
Abstract
Neuropathic pain, a prevalent chronic condition in clinical settings, has attracted widespread societal attention. This condition is characterized by a persistent pain state accompanied by affective and cognitive disruptions, significantly impacting patients’ quality of life. However, current clinical therapies fall short of addressing its complexity. Thus, exploring the underlying molecular mechanism of neuropathic pain and identifying potential targets for intervention is highly warranted. The transient receptor potential (TRP) receptors, a class of widely distributed channel proteins, in the nervous system, play a crucial role in sensory signaling, cellular calcium regulation, and developmental influences. TRP ion channels are also responsible for various sensory responses including heat, cold, pain, and stress. This review highlights recent advances in understanding TRPs in various rodent models of neuropathic pain, aiming to uncover potential therapeutic targets for clinical management.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Overview of Neuropathic Pain
Pain, deemed the fifth vital sign, serves as a protective mechanism in response to tissue injury, triggering rapid danger signals and promoting healing [1]. However, when pain transitions from acute to chronic, the persistent distressing sensation significantly impacts the quality of life. Neuropathic pain, a prevalent chronic condition, arises from lesions or diseases affecting the peripheral and/or central somatosensory nervous system [2]. Recently, it was redefined by the International Association for the Study of Pain as “Chronic neuropathic pain is chronic pain caused by a lesion or disease of the somatosensory nervous system”. Moreover, it can be categorized into peripheral and central neuropathic pain [3], stemming from many pathogenic factors, such as cancer, diabetes, viral infections, chemotherapy, and traumatic nerve injury [4]. Clinical presentations range from nerve compression (e.g., due to a tumor [5] or disc rupture in the spine, resulting in low back and/or leg pain [6], or nerve compression in the wrist, resulting in carpal tunnel syndrome) [7], nerve damage caused by diseases that affect the nerves (e.g., diabetes, herpes zoster, or chemotherapeutics) [8,9,10], to central neuropathic pain associated with conditions like post-stroke pain [11], multiple sclerosis-related pain [12], Parkinson’s disease-related pain [13] or spinal cord disease-related pain [14]. Characteristic symptoms of neuropathic pain include persistent spontaneous pain, hyperalgesia, and allodynia [15, 16]. Patients with chronic pain often experience psychological distress such as insomnia [17], depression [18], mental disorders, and even suicidal tendencies [19]. Millions of individuals worldwide suffer from neuropathic pain, significantly impacting their quality of life [20]. Current clinical treatments include opioids, calcium ion modifiers (pregabalin or gabapentin), and antidepressants [21]. However, these pharmacological interventions often lack stability in pain control and can lead to drug resistance, addiction, and side effects. Interventional therapies such as nerve pulse modulation exist [22], but struggle with recurrence control. Given the limitations in available therapeutic options, the pursuit of new targets remains essential.
The mechanisms of neuropathic pain are complex, involving alterations in neuronal cell membrane ion channel activities (Na+ [23, 24], K+ [25], and Ca2+ [26]), immune cell proliferation and activation (microglia [1, 27], astrocytes [28], and macrophages [29]), and dysregulation of the immune microenvironment [30] within the nervous systems. Among these mechanisms, TRP receptors, widely present in sensory neurons, play a pivotal role. They respond to various stimuli, such as temperature, pressure, and chemicals [31], converting them into electrophysiological signals that elevate intracellular calcium ion levels, thereby initiating action potentials and neurotransmitter release [32, 33]. In the initial stages, before apparent nerve damage, abnormalities in electrical activity, synaptic alterations, and chronic inflammation arise [34, 35]. Additionally, TRP receptors expressed in neuro-immune cells such as microglia, macrophages, and astrocytes influence intracellular ion levels and modulate the neuroinflammatory response via the altered proliferative state [36,37,38]. Moreover, tissue damage exacerbated this response, leading to abnormal production of pro-inflammatory mediators [39] and intensifying neuro-immune interactions [29, 40, 41]. Therefore, TRP receptors emerge as promising potential targets for neuropathic pain therapy.
Biofunction of TRP Ion Channels in Pain
TRP proteins, as transmembrane entities, are widely distributed in the nervous system. Comprised of four subunits, forming a tetramer, each subunit boasts six transmembrane domains. The fifth and sixth domains create a non-selective cation channel pore [42], defining the TRP ion channels’ structure. In mammals, TRP channels constitute six subfamilies: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPA (ankyrin), TRPP (polycystin), and TRPML (mucolipin) [43]. They possess a bio-ability to detect and react to a range of stimuli, such as temperature, pressure, pH, and chemical substances, thereby regulating intracellular calcium concentration and membrane potential [44, 45]. These ion channels significantly influence diverse physiological and pathological processes, spanning sensory signaling, cell proliferation, cell death, inflammation, pain, respiratory system diseases, and neurodegenerative conditions [31]. Within the TRP subfamilies, which vary in activation mechanisms and functional properties, TRPC, TRPV, TRPA, and TRPM stand out as potential targets for pain treatment.
The TRPC subfamily includes seven members (TRPC1-7), which are activated by diacylglycerol (DAG) produced by phospholipase C or G protein-coupled receptors (GPCRs) [46]. Their roles involve mediating calcium entry and amplifying intracellular calcium signals [47]. In cases of nerve injury or dysfunction, changes in TRPC channel expression and function in the nervous systems contribute to persistent or recurrent neuropathic pain, impacting neuronal excitability, plasticity, and neuroinflammation [48]. Notably, in peripheral nerve injury models, TRPC1, TRPC4, TRPC5, and TRPC6 overexpression in the injured sciatic nerve was associated with tactile hypersensitivity [49, 50].
The TRPV subfamily in mammals, comprising six members (TRPV1-6), exhibits responsiveness to diverse physical and chemical stimuli, particularly TRPV1-4’s thermal activation [51]. Therefore, they play a vital role in sensing temperature, pain, and inflammation. Highly expressed in the spinal dorsal horn and sciatic nerve, TRPVs correlate with spontaneous pain and tactile hypersensitivity [52]. For instance, TRPV1 responds to capsaicin and high temperatures, influencing thermal sensation and pain signal transmission [53]. TRPV4, on the other hand, conducts cutaneous tactile pressure sensation via class A and C fibers and facilitates mechanical stimuli perception [54, 55].
The TRPM subfamily, comprising eight members (TRPM1-8), responds readily to various physical and chemical stimuli, such as temperature and menthol. These channels are primarily related to sensation, neuroprotection, cell proliferation, and cell death [56, 57]. TRPM8, recognized as the primary cold receptor in mammals, is activated at temperatures below 28 °C [58], contributing to cold sensation and neuroprotection [59]. Similarly, TRPM2 and TRPM3 exhibit sensitivity to mechanical and thermal stimuli, holding potential as therapeutic targets [60, 61].
The TRPA family, with seven members where TRPA1 is exclusive to humans, is activated by endogenous inflammatory mediators and exogenous harmful stimuli, contributing to the development of acute and chronic pain and inflammation [62, 63]. TRPA1 can also be activated by mustard oil and mechanical force [64]. It acts as a sensor for temperature, chemical, and mechanical stimuli, mediating biological effects like pain stimulation and playing a critical role in nociceptive responses [65, 66].
TRP channels are intricate and multifaceted proteins crucial for perceiving and regulating the intracellular and extracellular environment. In sensory transduction, TRP is commonly expressed in C- and Aδ sensory nerve fibers that sense mechanical tension and temperature variation (Table 1). Understanding the molecular mechanisms underlying pain signal transduction through TRP channels offers new avenues for developing novel analgesics [79, 80], emphasizing the need for more in-depth research into these channels to explore neuropathic pain mechanisms and potential treatments.
Diverse Models of Neuropathic Pain
To identify therapeutic targets and develop novel treatments, several animal models of neuropathic pain have been established that mimic neuronal tissue damage or nervous system dysfunction. This review specifically focuses on common models of peripheral neuropathic pain rather than central neuropathic pain. Peripheral neuropathic pain holds clinical significance due to its high prevalence across various chronic diseases, and extensive research has been conducted in this domain. Commonly studied peripheral neuropathic pain models include chronic constriction injury of the sciatic nerve (CCI), spared nerve injury (SNI), spinal nerve ligation (SNL), diabetic neuropathic pain (DNP), and chemotherapy-induced peripheral neuropathy (CIPN) [81] (Fig. 1). Rodent models serve as valuable tools for controlling experimental variables and mimicking molecular and cellular mechanisms of human diseases. However, they present certain challenges: (1) Rodent models are not completely consistent with the phenotypic and clinical features of human diseases, such as pain sensitivity and drug response [82]. (2) The method of pain assessment in rodent models relies on objective indicators such as behavior, physiology, and neuro-electrophysiology, neglecting subjective factors such as emotion, cognition, and societal impact associated with human diseases [83]. Therefore, such differences need to be considered.

Diverse models of peripheral neuropathic pain. A The CCI model is established by ligating the main trunk of the sciatic nerve. B Ligation of the distal dorsal root ganglion used to build the SNL model. C Peripheral nerve sensitization (DNP/CIPN) is caused by injections of STZ or chemotherapeutic drugs. D The SNI model ligates the tibial nerve and the common peroneal nerve, leaving the sural nerve intact
The CCI model, widely employed in studying peripheral nerve neuropathic pain, involves surgically detaching the biceps femoris muscle under anesthesia to expose and isolate the sciatic nerve trunk. Three or four loose chromic gut (or 4–0 silk ligatures, spaced 1 mm apart) are placed around the sciatic nerve until a slight twitch is observed in the lower limbs [84]. Behavioral changes, including mechanical and thermal hyperalgesia, chemical hyper-reactivity, and cold allodynia, worsen within a week and peak in the second-week post-surgery, with the most severe pain-related behaviors persisting for more than two months [84, 85]. In the SNL model, ligation of the L5 and L6 spinal nerves triggers mechanical and thermal hyperalgesia and spontaneous pain within 24–48 h, persisting approximately four months [86, 87]. The SNI model involves ligating the common peroneal and tibial nerves using a 5–0 silk suture. Their distal ends were transected, but the sural nerve was preserved [88]. This model induces mechanical and thermal hyperalgesia and allodynia within four days after the injury, persisting for several weeks (up to a half year) [89]. The advantage of the SNI model is the robustness of the response without significant lameness in the lower limb. Nociception and hyperresponsiveness of C fibers in a DNP model are induced by the subcutaneous injection of streptozotocin (STZ), persisting for approximately 2–3 weeks [90, 91]. Similarly, in the CIPN model, animals were induced to develop thermal and mechanical hypersensitivity through chemotherapeutic drugs such as paclitaxel [92], cisplatin [93], and oxaliplatin [94]. Numerous studies demonstrate the critical roles of TRPs in different neuropathic pain models. (Table 2) Here, we briefly review the recent advancements regarding TRP receptors in these described neuropathic pain models.
TRPs in Different Neuropathic Pain Models
CCI Model
In studies exploring nociception within the CCI model, multiple TRP receptors have demonstrated significant involvement. TRPM8, crucial for cold sensation post-injury, exhibits increased expression in the dorsal root ganglion, contributing to the generation of cold sensory hypersensitivity [95]. Both TRPM8 antagonist and TRPM8 knockout models significantly alleviate mechanical and thermal sensitization [77, 95]. TRPM2 and TRPM3 have also shown sensitization to these stimuli [75, 76]. Similarly, Wang et al. investigated the mechanism of TRPA1-induced stimulation of the progression of neuropathic pain via Mas-related GPCR D (MrgprD) regulation, revealing that TRPA1 can activate MrgprD via the PKA pathway to amplify sensitivity [96]. Notably, the pain hypersensitivity induced by alanine injection into the plantar surface of TRPA1 knockout mice was significantly attenuated. Traditional Chinese medicine studies involving baicalin exhibited analgesic effects via a dose-dependent reduction in mechanical and thermal injury reactions [70]. The potential mechanism may be related to the inhibition of TRPV1 overexpression in the dorsal root ganglion and the phosphorylation of ERK, thus ameliorating peripheral neuroinflammation [70]. TRP channels have been observed to influence the oxidative stress response in the CCI model. Elevated ROS expression and TRPV1/TRPA1 overexpression in dorsal root ganglion neurons suggest oxidative stress injury in local tissues triggered by the CCI model [71]. Notably, the inhibition of TRPV1/TRPA1 expression significantly reduced thermal and mechanical sensitization. Additionally, TRPM2 was associated with iNOS expression, NO, and ROS generation [75]. A previous study [69] showed increased TRPC6 expression in the dorsal root ganglion in the CCI model. Continuous intraperitoneal dexmedetomidine injection significantly reduced TRPC6 and inflammatory cytokine expression, alleviating mechanical nociception and thermal sensitization.
SNI Model
The SNI model closely mimics clinical neuropathic pain features. Staaf et al. [97]. explored the differential regulation of TRP channels in an SNI rat model. Among the members of the TRP channel, TRPML3 showed significant changes between the surgical and control groups [97]. In situ hybridization revealed increased TRPML3 expression across neurons of different sizes. Additionally, mRNA levels of TRPM6, TRPM8, TRPV1, TRPA1, TRPC3, TRPC4, and TRPC5 were decreased in the SNI model [97]. Thus, these observations suggest that TRPML3 is associated with the development of neuropathic pain. Similarly, Poulson et al. [78]. Discovered that normal mice and nude mice have comparable sensitivity to mechanical stimuli, but showed variation in their reaction to cold. Compared to normal mice, naked mole rats showed increased TRPA1 mRNA expression and decreased TRPM8. Moreover, TRPM2 knockout mice, compared to wild-type mice, demonstrated no basal sensitivity differences to mechanical and thermal stimulation but played a key role in injurious stimuli [60]. Elevated TRPC6 expression in the dorsal horn of SNI model rats correlated with microglial proliferation and increased inflammatory factors [68]. It is worth noting that TRPC6 co-localized with microglia. The administration of larixyl acetate, a TRPC6 inhibitor, significantly inhibited microglial activation and p38 phosphorylation in the spinal cord dorsal horn, alleviating inflammatory response and pain behavior [68]. Furthermore, TRPV1 receptors in SNI rats have been studied for their role in regulating neural activity and neuropathic pain following nerve injury. Yamamoto et al. demonstrated the role of TRPV1 receptor-expressing fibers in spinal ventral root discharges and mechanical pain in a rat model of SNI [98]. Discharges were observed in the ventral roots of the spinal cord on the first day after surgery, which persisted for two weeks. These discharges were positively correlated with the development of mechanical pain. In rats with selective ablation of TRPV1-positive fibers with resiniferatoxin, mechanical hyperalgesia was abolished together with the discharges, while allodynia was unaffected, suggesting different nerve fibers contribute to the two processes [98]. Overall, TRPV1 is speculated to play a crucial role in regulating neural activity and neuropathic pain post-nerve injury.
SNL Model
In the SNL model, TRPM8 and TRPA1 receptors take center stage. Ji et al. [73]. Highlighted the role of TRPA1 in primary sensory neurons, correlating increased sensitivity to cold stimuli post-surgery with elevated TRPA1 expression in the dorsal root ganglia. This receptor, transported from the dorsal root ganglion to peripheral axons, localizes in unmyelinated and finely myelinated cutaneous axons, impacting physiological functions. TRPA1 antagonists significantly reduced the electrical activity of Aδ fibers and alleviated nociceptive hypersensitivity. In another study, ICLIN, an agonist of TRPM8 and TRPA1, increased calcium ion concentration in the dorsal root ganglia [99]. Suppression of those expression lessened abnormal neuronal firing and reduced sensitivity to mechanical and cold stimuli. Moreover, neuroinflammation and nerve injury-induced cold sensory hypersensitivity were suppressed by the intrathecal administration of an anti-nerve growth factor, p38MAPK inhibitor, and TRPA1 antagonist [74], indicating that NGF induces TRPA1 overexpression in sensory neurons through p38 activating, serving as an essential factor in cold nociceptive hypersensitivity. However, Hagenacker et al. [100]. Found no significant difference in the effect of ICLIN on voltage-gated calcium channels in uninjured and SNL-injured DRG neurons using the whole-cell membrane clamp technique. Neither local nor intrathecal application of ICLIN had an effect on tactile hypersensitivity or thermal hyperalgesia after SNL in vivo. Therefore, they concluded that ICLIN, an agonist of TRPM8 and TRPA1, does not regulate voltage-gated calcium channels in DRG neurons or mediate the pathogenesis of cold hypersensitivity. Collectively, these studies imply that TRPM8 and TRPA1 are strongly associated with mechanical pain and cold sensitization; however, further studies are warranted in the SNL model.
DNP Model
The complex pathogenesis of DNP involves ion channel activation leading to neuronal hyperexcitability under hyperglycemic conditions [101]. In the DNP model, the expression level of TRPM8 increased in DRG neurons, while the expression level of TRPA1 showed no significant changes [102]. Nefopam pretreatment effectively reduced TRPM8 expression and inhibited STZ-induced mechanical and cold sensory hypersensitivity. However, painful neuropathy associated with diabetes has been linked to TRPA1 in animal studies [103, 104]. TRPA1 knockout diabetic neuropathic rats exhibited hyperglycemia and mechanical allodynia, suggesting TRPA1 gene deletion’s crucial role in diabetic neuropathy development [105]. Additionally, TRPM2 and TRPV1-mediated calcium influx and oxidative stress contribute to peripheral neuralgia. Kahya et al. [106]. Found that TRPM2 and TRPV1 were involved in neuronal death induced by Ca2+ efflux. Melatonin and selenium mitigate diabetes-induced oxidative stress, apoptosis, and calcium ion concentration by modulating TRPM2 and TRPV1 in the dorsal root ganglia and hippocampus. Similarly, on intrathecal injection of Alpha-lipoic acid, TRPV1 expression can be inhibited through the NF-κB pathway, which effectively relieves pain hypersensitivity and neuronal excitability [101]. The role of TRPC6 also has been closely scrutinized. It is increased within the dorsal root ganglion in the DNP model. Hydrogen sulfide (H2S) synthesized by cystathionine beta-synthase (CBS) enzymes activates/sensitizes TRPV1, TRPA1, and TRPC channels, leading to nociceptive hypersensitivity and peripheral nerve fiber loss [107]. Inhibitors of CBS enzymes or TRP channel blockers present potential treatments for diabetic peripheral neuropathic pain. Furthermore, Miao et al. [108] reported a time-dependent expression of BDNF and TRPC6 in the dorsal root ganglia and spinal cord of DNP rats, which was closely related to the generation of mechanical abnormality. Moreover, the inhibition of TRPC6 expression attenuated intraneuronal calcium influx and reversed anaphylaxis.
CIPN Model
CIPN is a prevalent side effect of various chemotherapy drugs like cisplatin, oxaliplatin, and paclitaxel, causing symptoms such as numbness, aching, burning, or tingling in the affected limb [109].
Using models of cisplatin/oxaliplatin-induced painful neuropathy, Leo et al. [110]. Demonstrated that TRPA1/TRPV1 expression was increased within dorsal root ganglion neurons, promoting calcium ion concentrations in the cytoplasm and mitochondria. This results in mitochondrial dysfunction and reduced antioxidant capacity, contributing significantly to neuropathic pain following chemotherapy. Similarly, compounds like LPP1 and pregabalin can inhibit TRPV1 and TRPA1 expression, reducing abnormal tactile and thermal sensitization induced by oxaliplatin/paclitaxel chemotherapeutic agents. However, their effects on cold sensitization are limited [111]. In other models of neuropathic pain induced by oxaliplatin, acute cold sensory hypersensitivity arises after four days of treatment, and increased TRPA1 and TRPV1 expression in small DRG neurons and increased TRPM8 expression in medium-sized DRG neurons was reported [112]. The alteration in the expression of TRP channels emerges as a potential therapeutic target for treating oxaliplatin-induced peripheral neuropathic pain. The herbal medicine Cinobufacini demonstrated the inhibition of oxaliplatin-induced TRPV1 upregulation in the dorsal root ganglion and spinal astrocyte activation, thereby reducing mechanical pain and thermal hypersensitivity in rats [113]. Additionally, the significance of TRPM8 cannot be understated. Bertamino et al. [114]. Screened the tryptophan derivative 14 and demonstrated through in vivo studies its potential analgesic effects by alleviating TRPM8-mediated hypersensitive response to cold perception. Moreover, oxaliplatin may contribute to the development of neuropathic pain by depolarizing IB4 neurons through the TRPM8 channel [115]. Membrane depolarization can be blocked by utilizing TRPM8 antagonists, preventing oxaliplatin-induced neuropathic pain. Accordingly, TRPM8 expression increases within the dorsal root ganglion in oxaliplatin-induced nociceptive hypersensitivity, and the administration of an L-type calcium channel antagonist effectively alleviates cold nociceptive hypersensitivity via the NFAT/TRPM8 pathway [116].
In the model of paclitaxel-induced peripheral neuralgia, increased TRPV4 expression in sensory nerve fibers [54] contributes to nociceptive hypersensitivity, while enhanced osmotic conductance in nociceptors depends on integrin/Src tyrosine kinase signaling. Administering TRPV4 antisense helps mitigate mechanical hypersensitivity [54]. Additionally, there appears to be an association between TRPV4 and stretch-activated ion channels that contribute to mechanical pain transmission mechanisms [50]. TRPV4 collaborates with TRPC1/TRPC6 to facilitate mechanical hypersensitivity and enhance the sensitivity of primary afferent nociceptors to alleviate neuropathic pain caused by paclitaxel [50]. TRPV1 has been established as a critical receptor in paclitaxel-induced hypersensitivity to pain. It was increased in small and medium DRG neurons after paclitaxel treatment, revealing a potential role of TRPV1 in thermal nociceptive hypersensitivity [117]. Ba et al. [118] used warfarin to mitigate the mechanical and thermo-sensory hypersensitivity caused by paclitaxel, which was achieved by inhibiting TRPV1 upregulation, spinal astrocyte activation, and reducing the production of inflammatory factors such as TNF-α and IL-1β. Moreover, Chen et al. [72] also observed an increasing activity of mast cell trypsin in the spinal cord, dorsal root ganglia, and peripheral tissues of mice during paclitaxel treatment. The protease receptor triggered the activation of phospholipase C, protein kinase A, and protein kinase C, along with the activation of downstream TRPV1, TRPV4, and TRPA1 signaling pathways [72]. Targeting this signaling molecule may provide novel avenues for the treatment of paclitaxel-induced neuropathy. PAX6/TRPC6 in DRG neurons may be considered a common signaling pathway for mechanical nociceptive hypersensitivity after oxaliplatin, paclitaxel, or bortezomib application [119]. Notably, epigenetic mechanisms, specifically DNA hypomethylation of the PAX6 gene, facilitate the upregulation of the TRPC6 pathway implicated in peripheral neuropathic pain.
In peripheral neuropathy induced by vincristine, increased TRPV1 expression was observed in the dorsal root ganglion and was mainly concentrated in small B4+ neurons. TRPV1 antagonists significantly inhibited nociceptive hypersensitivity [120]. Khan et al. [121] reported that substances like white mandrake exhibit neuroprotective effects by attenuating the levels of inflammatory cytokines and oxidative stress via the downregulation of TRPV1 and TRPM8 expression. This effect mitigates vincristine-induced mechanical pain, as well as cold and heat sensory sensitization. Similarly, in a stavudine-induced neuropathic pain model, niflumic acid alleviated nociceptive hypersensitivity in rats by directly antagonizing the TRPV1 pathway and blocking oxidative stress and inflammatory responses [122]. Studies across various chemotherapeutic drug-induced peripheral neuropathic pain models underscore the important role of TRP channels, establishing them as potential therapeutic targets for alleviating peripheral neuropathic pain.
Conclusions and Perspectives
In this review, we have described various models of neuropathic pain and elucidated the analgesic mechanism orchestrated by TRP channels (Fig. 2). Chronic neuropathic pain, often associated with damage or dysfunction in the somatosensory nervous system, [16, 123] poses challenges in obtaining clinical samples due to its specific neural nature. Animal models provide a simplified representation of human neuropathic pain, inclusive of the spatial, temporal, and biological complexity of human neuropathic pain, making them a valuable tool in pain research. However, caution is imperative when translating findings from animals to humans, considering factors like genetic heterogeneity, treatment interventions, disease progression, and pharmacokinetics. Despite these limitations, rodent pain models remain invaluable, with peripheral nerve injury-induced neuropathic pain currently being the most prevalent. Models like SNL, SNI, and CCI induce regional pain associated with single-branch neuropathy. Additionally, STZ-induced neuropathic pain in diabetes mellitus and chemotherapeutic drug-induced neuropathic pain models causing peripheral neuropathy are widely employed.

Overview TRP channels that have been implicated in different models of chronic neuropathic pain. The black dots represent the source of the tissue
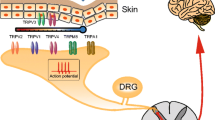
The development and maintenance of neuropathic pain involve multifaceted factors within the peripheral and central nervous systems, encompassing altered ion channel activity [44, 124], immune cell activation [29, 125], and immune regulation imbalances [126]. TRP channels play a key role as sensors and modulators in neuropathic pain, influencing pain signal generation and transmission by modulating neuronal excitability, plasticity, and neuroinflammatory responses [127, 128] (Fig. 3). Different TRP subfamilies and subtypes exhibit varying sensitivities and functional characteristics, potentially interacting to form complex signaling networks.

Potential mechanisms of TRP receptors in neuropathic pain. TRP, as transmembrane entities, are widely distributed in the nervous system. Here we take microglia as an example (not limited to this type of cell). Tissue damage leads to receptor activation. They can affect intracellular ion concentrations and abnormal excitability. Similarly, they will interfere with immune cell activation and proliferation, as well as neuroinflammatory responses via downstream pathways (such as ERK/ JAK p38MAPK/ NF-κB). Inhibitors (shown in the figure, Baicalin, etc.) suppress TRP expression and attenuate pain sensitization. Only some of the TRP receptors are listed here
This review presents novel insights into the molecular mechanisms by which TRP channels contribute to chronic neuropathic pain, establishing them as potential therapeutic targets. Given the important and diverse roles of TRP channels in neuropathic pain, they provide opportunities and approaches for the development of novel analgesics [129]. Receptors such as TRPA1, TRPV1, TRPM8, and TRPC6 have demonstrated unique roles in neuropathic pain, and several agonists or antagonists targeting these TRP channels have entered clinical trials or the market for treating various pain conditions, dermatologic diseases, and respiratory disorders [80, 130, 131]. However, challenges such as TRP channel selectivity and side effects remain, necessitating highly selective drug development to avoid interference with other stimuli [132]. TRPV1 has also been demonstrated to be sensitive to high temperature, low pH, and capsaicin treatment [133]. Meanwhile, TRPA1 activity exhibits significant species-related differences, which impede the efficacy of preclinical studies in predicting clinical efficacy [134]. Furthermore, in clinical trials, TRPV1 inhibitors elevated body temperature in participants and reduced their ability to perceive harmful heat [135]. Nonetheless, the study of TRP channels is of clinical significance from both basic research and drug discovery perspectives, necessitating continued in-depth exploration.
Central mechanisms underlying neuropathic pain, especially TRP-related studies in the central nervous system, are less explored. Understanding altered neuronal excitability, neural-immune cell interactions, and relevant brain nuclei activation remains an area for further investigation [136,137,138]. Gene editing technologies, such as knockout animals, allow one to study the role of specific genes in the development and maintenance of pain, as well as identify novel therapeutic targets. It can inactivate or replace the function of a target gene within an experimental animal, thus helping researchers to understand the normal function of the gene and the effect of gene deletion or mutation on pain perception. In conclusion, we highlight recent advances in understanding TRPs in various rodent models of neuropathic pain, and future research should explore TRP channels in lesser-utilized models of neuropathic pain, particularly those stemming from CNS disease, while also focusing on developing safer and more effective TRP channel modulators.
Data Availability
The data that support the findings of this study are available from the corresponding author, [Yun Wang], upon reasonable request.
References
Inoue K, Tsuda M (2018) Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat Rev Neurosci 19:138–152
Ochoa JL (2009) Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 72:1282–1283
Scholz J et al (2019) The IASP classification of chronic pain for ICD-11: Chronic neuropathic pain. Pain 160:53–59
Attal N, Bouhassira D, Colvin L (2023) Advances and challenges in neuropathic pain: a narrative review and future directions. Br J Anaesth 131:79–92
Yoon SY, Oh J (2018) Neuropathic cancer pain: Prevalence, pathophysiology, and management. Korean J Intern Med 33:1058–1069
Van Der Windt DAWM et al (2008) Physical examination for lumbar radiculopathy due to disc herniation in patients with low-back pain. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD007431
Doughty CT, Bowley MP (2019) Entrapment neuropathies of the upper extremity. Med Clin North Am 103:357–370
Calcutt NA (2020) Diabetic neuropathy and neuropathic pain: a (con)fusion of pathogenic mechanisms. Pain. https://doi.org/10.1097/j.pain.0000000000001922
Sampathkumar P, Drage LA, Martin DP (2009) Herpes zoster (Shingles) and postherpetic neuralgia. Mayo Clin Proc 84:274–280
Colvin LA (2020) Europe PMC funders group chemotherapy-induced peripheral neuropathy (CIPN): where are we now ? Pain 160:1–22
Klit H, Finnerup NB, Jensen TS (2009) Central post-stroke pain: clinical characteristics, pathophysiology, and management. The Lancet Neurol 8:857–868
Maayah ZH, Takahara S, Ferdaoussi M, Dyck JRB (2020) The anti-inflammatory and analgesic effects of formulated full-spectrum cannabis extract in the treatment of neuropathic pain associated with multiple sclerosis. Inflamm Res 69:549–558
Ford B (2010) Pain in Parkinson’s disease. Mov Disord 25:98–103
Shiao R, Lee-Kubli CA (2018) Neuropathic pain after spinal cord injury: challenges and research perspectives. Neurotherapeutics 15:635–653
Jensen TS, Finnerup NB (2014) Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol 13:924–935
Borzan J, Meyer RA (2009) Neuropathic pain. Encyclopedia Neurosci. https://doi.org/10.1016/B978-008045046-9.01926-4
Andersen ML, Araujo P, Frange C, Tufik S (2018) Sleep disturbance and pain: a tale of two common problems. Chest 154:1249–1259
Yang C et al (2019) Key role of gut microbiota in anhedonia-like phenotype in rodents with neuropathic pain. Transl Psychiatry. https://doi.org/10.1038/s41398-019-0379-8
Chen C et al (2023) Chronic pain conditions and risk of suicidal behavior: a 10-year longitudinal co-twin control study. BMC Med 21:1–11
Gilron I, Baron R, Jensen T (2015) Neuropathic pain: Principles of diagnosis and treatment. Mayo Clin Proc 90:532–545
Beydoun A (2003) Neuropathic pain: from mechanisms to treatment strategies. J Pain Symptom Manage 25:1–114
Yeh KY et al (2021) A dual-mode multifunctional pulsed radio-frequency stimulator for trigeminal neuralgia relief and its animal model. IEEE Trans Biomed Circuits Syst 15:719–730
Li Y et al (2018) Drg voltage-gated sodium channel 1.7 is upregulated in paclitaxel-induced neuropathy in rats and in humans with neuropathic pain. J Neurosci 38:1124–1136
Li N et al (2020) Upregulation of transcription factor 4 downregulates NaV1.8 expression in DRG neurons and prevents the development of rat inflammatory and neuropathic hypersensitivity. Exp Neurol 327:113240
Busserolles J, Tsantoulas C, Eschalier A, García JAL (2016) Potassium channels in neuropathic pain: advances, challenges, and emerging ideas. Pain 157:S7–S14
Bourinet E, Francois A, Laffray S (2016) T-type calcium channels in neuropathic pain. Pain 157:S15–S22
Guan Z et al (2015) Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci 19:94–101
Takeda I et al (2022) Controlled activation of cortical astrocytes modulates neuropathic pain-like behaviour. Nat Commun 13:1–12
Yu X et al (2020) Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain. Nat Commun 11:1–12
Pinho-Ribeiro FA, Verri WA, Chiu IM (2017) Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol 38:5–19
Venkatachalam K, Montell C (2007) TRP channels. Annu Rev Biochem 76:387–417
Sappington RM et al (2015) Activation of transient receptor potential vanilloid-1 (TRPV1) influences how retinal ganglion cell neurons respond to pressure-related stress. Channels 9:102–113
Harteneck C (2003) Proteins modulating TRP channel function. Cell Calcium 33:303–310
Taylor BK (2001) Pathophysiologic mechanisms of neuropathic pain. Curr Pain Headache Rep. https://doi.org/10.1007/s11916-001-0083-1
Sommer C, Leinders M, Üçeyler N (2018) Inflammation in the pathophysiology of neuropathic pain. Pain. https://doi.org/10.1097/j.pain.0000000000001122
Nishimoto R et al (2021) Thermosensitive TRPV4 channels mediate temperature-dependent microglia movement. Proce Natl Acad Sci USA. https://doi.org/10.1073/pnas.2012894118
Wu J et al (2023) Function of TRP channels in monocytes/macrophages. Front Immunol 14:1–11
Scimemi A (2013) A TRP among the astrocytes. J Physiol 591:9–15
Wedel S et al (2022) SAFit2 reduces neuroinflammation and ameliorates nerve injury-induced neuropathic pain. J Neuroinflammation 19:1–21
Chen G, Zhang YQ, Qadri YJ, Serhan CN, Ji RR (2018) Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron 100:1292–1311
Jiang BC et al (2016) CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Investig 126:745–761
Ramsey IS, Delling M, Clapham DE (2006) An introduction to TRP channels. Annu Rev Physiol 68:619–647
Gees M, Colsoul B, Nilius B (2010) The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a003962
Julius D (2013) TRP channels and pain. Annu Rev Cell Dev Biol. https://doi.org/10.1146/annurev-cellbio-101011-155833
Clapham DE (2003) TRP channels as cellular sensors. Nature 426:517–524
Rubaiy HN (2019) Treasure troves of pharmacological tools to study transient receptor potential canonical 1/4/5 channels. Br J Pharmacol 176:832–846
Ratté S, Karnup S, Prescott SA (2018) Nonlinear relationship between spike-dependent calcium influx and TRPC channel activation enables robust persistent spiking in neurons of the anterior cingulate cortex. J Neurosci 38:1788–1801
Yang F, Sivils A, Cegielski V, Singh S, Chu XP (2023) Transient receptor potential (TRP) channels in pain, neuropsychiatric disorders, and epilepsy. Int J Mol Sci. https://doi.org/10.3390/ijms24054714
Chu WG et al (2020) TRPC1/4/5 channels contribute to morphine-induced analgesic tolerance and hyperalgesia by enhancing spinal synaptic potentiation and structural plasticity. FASEB J 34:8526–8543
Alessandri-Haber N, Dina OA, Chen X, Levine JD (2009) TRPC1 and TRPC6 channels cooperate with TRPV4 to mediate mechanical hyperalgesia and nociceptor sensitization. J Neurosci 29:6217–6228
Benham CD, Gunthorpe MJ, Davis JB (2003) TRPV channels as temperature sensors. Cell Calcium 33:479–487
Quartu M et al (2014) Bortezomib treatment produces nocifensive behavior and changes in the expression of TRPV1, CGRP, and substance P in the Rat DRG, spinal cord, and sciatic nerve. BioMed Res Int. https://doi.org/10.1155/2014/180428
Kwon DH et al (2021) Heat-dependent opening of TRPV1 in the presence of capsaicin. Nat Struct Mol Biol. https://doi.org/10.1038/s41594-021-00616-3
Alessandri-Haber N et al (2004) Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J Neurosci 24:4444–4452
Premkumar LS, Abooj M (2013) TRP channels and analgesia. Life Sci 92:415–424
Jimenez I et al (2020) TRPM channels in human diseases. Cells. https://doi.org/10.3390/cells9122604
McNulty S, Fonfria E (2005) The role of TRPM channels in cell death. Pflugers Arch 451:235–242
Pérez De Vega MJ, Gómez-Monterrey I, Ferrer-Montiel A, González-Muñiz R (2016) Transient receptor potential melastatin 8 channel (TRPM8) modulation cool entryway for treating pain and cancer. J Med Chem 59:10006–10029
Dhaka A et al (2007) TRPM8 is required for cold sensation in mice. Neuron 54:371–378
Haraguchi K et al (2012) TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J Neurosci 32:3931–3941
Aloi VD et al (2023) TRPM3 as a novel target to alleviate acute oxaliplatin-induced peripheral neuropathic pain. Pain 164:2060–2069
Monteiro S, de Araujo D, Nassini R, Geppetti P, De Logu F (2020) TRPA1 as a therapeutic target for nociceptive pain. Expert Opin Ther Tar 24:997–1008
Talavera K et al (2020) Mammalian transient receptor potential TRPA1 channels: from structure to disease. Physiol Rev 100:803
Pop C et al (2020) Effects of Lycium barbarum L. Polysaccharides on inflammation and oxidative stress markers in a pressure overload-induced heart failure rat model. Molecules 25:1–10
Harrison RS, Sharpe PC, Singh Y, Fairlie DP (2007) Amyloid peptides and proteins in review. Rev Physiol Biochem Pharmacol 159:1–77
Gouin O et al (2017) TRPV1 and TRPA1 in cutaneous neurogenic and chronic inflammation: pro-inflammatory response induced by their activation and their sensitization. Protein Cell 8:644–661
Garrison SR, Dietrich A, Stucky CL (2012) TRPC1 contributes to light-touch sensation and mechanical responses in low-threshold cutaneous sensory neurons. J Neurophysiol 107:913–922
Wang J et al (2020) The analgesic action of larixyl acetate, a potent TRPC6 inhibitor, in rat neuropathic pain model induced by spared nerve injury. J Neuroinflammation 17:1–20
Xu S et al (2022) Dexmedetomidine alleviates neuropathic pain via the TRPC6-p38 MAPK pathway in the dorsal root ganglia of rats. J Pain Res 15:2437–2448
Wang Z, Ling D, Wu C, Han J, Zhao Y (2020) Baicalin prevents the up-regulation of TRPV1 in dorsal root ganglion and attenuates chronic neuropathic pain. Vet Med Sci 6:1034–1040
Yang B et al (2021) Higenamine attenuates neuropathic pain by inhibition of NOX2/ROS/TRP/P38 mitogen-activated protein kinase/NF-ĸB signaling pathway. Front Pharmacol 12:1–15
Chen Y, Yang C, Wang ZJ (2011) Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience 193:440–451
Ji G, Zhou S, Carlton SM (2008) Intact Aδ-fibers up-regulate transient receptor potential A1 and contribute to cold hypersensitivity in neuropathic rats. Neuroscience 154:1054–1066
Obata K et al (2010) TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury (Journal of Clinical Investigation (2005) 115, (2393–2401)). J Clin Investig 120:394
Wang H, Song T, Wang W, Zhang Z (2019) TRPM2 participates the transformation of acute pain to chronic pain during injury-induced neuropathic pain. Synapse. https://doi.org/10.1002/syn.22117
Su S, Yudin Y, Kim N, Tao YX, Rohacs T (2021) TRPM3 channels play roles in heat hypersensitivity and spontaneous pain after nerve injury. J Neurosci 41:2457–2474
De Caro C et al (2018) Antinociceptive effect of two novel transient receptor potential melastatin 8 antagonists in acute and chronic pain models in rat. Br J Pharmacol 175:1691–1706
Poulson SJ et al (2020) Naked mole-rats lack cold sensitivity before and after nerve injury. Mol Pain. https://doi.org/10.1177/1744806920955103
Dai Y (2016) TRPs and pain. Semin Immunopathol 38:277–291
Marwaha L et al (2016) TRP channels: potential drug target for neuropathic pain. Inflammopharmacology 24:305–317
Jaggi AS, Jain V, Singh N (2011) Animal models of neuropathic pain. Fundam Clin Pharmacol 25:1–28
Zhang XL, Lee KY, Priest BT, Belfer I, Gold MS (2015) Inflammatory mediator-induced modulation of GABAA currents in human sensory neurons. Neuroscience 310:401–409
Ririe DG, Liu B, Clayton B, Tong C, Eisenach JC (2008) Electrophysiologic characteristics of large neurons in dorsal root ganglia during development and after hind paw incision in the rat. Anesthesiology 109:111–117
Bennett GJ, Xie YK (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33:87–107
De Vry J, Kuhl E, Franken-Kunkel P, Eckel G (2004) Pharmacological characterization of the chronic constriction injury model of neuropathic pain. Eur J Pharmacol 491:137–148
Ho Kim S, Mo Chung J (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50:355–363
Yoon C, Young Wook Y, Heung Sik N, Sun Ho K, Jin Mo C (1994) Behavioral signs of ongoing pain and cold allodynia in a rat model of neuropathic pain. Pain 59:369–376
Boccella S et al (2018) Spared nerve injury as a long-lasting model of neuropathic pain. Methods Mol Biol 1727:373–378
Bourquin AF et al (2006) Assessment and analysis of mechanical allodynia-like behavior induced by spared nerve injury (SNI) in the mouse. Pain 122(14):e1-14.e14
Courteix C, Eschalier A, Lavarenne J (1993) Streptozocin-induced diabetic rats: behavioural evidence for a model of chronic pain. Pain 53:81–88
Burchiel KIMJ, Russell LC, Lee RP, Sima AAF (1985) Spontaneous activity of primary afferent neurons in diabetic BB/Wistar rats. Diabetes. https://doi.org/10.2337/diab.34.11.1210
Flatters SJL, Bennett GJ (2006) Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain 122:245–257
Tredici G, Tredici S, Fabbrica D, Minoia C, Cavaletti G (1998) Experimental cisplatin neuronopathy in rats and the effect of retinoic acid administration. J Neurooncol 36:31–40
Ling B, Authier N, Balayssac D, Eschalier A, Coudore F (2007) Behavioral and pharmacological description of oxaliplatin-induced painful neuropathy in rat. Pain 128:225–234
Colburn RW et al (2007) Attenuated cold sensitivity in TRPM8 null mice. Neuron 54:379–386
Wang C et al (2019) Facilitation of MrgprD by TRP-A1 promotes neuropathic pain. FASEB J 33:1360–1373
Staaf S, Oerther S, Lucas G, Mattsson JP, Ernfors P (2009) Differential regulation of TRP channels in a rat model of neuropathic pain. Pain 144:187–199
Yamamoto S, Ohsawa M, Ono H (2013) Contribution of TRPV1 receptor-expressing fibers to spinal ventral root after-discharges and mechanical hyperalgesia in a spared nerve injury (SNI) rat model. J Pharmacol Sci 121:9–16
Brignell JL, Chapman V, Kendall DA (2008) Comparison of icilin- and cold-evoked responses of spinal neurones, and their modulation of mechanical activity, in a model of neuropathic pain. Brain Res 1215:87–96
Hagenacker T, Lampe M, Schäfers M (2014) Icilin reduces voltage-gated calcium channel currents in naïve and injured DRG neurons in the rat spinal nerve ligation model. Brain Res 1557:171–179
Zhang BY et al (2020) Alpha-lipoic acid downregulates TRPV1 receptor via NF-κB and attenuates neuropathic pain in rats with diabetes. CNS Neurosci Ther 26:762–772
Nam JS et al (2014) Effects of nefopam on streptozotocin-induced diabetic neuropathic pain in rats. Korean J Pain 27:326–333
Huang Q, Chen Y, Gong N, Wang YX (2016) Methylglyoxal mediates streptozotocin-induced diabetic neuropathic pain via activation of the peripheral TRPA1 and Nav1.8 channels. Metabolism 65:463–474
Wei H, Hämäläinen MM, Saarnilehto M, Koivisto A, Pertovaara A (2009) Attenuation of mechanical hypersensitivity by an antagonist of the TRPA1 ion channel in diabetic animals. Anesthesiology 111:147–154
Reese RM et al (2020) Behavioral characterization of a CRISPR-generated TRPA1 knockout rat in models of pain, itch, and asthma. Sci Rep 10:1–11
Kahya MC, Nazıroğlu M, Övey İS (2017) Modulation of diabetes-induced oxidative stress, apoptosis, and Ca2+ Entry Through TRPM2 and TRPV1 channels in dorsal root ganglion and hippocampus of diabetic rats by melatonin and selenium. Mol Neurobiol 54:2345–2360
Roa-Coria JE et al (2019) Possible involvement of peripheral TRP channels in the hydrogen sulfide-induced hyperalgesia in diabetic rats 11 Medical and Health Sciences 1109 Neurosciences. BMC Neurosci 20:1–17
Miao B et al (2021) The implication of transient receptor potential canonical 6 in BDNF-induced mechanical allodynia in rat model of diabetic neuropathic pain. Life Sci 273:119308
Wolf SL et al (2012) The relationship between numbness, tingling, and shooting/burning pain in patients with chemotherapy-induced peripheral neuropathy (CIPN) as measured by the EORTC QLQ-CIPN20 instrument, N06CA. Support Care Cancer 20:625–632
Leo M et al (2020) Platinum-based drugs cause mitochondrial dysfunction in cultured dorsal root ganglion neurons. Int J Mol Sci 21:1–8
Sałat K et al (2014) Antiallodynic and antihyperalgesic activity of 3-[4-(3-trifluoromethyl- phenyl)-piperazin-1-yl]-dihydrofuran-2-one compared to pregabalin in chemotherapy-induced neuropathic pain in mice. Pharmacol Biochem Behav 122:173–181
Chukyo A et al (2018) Oxaliplatin-induced changes in expression of transient receptor potential channels in the dorsal root ganglion as a neuropathic mechanism for cold hypersensitivity. Neuropeptides 67:95–101
Hao Y et al (2019) Huachansu suppresses TRPV1 up-regulation and spinal astrocyte activation to prevent oxaliplatin-induced peripheral neuropathic pain in rats. Gene 680:43–50
Bertamino A et al (2018) Identification of a potent tryptophan-based TRPM8 antagonist with in vivo analgesic activity. J Med Chem 61:6140–6152
Wu B et al (2021) Oxaliplatin depolarizes the IB4– dorsal root ganglion neurons to drive the development of neuropathic pain through TRPM8 in mice. Front Mol Neurosci 14:1–13
Kawashiri T et al (2012) L type Ca2+ channel blockers prevent oxaliplatin-induced cold hyperalgesia and TRPM8 overexpression in rats. Mol Pain 8:7
Hara T et al (2013) Effect of paclitaxel on transient receptor potential vanilloid 1 in rat dorsal root ganglion. Pain 154:882–889
Ba X et al (2018) Cinobufacini protects against paclitaxel-induced peripheral neuropathic pain and suppresses TRPV1 up-regulation and spinal astrocyte activation in rats. Biomed Pharmacother 108:76–84
Tu Y-P et al (2020) Swabs collected by patients or health care workers for SARS-CoV-2 testing. N Engl J Med 383:494–496
Chiba T et al (2017) Vincristine-induced peripheral neuropathic pain and expression of transient receptor potential vanilloid 1 in rat. J Pharmacol Sci 133:254–260
Khan A et al (2021) Suppression of TRPV1/TRPM8/p2y nociceptors by withametelin via downregulating mapk signaling in mouse model of vincristine-induced neuropathic pain. Int J Mol Sci. https://doi.org/10.3390/ijms22116084
Marwaha L et al (2016) Niflumic acid, a TRPV1 channel modulator, ameliorates stavudine-induced neuropathic pain. Inflammopharmacology 24:319–334
Van Hecke O, Austin SK, Khan RA, Smith BH, Torrance N (2014) Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 155:654–662
Hargus NJ, Patel MK (2007) Voltage-gated Na+ channels in neuropathic pain. Expert Opin Investig Drugs 16:635–646
Inoue K, Tsuda M (2018) Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potentialfile:///C:/Users/10548/Desktop/DRG/Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy.pdf. Nat Rev Neurosci 19:138–152
Mollazadeh H et al (2019) Immune modulation by curcumin: the role of interleukin-10. Crit Rev Food Sci Nutr. https://doi.org/10.1080/104083981358139
Hong Z et al (2016) Transient receptor potential vanilloid 4-induced modulation of voltage-gated sodium channels in hippocampal neurons. Mol Neurobiol 53:759–768
Egaña-Huguet J et al (2021) Lack of the transient receptor potential vanilloid 1 shifts cannabinoid-dependent excitatory synaptic plasticity in the dentate gyrus of the mouse brain hippocampus. Front Neuroanat 15:1–12
Premkumar LS (2014) Transient receptor potential channels as targets for phytochemicals. ACS Chem Neurosci 5:1117–1130
Mahmoud O, Soares GB, Yosipovitch G (2023) Transient receptor potential channels and Itch. Int J Mol Sci. https://doi.org/10.3390/ijms24010420
Müller I et al (2022) Transient receptor potential (TRP) channels in airway toxicity and disease: an update. Cells. https://doi.org/10.3390/cells11182907
Fernández-Carvajal A, González-Muñiz R, Fernández-Ballester G, Ferrer-Montiel A (2020) Investigational drugs in early phase clinical trials targeting thermotransient receptor potential (thermoTRP) channels. Expert Opin Investig Drugs 29:1209–1222
Rosenbaum T, Islas LD (2023) Molecular physiology of TRPV channels: controversies and future challenges. Annu Rev Physiol 85:293–316
Chen J et al (2013) Species differences and molecular determinant of TRPA1 cold sensitivity. Nat Commun 4:1–7
Moran MM (2018) TRP channels as potential drug targets. Annu Rev Pharmacol Toxicol 58:309–330
Palazzo E et al (2002) Interaction between vanilloid and glutamate receptors in the central modulation of nociception. Eur J Pharmacol 439:69–75
Enrich-Bengoa J et al (2022) TRPV2: a key player in myelination disorders of the central nervous system. Int J Mol Sci. https://doi.org/10.3390/ijms23073617
Gualdani R, Gailly P (2020) How TRPC channels modulate hippocampal function. Int J Mol Sci 21:1–19
Acknowledgements
The authors thank Home for Researchers (www.home-for-researchers.com) for English language editing. Figures were created by BioRender (Publication Permissions: JA264FWIEQ; KP264FWQ0C; MF264JVU2N).
Funding
This work was supported by the National Natural Science Foundation of China (81771181, 81571065, 82171217) and the Beijing Natural Science Foundation (7202053).
Author information
Authors and Affiliations
Contributions
All authors made a significant contribution to the work. SX: literature search and selection, content writing, figure and table creation. YW: conceptualization, structure design, content revision and funding acquisition.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xu, S., Wang, Y. Transient Receptor Potential Channels: Multiple Modulators of Peripheral Neuropathic Pain in Several Rodent Models. Neurochem Res 49, 872–886 (2024). https://doi.org/10.1007/s11064-023-04087-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-04087-4