustaining corrosion attack can occur, the Rapp–Goto Criterion:
A negative solubility gradient in the oxide solubility at the oxide/scale interface results in dissolution of the protective oxide scale and reprecipitation of the oxide as discontinuous non-protective particles in regions of the molten salt deposit where the solubility is lower. It is important to note that, in cases of acidic fluxing, if the acidifying species is coming from the vapor phase (e.g., high pSO3 or V coming from the fuel), a positive solubility gradient will be established and self-sustaining fluxing does not occur. In many cases (e.g., basic and synergistic fluxing), rapid interface reactions can occur with little or no influence from the gas phase and produce the negative solubility gradient(s).
Hot corrosion can occur at temperatures near 700 °C by a variety of mechanisms if a phase which allows rapid transport is formed. This includes compounds such as Na2MoO4 (even in atmospheres without SO3), MSO4–Na2SO4 solutions or metastable nanostructured phases. Calling all corrosion in this temperature regime “Type II” can be misleading with regard to mechanism. The term gas-phase-induced acidic fluxing, in many cases, is a misnomer in that the gas serves only to produce the rapid transport phase and the underlying cause of the corrosion is actually synergistic fluxing.
The corrosive species responsible for fireside corrosion of ferrous alloys is a liquid (Na,K)2SO4–Fe2(SO4)3 solution and not alkali iron trisulfate. The propagation mechanism involves a synergistic dissolution process, whereby simultaneous basic and acidic dissolution of protective Cr2O3 and Fe2O3 disrupts protective oxide formation and locally produces negative solubility gradients at the oxide/salt interface. The dissolved Fe2O3 and Cr2O3 precipitate where there is lower solubility. Thus, fireside corrosion in coal ash involves the fundamental process of degradation by liquid alkali sulfate solutions, which is modified by the presence of Fe2O3 and other oxides, which are present in the ash.
References
W. T. Reid, External Corrosion and Deposits Boilers and Gas Turbines, (American Elsevier Publishing Company Inc., New York, 1971).
J. Stringer, Annual Review of Material Science 7, 1977 (477–509).
I. G. Wright and S. C. Kung, Materials at High Temperatures 35, 2018 (316).
F. S. Pettit, Oxidation of Metals 76, 2011 (1).
N. L. Ndamka, R. G. Wellman and J. R. Nicholls, Materials at High Temperatures 33, 2016 (44).
G. P. Huffman and F. E. Huggins, Reactions and Transformations of Coal Mineral Matter at Elevated Temperatures. US Steel Corporation Technical Center, Monroeville, PA. The American Chemical Society, Vol. 188. No. 1155, 1984.
J. G. Tschinkel, Corrosion 28, 1972 (161).
N. S. Bornstein, JOM 48, 1996 (37).
N. S. Bornstein, Materials Science Forum 251–254, 1997 (127).
J. Smeggil and J. G. Smeggil, Hot Corrosion of Turbine Materials in the Presence of Mixed Sulfates, (ONR Final Technical Report, UTRC, East Hartford, CT, 2000).
M. P. Borom, C. A. Johnson and L. A. Peluso, Surface and Coatings Technology 86–87, 1996 (116).
K.-Y. Jung, Ph. D Thesis, University of Pittsburgh, 2009.
W. Braue, The Journal of Materials Science 44, 2009 (1164–1675).
W. Braue, P. Mechnich and P. W. M. Peters, Materials at High Temperatures 28, 2011 (315).
W. Braue and P. Mechnich, Journal of the American Ceramic Society 94, 2011 (4483).
N. Bohna, Ph. D Thesis, Univ. of Pittsburgh, 2015.
V. Tolpygo, Oxidation of Metals 88, 2017 (87).
B. S. Lutz, R. W. Jackson, N. M. Abdul-Jabbar, V. Tolpygo and C. G. Levi, Oxidation of Metals 88, 2017 (73).
D. A. Shifler, Materials at High Temperatures 35, 2018 (225).
P. Hancock, Materials Science and Technology 3, 1987 (536).
S. Pahlavanyali, H. T. Pang, F. Li, S. Bagnali and C. Rae, Materials Science and Technology 30, 2014 (1890).
M. A. DeCrescente and N. S. Bornstein, Corrosion 24, 1968 (127).
P. Knutsson, H. Lai and K. Stiller, Corrosion Science 73, 2013 (230).
M. G. Lawson, F. S. Pettit and J. R. Blachere, Journal of Materials Research 8, 1993 (1964).
A. Rahmel, W. Jaeger and Z. Anorg, Chem. 303, 1960 (90).
K. L. Luthra, Metallurgical Transactions A 13A, 1982 (1647).
K. T. Chiang, G. H. Meier and R. A. Perkins, Journal of Materials for Energy Systems 6, 1984 (71).
N. Birks, G. H. Meier and F. S. Pettit, Introduction to the High Temperature Oxidation of Metals, (Cambridge University Press, Cambridge, 2006).
R. A. Rapp, Corrosion 42, 1986 (568).
R. A. Rapp and K. S. Goto, in Molten Salts, eds. J. Braunstein and J. R. Selman (Electrochemical Society, Pennington, 1981), p. 81.
N. Otsuka and R. A. Rapp, Journal of Electrochemical Society 137, 1990 (46).
Y. S. Hwang and R. A. Rapp, Journal of Electrochemical Society 137, (4), 1990 (1276).
L. Longa-Nava, Y. S. Zhang, M. Takemoto and R. A. Rapp, Corrosion Science 52, 1996 (680).
B. S. Lutz, J. M. Alvarado-Orozco, L. Garcia-Fresnillo and G. H. Meier, Oxidation of Metals 88, 2017 (599).
K. L. Luthra, in High Temperature Corrosion, ed. R. A. Rapp (NACE Houston 1983), p. 507.
K. L. Luthra, Metallurgical and Materials Transactions A 13A, 1982 (1843).
K. L. Luthra, Metallurgical and Materials Transactions A 13A, 1982 (1853).
J. M. Alvarado-Orozco, J. E. Garcia-Herrera, B. Gleeson, F. S. Pettit and G. H. Meier, Oxidation of Metals 90, 2018 (527).
J. E. Garcia-Herrera, L. Garcia-Fresnillo and G. H. Meier, Univ. of Pittsburgh (unpublished research).
W.-J. Zhang and R. Sharghi-Moshtaghin, Metallurgical and Materials Transactions A 49A, 2018 (4362).
E. Kistler, W. T. Chen, G. H. Meier and B. Gleeson, Materials and Corrosion 70, 2019 (1346).
K. Y. Jung, F. S. Pettit and G. H. Meier, Materials Science Forum 595–598, 2008 (805).
T. Gheno, G. H. Meier and B. Gleeson, Oxidation of Metals 84, 2015 (185).
P. T. Brennan, Ph. D Thesis, Univ. of Pittsburgh, 2015.
B. S. Lutz, G. R. Holcomb and G. H. Meier, Oxidation of Metals 84, 2015 (353).
I. G. Wright and J. P. Shingledecker, Materials at High Temprature 32, 2015 (426).
Acknowledgements
The author gratefully acknowledges the contributions of many students and postdocs and collaboration over the years with his University colleague, Fred Pettit, who provided a wealth of knowledge of hot corrosion, and his late colleague, Roger Perkins, who provided his insight and cleverness in experimental design. Prof. Brian Gleeson is acknowledged for many helpful comments on the manuscript. Much of the work described was supported by the Office of Naval Research, most recently, under ONR Contract N00014-10-1-0661, David A. Shifler, Scientific Monitor.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares he has no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Meier, G.H. Invited Review Paper in Commemoration of Over 50 Years of Oxidation of Metals: Current Aspects of Deposit-Induced Corrosion. Oxid Met 98, 1–41 (2022). https://doi.org/10.1007/s11085-020-10015-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-020-10015-6