
Abstract
Transient receptor potential vanilloid 4 (TRPV4) is reported to control the resting membrane potential and increase excitability in many types of cells. Voltage-gated sodium channels (VGSCs) play an important role in initiating action potentials in neurons. However, whether VGSCs can be modulated by the activation of TRPV4 in hippocampal pyramidal neurons remains unknown. In this study, we tested the effect of TRPV4 agonists (GSK1016790A and 4α-PDD) on voltage-gated sodium current (I Na) in hippocampal CA1 pyramidal neurons and the protein levels of α/β-subunit of VGSCs in the hippocampus of mice subjected to intracerebroventricular (icv.) injection of GSK1016790A (GSK-injected mice). Herein, we report that I Na was inhibited by acute application of GSK1016790A or 4α-PDD. In the presence of TRPV4 agonists, the voltage-dependent inactivation curve shifted to the hyperpolarization, whereas the voltage-dependent activation curve remained unchanged. The TRPV4 agonist-induced inhibition of I Na was blocked by the TRPV4 antagonist or tetrodotoxin. Moreover, blocking protein kinase A (PKA) markedly attenuated the GSK1016790A-induced inhibition of I Na, whereas antagonism of protein kinase C or p38 mitogen-activated protein kinase did not change GSK1016790A action. Finally, the protein levels of Nav1.1, Nav1.2, and Nav1.6 in the hippocampus increased in GSK-injected mice, whereas those of Nav1.3 and Navβ1 remained nearly unchanged. We conclude that I Na is inhibited by the acute activation of TRPV4 through PKA signaling pathway in hippocampal pyramidal neurons, but protein expression of α-subunit of VGSCs is increased by sustained TRPV4 activation, which may compensate for the acute inhibition of I Na and provide a possibility for hyper-excitability upon sustained TRPV4 activation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transient receptor potential vanilloid 4 (TRPV4), which was first identified as a cellular osmolarity sensor, is a member of the transient receptor potential (TRP) ion channel superfamily [1]. In addition to its sensitivity to hypotonic stimulation, TRPV4 can be activated by multiple stimuli, including moderate heat, cell swelling, and endogenous chemicals, such as anandamide, arachidonic acid, and the epoxyeicosatrienoic acid metabolites of the latter, as well as by a growing number of exogenous chemical ligands [2]. TRPV4 is broadly expressed in the peripheral nervous system (PNS, including dorsal root ganglion neurons and trigeminal ganglion (TG) neurons) and the central nervous system (CNS, including the hippocampus, cortex, thalamus, and cerebellum) [1]. As a calcium-selective channel, the activation of TRPV4 mediates calcium influx and is responsible for increasing the intracellular calcium concentration ([Ca2+] i ), which plays an important role in regulating physiological functions and is involved in multiple pathological conditions [3]. For example, TRPV4 is responsible for mechanical hyperalgesia through the Ca2+-entry-induced release of substance P and calcitonin gene-related peptide from the dorsal horn of the rat spinal cord [4]. Activation of TRPV4, together with TRPA1, stimulates Ca2+-dependent release of ATP in human odontoblast-like cells [5]. Because of the inward current induced by TRPV4 activation, application of a TRPV4 agonist increases the spontaneous firing rate in mouse retinal ganglion cells, and the number of evoked action potentials (APs) was markedly increased by TRPV4 activation in TG neurons [6, 7]. In the CNS, the hippocampal resting membrane potential is regulated by TRPV4 at the physiological body temperature [8]. Moreover, activation of TRPV4 enhances hippocampal excitatory synaptic transmission, which may also facilitate neuronal discharge [9]. However, the mechanisms underlying TRPV4-mediated modulation of hippocampal neuronal excitability required further clarification.
Voltage-gated sodium channels (VGSCs) are important for the initiation and propagation of APs in neurons and other excitable cells [10]. Based on their sensitivity to tetrodotoxin (TTX), VGSCs can be divided into TTX-sensitive (TTX-S) channels and TTX-resistant (TTX-R) channels [10]. VGSC proteins in mammalian brain are composed of a 260-kDa α-subunit associated with auxiliary β-subunits (β1–β4) of 33–36 kDa in size. The α-subunit is the functional subunit of VGSCs, allowing sodium ion influx, and four α-subunits (Nav1.1, Nav1.2, Nav1.3, and Nav1.6) have been characterized in the CNS [11]. The above four Na+ channels share similar sensitivity to TTX and can be blocked by nanomolar concentrations of TTX. It is known that the function of VGSCs in neurons can be regulated by physiological signals through intracellular signaling pathways. For example, activation of protein kinase A (PKA) or protein kinase C (PKC) reduces sodium currents and the generation of APs [12]. Additional modulation of VGSCs by the activation of p38 mitogen-activated protein kinase (p38 MAPK) has also been reported in neurons and transfected mammalian cells [13, 14]. It has been reported that TRPV4 is involved in the modulation of VSGCs (TTX-R and TTX-S) in TG neurons through the regulation of various intracellular signaling pathways [15, 16]. In addition to VGSCs, voltage-gated potassium and calcium channels, TRPV1, and glutamate receptors are also modulated by TRPV4 activation [9, 17–20].
VGSCs are responsible for the rising phase of APs in the neuronal membrane. Therefore, in this study, we examined whether voltage-gated sodium current and α/β-subunit expression in hippocampal neurons could be modulated by the activation of TRPV4 and further explored the mechanisms underlying the action of TRPV4.
Materials and Methods
Animals
Male mice (ICR, Oriental Bio Service Inc., Nanjing, China) were used. Care of animals conformed to standards established by the National Institutes of Health. All animal experiments were carried out in accordance with the Guidelines for Laboratory Animal Research of Nanjing Medical University, and all animal protocols were performed with the approval of the Institutional Animal Care and Use Committee of Nanjing Medical University. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Slice Preparation
To prepare brain slices, 3-week-old mice were decapitated under deep anesthesia with ethyl ether, and the brains were removed rapidly. Coronal brain slices (400 μm) were cut using a vibrating microtome (Microslicer DTK 1500, Dousaka EM Co, Kyoto, Japan) in ice-cold modified artificial cerebrospinal fluid (mACSF) composed of (in mM) 126 NaCl, 1 CaCl2, 2.5 KCl, 1 MgCl2, 26 NaHCO3, 1.25 KH2PO4, and 20 d-glucose, oxygenated with a gas mixture of 95 % O2/5 % CO2. After 1 h of recovery, hippocampal slices were transferred to a recording chamber.
Electrophysiological Recording
Whole-cell patch clamp recordings were performed at room temperature (22–23 °C). Hippocampal neurons were viewed with an upright microscope equipped with an infrared-sensitive camera (DAGE-MTI, IR-1000). Voltage-gated sodium current (I Na) was recorded using an EPC-10 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany), sampled at 10 kHz, and filtered (Bessel) at 2.9 kHz. The capacitance and series resistance were compensated (>90 %) before recording. Data obtained from neurons in which uncompensated series resistance resulted in voltage-clamp errors >5 mV were not used for further analysis. Liquid junction potentials were compensated before patching. When the external solution was changed, measurements of the changes in liquid junction potentials were <2 mV and were not corrected. The glass pipettes [No. 64-0817(G85150T-3), Warner Instruments Inc., Hamden, CT, USA] with a resistance of 1–3 MΩ when filled with pipette solution were used.
To record I Na, the holding potential was −80 mV. The voltage-dependent activation curve (G–V curve) of sodium current was measured by 20-ms depolarizing pulses from −80 to +40 mV stepping by 10 mV with interval of 2 s. The voltage-dependent inactivation curve (inactivation–voltage curve) was obtained by double pulses: preconditioning pulses (20 ms) from −100 to +20 mV in 10-mV steps and following 0-mV test pulse (20 ms) with interval of 4 s. The pipette solution contained (in mM) 130 CsCl, 10 NaCl, 1 CaCl2, 2 MgCl2, 10 EGTA, 10 HEPES, and 5 Tris-ATP at pH 7.3. The external solution was composed of (in mM): 5 NaCl, 2.5 KCl, 1 MgCl2, 1 CaCl2, 26 NaHCO3, 1.25 KH2PO4, and 20 d-glucose, 3 4-AP, 10 TEA–Cl, 105 choline–Cl, and 0.1 GdCl2, oxygenated with a gas mixture of 95 % O2/5 % CO2.
Drug Treatment
The TRPV4 agonist GSK1016790A was intracerebroventricularly (icv.) injected. Mice were anesthetized with 2 % chloral hydrate (20 ml/kg) and then placed in a stereotactic device (Kopf Instruments, Tujunga, CA). A 23-G stainless-steel guide cannula (Plastics One, Roanoke, VA, USA) was first implanted into the right lateral ventricle (0.3 mm posterior, 1.0 mm lateral, and 2.5 mm ventral to the bregma) using a stepper-motorized microsyringe (Stoelting, Wood Dale, IL, USA) and anchored to the skull with four stainless steel screws and dental cement. Two days after implantation, GSK1016790A (500 nM/2 μl/mouse) was injected once daily for 3 days using a 26-G stainless-steel needle. Control mice were given an equal volume of vehicle. The injection site was confirmed in preliminary experiments. Neither needle insertion nor saline injection significantly affected survival.
Western Blot
Western blot analysis was performed 3 days after GSK1016790A injection. After mice were decapitated, hippocampi were quickly removed and then homogenized in lysis buffer containing 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, 10 mM NaF, 1 mM sodium orthovanadate, 1 % Triton X-10 0, 0.5 % sodium deoxycholate, 1 mM phenyl-methylsulfonyl fluoride, and protease inhibitor cocktail (Complete; Roche, Mannheim, Germany). Protein concentrations were determined using BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Samples of total protein were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were incubated with 5 % bovine serum albumin or 5 % nonfat dried milk in Tris-buffered saline containing 0.1 % Tween 20 (TBST) for 60 min at room temperature and then with anti-Nav1.1 (catalogue no. ASC-001, 1:200, Alomone labs), anti-Nav1.2 (catalogue no. ASC-002, 1:200, Alomone labs), anti-Nav1.3 (catalogue no. ASC-004, 1:200, Alomone labs), anti-Nav1.6 (catalogue no. ASC-009, 1:200, Alomone labs), anti-Navβ1 (catalogue no. ASC-041, 1:400, Alomone labs), or anti-GAPDH (catalogue no. 22556, 1:5000; Abcam) antibodies at 4 °C overnight. After washes with TBST, the membranes were incubated with an HRP-labeled secondary antibody and developed using the ECL detection kit (Amersham Biosciences, Piscataway, NJ, USA). Western blot bands were scanned and analyzed with the Image J software (National Institutes of Health). Hippocampal samples collected from 3 mice were considered a set for western blot analysis, and the summarized data represent the average of three experimental sets.
Data Analysis
Data were analyzed using PulseFit (HEKA Elektronik) and Stata 7.0 software (STATA Corporation, USA). All data are presented as the mean ± SEM, and the significance was indicated as p < 0.05 and p < 0.01 tested by paired or unpaired Student’s t tests. In the present study, after testing the effect of GSK1016790A or 4α-PDD on I Na, 500 nM GSK1016790A was applied to the same neuron to test whether the neuron had TRPV4 receptors. All analyzed data came from the neurons in which GSK1016790A-evoked current could be recorded (Supplementary Fig. 1). The amplitude of sodium current was calculated as the peak current using the same depolarizing stimulation in the same neuron. G–V curve and inactivation–voltage curve were fitted using Boltzmann functions: G/G max = 1/(1 + exp(V 0.5 – V m)/k) or I/I max = 1/(1 + exp(V 0.5 – V m)/k). V 0.5 is the membrane potential (V m) at which 50 % of activation or inactivation was observed and k is the slope of the function. The dose–response curve was fitted using Hill equation, in which I peak = I peakmax/[1 + (EC50/C)n], with n being the Hill coefficient and EC50 being the concentration that produced a 50 % effect.
Chemicals
TTX was obtained from Enzo Life Science (Ann Arbor, MI, USA). Other chemicals, unless stated, were obtained from Sigma Chemical Company. GSK1016790A, 4α-PDD, HC-067047, d-sphingosine, bisindolylmaleimide II (BIM), phorbol 12-myristate 13-acetate (PMA), N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride (H-89), (9S,10S,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1, 6]benzodiazocine-10-carboxylic acid hexyl ester (KT 5720), and 8-bromoadenosine 3′,5′-cyclic monophosphate sodium salt (8-Br-cAMP) were prepared as stock solutions in dimethyl sulfoxide (DMSO). The final concentration of DMSO in the bath chamber or pipette solution was <0.1 %. d-Sphingosine, H-89 and KT5720 were added to the pipette solution, whereas GSK1016790A, 4α-PDD, HC-067047, BIM, PMA, 8-Br-cAMP, anisomycin, and SB203580 were added to the bath solution. The concentrations of the above drugs were chosen based on previous reports [9, 13, 15, 18].
Results
Effect of TRPV4 Agonists on I Na in Hippocampal CA1 Pyramidal Neurons
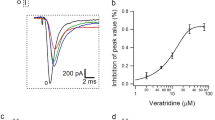
The acute effect of TRPV4-activaion on I Na was examined by bath application of TRPV4 agonists (GSK1016790A and 4α-PDD) for at least 8–10 min. As shown in Fig. 1a, the amplitude of I Na decreased markedly in the presence of GSK1016790A, and the decrease in I Na was largely recovered after GSK1016790A was washed out. On the average, I Na was reduced by 51.82 ± 5.17 % from −67.74 ± 5.71 pA/pF to −33.91 ± 3.01 pA/pF when GSK1016790A (500 nM) was added to the external solution (n = 20, paired t test, p < 0.01). G–V curve of I Na did not shift during GSK1016790A treatment (n = 8, paired t test, p > 0.05), whereas inactivation–voltage curve markedly shifted to hyperpolarization after GSK1016790A treatment (n = 8, paired t test, p < 0.01) (Fig. 1c, d). Figure 1e shows that at concentrations ranging from 0.01 nM to 3 μM, GSK1016790A inhibited I Na in a dose-dependent manner. Because GSK1016790A showed significant inhibition of I Na at the concentration of 500 nM, this concentration was used in the subsequent experiments.

Effect of GSK1016790A on I Na in hippocampal CA1 pyramidal neurons. a Typical recordings show that I Na was inhibited from −1.49 to −0.78 nA by the application of GSK1016790A (500 nM), and the current recovered to −1.31 nA after washout. b The voltage–current relationship (I–V curve) is shown before and during GSK1016790A treatment. c G–V curve was nearly unchanged in the presence of GSK1016790A. For control and GSK1016790A, V 0.5 was −46.88 ± 2.03 mV and −45.03 ± 1.19 mV, respectively (n = 8, paired t test, p > 0.05); k was 6.87 ± 1.51 and 5.24 ± 1.76 (n = 8, paired t test, p > 0.05), respectively. Data were transformed from the I–V data shown in b. d In the presence of GSK1016790A, inactivation–voltage curve of I Na markedly shifted to hyperpolarization. For control and GSK1016790A, V 0.5 was −50.14 ± 1.56 mV and −65.16 ± 3.67 mV, respectively (n = 8, paired t test, p < 0.01); k was −9.37 ± 1.59 and −12.37 ± 1.06, respectively (n = 8, paired t test, p < 0.01). e Plots showing the inhibition of I Na by GSK1016790A at concentrations of 0.01, 0.03, 0.1, 0.5, 1, and 3 μM. The dose–response curve is fitted to Hill equation with EC50 being 64.24 μM and n being 1.06
Another TRPV4 agonist, 4α-PDD, was also tested and I Na was inhibited by 25.13 ± 2.14 % from −66.99 ± 4.0891 pA/pF to −50.82 ± 3.07 pA/pF after application of 4α-PDD (30 μM) (n = 10, paired t test, p < 0.01) (Fig. 2a, b). Similar to the effect of GSK1016790A on I Na, in the presence of 4α-PDD, G–V curve of I Na did not shift (n = 8, paired t test, p > 0.05), whereas inactivation–voltage curve shifted to hyperpolarization (n = 8, paired t test, p < 0.01) (Fig. 2c, d).

Effect of 4α-PDD on I Na in hippocampal CA1 pyramidal neurons. a Typical recordings show that I Na was reversibly inhibited by application of 4α-PDD (30 μM). The amplitude of I Na was −1.29 nA, −0.75 nA, and −1.17 nA before, during, and after 4α-PDD-treatment, respectively. b I–V curve before and during 4α-PDD treatment. c In the presence of 4α-PDD, G–V curve did not shift, with V 0.5 (control, −46.45 ± 1.67 mV; 4α-PDD, −45.75 ± 1.73 mV) and k (control, 6.34 ± 0.83; 4α-PDD, 5.83 ± 1.07) remaining unchanged (n = 8, paired t test, p > 0.05 in each case). Data were transformed from the I–V data shown in b. d Inactivation–voltage curve of I Na markedly shifted to hyperpolarization after the application of 4α-PDD, with V 0.5 being −50.09 ± 1.59 mV and −59.40 ± 2.26 mV (n = 8, paired t test, p < 0.01), and k being −9.21 ± 1.79 and −9.36 ± 1.98 for control and 4α-PDD, respectively (n = 8, paired t test, p < 0.01 in each case)
Effect of HC-067047 and TTX on GSK1016790A-Induced Inhibition of I Na
I Na was hardly affected by application of the TRPV4 specific antagonist HC-067047 (10 μM) (n = 6, paired t test, p > 0.05). However, it is notable that GSK1016790A- or 4α-PDD-induced inhibition of I Na was blocked completely by pre-application of HC-067047 (unpaired t test, p < 0.01 in each case) (Fig. 3a). Collectively, the above results indicate that I Na is inhibited by acute activation of TRPV4.

Effects of HC-067047 and TTX on the inhibition of I Na by TRPV4 agonists. a After pre-application of the TRPV4 antagonist HC-067047, the inhibition of I Na by GSK1016790A was markedly attenuated from 51.82 ± 5.17 % to 2.07 ± 1.07 % (n = 28, unpaired t test, p < 0.01), and the inhibition by 4α-PDD was reduced from 24.39 ± 2.13 % to 1.07 ± 0.92 % (n = 18, unpaired t test, p < 0.01); **p < 0.01 vs. agonist. b I Na was markedly reduced by the application of TTX, and with TTX in the bath solution, the current was nearly unchanged by the application of GSK1016790A (TTX, −0.96 ± 0.77 pA/pF; GSK1016790A + TTX, −1.03 ± 0.56 pA/pF; n = 7, paired t test, p > 0.05) or 4α-PDD (TTX, −1.01 ± 0.34 pA/pF; 4α-PDD + TTX, −1.07 ± 0.87 pA/pF; n = 8, paired t test, p > 0.05); **p < 0.01 vs. control
Based on their sensitivity to TTX, VGSCs can be divided into TTX-S and TTX-R channels [10]. Here, I Na was reduced by 98.03 ± 1.74 % from −67.74 ± 5.71 pA/pF to 0.96 ± 0.77 pA/pF when TTX (0.3 μM) was added to the bath solution (n = 7, paired t test, p < 0.01), indicating that I Na was TTX-S channel. We found that after pre-application of TTX, the current was nearly unchanged by treatment with either GSK1016790A or 4α-PDD, implying that the acute activation of TRPV4 inhibits TTX-S channels (paired t test, p > 0.05 in each case) (Fig. 3b). GSK1016790A is a recently reported agonist of TRPV4 that exhibits greater efficiency and selectivity, and it was therefore used in the subsequent experiments.
Involvement of Intracellular Signaling Pathways in GSK1016790A-Induced Inhibition of I Na
The phosphorylation of VGSCs by intracellular signaling pathways has been proven to modulate sodium channel function. Activation of PKA or PKC is reported to decrease sodium current [12]. In the present study, I Na was reduced 11.51 ± 2.62 % (n = 7, paired t test, p < 0.05) and 18.08 ± 3.04 % (n = 6, paired t test, p < 0.05) by application of the PKA agonist 8-Br-cAMP (1 mM) or the PKC agonist PMA (1 μM), respectively. Consistently, I Na was increased 24.93 ± 3.51 % (n = 6, paired t test, p < 0.01) and 28.74 ± 3.51 % (n = 7, paired t test, p < 0.01) by application of the PKA antagonist H-89 (10 μM) or KT5720 (1 μM), respectively; it was increased 10.96 ± 2.09 % (n = 6, paired t test, p < 0.05) and 14.44 ± 3.22 % (n = 8, paired t test, p < 0.05) by application of the PKC antagonist BIM (1 μM) or d-sphingosine (20 μM), respectively. Here, after pre-application of the PKA antagonist H-89 or KT5720, I Na was reduced 30.54 ± 5.06 % (n = 9) and 26.46 ± 4.17 % (n = 8) by GSK1016790A treatment, respectively, which was markedly different from the inhibition induced by GSK1016790A alone (unpaired t test, p < 0.01 in each case) (Fig. 4a). By contrast, in the presence of the PKC antagonist BIM or d-sphingosine, I Na was reduced 43.67 ± 4.87 % (n = 11) and 47.09 ± 3.17 % (n = 10) by GSK1016790A, respectively, which was similar to GSK1016790A-induced inhibition (unpaired t test, p > 0.05 in each case) (Fig. 4b).

Involvement of PKA in GSK1016790A-inhibited I Na. a, b The GSK1016790A-induced inhibition of I Na was significantly attenuated by the pre-application of a PKA antagonist (H-89 or KT5720) (a), but not by the application of a PKC antagonist (BIM or d-sphingosine) (b); **p < 0.01 vs. GSK1016790A. c GSK1016790A-induced inhibition of I Na was not affected by either activation or inhibition of p38 MAPK. In the presence of anisomycin or SB203580, the inhibition of I Na by GSK1016790A was 51.73 ± 2.01 % (n = 10) and 50.01 ± 4.03 % (n = 11), which did not differ from the inhibition by GSK1016790A alone
In this study, we also tested the role of p38 MAPK in GSK1016790A-inhibited I Na. I Na was decreased by 12.13 ± 3.13 % (from −68.01 ± 7.48 pA/pF to −58.94 ± 5.09 pA/pF, n = 7, paired t test, p < 0.05) when anisomycin (10 μg/ml) was added to the bath solution for 30 min. Moreover, the inhibition of I Na by anisomycin was blocked by pre-application of SB203580 (10 μM), a specific inhibitor of p38 MAPK (SB203580: −64.23 ± 5.28 pA/pF, SB203580 + anisomycin: −61.09 ± 6.13 pA/pF, n = 6, paired t test, p > 0.05). In the presence of anisomycin, I Na was reduced from −59.94 ± 3.80 pA/pF to −28.95 ± 4.11 pA/pF by the application of GSK1016790A, and moreover, I Na was inhibited 50.01 ± 4.03 % by the co-application of SB203580 and GSK1016790A, both of which were not significantly different from that induced by GSK1016790A alone (unpaired t test, p > 0.05 in each case) (Fig. 4c). These results indicate that PKA signaling pathway was selectively responsible for the TRPV4-induced inhibition of I Na.
Effect of GSK1016790A on the Expression of Sodium Channel Subunits
The present study also examined the chronic effect of TRPV4 activation on sodium channel protein expression in mice that were injected with GSK1016790A (GSK-injected mice) for three consecutive days. This time point was chosen because our unpublished data demonstrate that the protein level of glutamate receptor in the hippocampus could be affected in mice treated with GSK1016790A for 3 days. In this study, unlike the inhibition of I Na by acute treatment with a TRPV4 agonist, the protein levels of Nav1.1, Nav1.2, and Nav1.6 increased in GSK-injected mice, compared with those in vehicle-injected mice. By contrast, the protein levels of Nav1.3 and Navβ1 were nearly unaffected by GSK1016790A injection (Fig. 5).

Expression of Na+ channel protein in GSK-injected mice. In GSK-injected mice, the protein levels of Nav1.1 (a), Nav1.2 (b), and Nav1.6 (c) increased, but those of Nav1.3 (d) and Navβ1 (e) did not change; **p < 0.01 vs. control
Discussion
AP firing in hippocampal neurons is crucial to maintain higher brain functions, such as learning and memory. Neurons possess various voltage-gated ion channels, among which VGSCs are crucial for controlling membrane electrical excitability, because they play an essential role in the initiation and propagation of APs [10]. The present study shows that VGSCs may be differentially modulated by acute or sustained activation of TRPV4 in the hippocampus: that is, I Na was inhibited reversibly by the acute application of TRPV4 agonists (Figs. 1 and 2), but the protein levels of Nav1.1, Nav1.2, and Nav1.6 increased upon sustained treatment with TRPV4 agonist (Fig. 5). Our data showed that I Na in hippocampal CA1 pyramidal neurons was inhibited by the application of TRPV4 agonists GSK1016790A and 4α-PDD. GSK1016790A is a recently described TRPV4 agonist, and the present study found a dose-dependent inhibition of I Na by GSK1016790A (Fig. 1e). The inhibition of I Na induced by GSK1016790A and 4α-PDD was almost completely blocked by pre-application of the TRPV4 specific antagonist HC-067047 (Fig. 3a), confirming the TRPV4-induced inhibition of VGSCs. In addition, inactivation-voltage curve of I Na shifted to hyperplolarization upon GSK1016790A or 4α-PDD treatment (Figs. 1d and 2d), whereas G–V curve showed almost no change (Figs. 1c and 2c). These results indicate that activation of TRPV4 could facilitate the inactivation of VGSCs without significantly affecting channel opening and that this facilitation of inactivation is likely responsible for the TRPV4-induced acute inhibition of I Na. In this study, after pre-application of TTX, the current was not changed by GSK1016790A or 4α-PDD (Fig. 3b), suggesting that the TRPV4-induced inhibition of I Na was mediated through TTX-S channels.
In the CNS, functional modulation of VGSCs by a wide variety of neurotransmitters is mediated mainly through the regulation of PKC- and PKA-dependent phosphorylation [12]. For example, acetylcholine and dopamine have been shown to alter the functional properties of VGSCs in neurons via the activation of PKC and PKA, respectively [21, 22]. In the present study, I Na was inhibited by applying agonist of either PKA or PKC, which was consistent with previous reports in hippocampal pyramidal neurons. In the presence of PKA antagonist, the inhibition of I Na by GSK1016790A was markedly blocked, compared with that in the absence of H-89 or KT5720 (Fig. 4a). By contrast, pre-incubation with PKC antagonist, either d-sphingosine or BIM, did not significantly affect GSK1016790A-induced inhibition of I Na (Fig. 4b). Activation of p38 MAPK has been reported to modulate VGSCs (Nav1.6) in hippocampal neurons. Here, with the activation of p38 MAPK, the inhibition of I Na by GSK1016790A was not enhanced, and moreover, GSK1016790A-induced inhibition of I Na was not attenuated by the inhibition of p38 MAPK (Fig. 4c). Collectively, these results suggest that PKA signaling pathway is specifically responsible for inhibition of I Na upon the acute activation of TRPV4. It has been reported that activating TRPV4 by hypotonic stimulation may enhance TTX-S current in TG neurons [16]. This discrepant modulation of TTX-S channels by TRPV4 activation may be attributed to the difference in VGSCs subtypes in the CNS and PNS. TTX-R and voltage-gated calcium channels can be modulated by TRPV4 under hypotonic stimulation through PKA and PKG signaling pathways in TG neurons [15, 18]. However, there is still insufficient evidence concerning how PKA or PKG signaling is regulated by TRPV4 activation, although the interaction of TRPV4 with the components of G-protein-coupled receptor signaling pathway is required for sensing physical stimuli [23]. Activation of TRPV4 induces Ca2+ influx, leading to increase of [Ca2+] i [3]. As the important second messengers, a crosstalk between Ca2+ and cyclic adenosine monophosphate (cAMP) signaling system has been reported in many types of cells [24–27]. TRPV4, acting as an osmotic cellular sensor, may mediate hypotonic stimulation-induced increase of intracellular cAMP level in the tilapia osmoreceptive prolactin cells [28]. TRPV1, another member of TRPV channel family and permeable to Ca2+, has been shown to mediate capsaicin-inhibited VGSCs via activating cAMP/PKA signaling in TG neurons [29]. Therefore, it is suggested that TRPV4-induced activation of PKA may be mediated through modulating intracellular cAMP level. This suggestion is tested, on the other hand, by the experiment that the GSK1016790A-induced inhibition of I Na was partially attenuated by the pre-application of BAPTA-AM, an intracellular Ca2+ chelator (Supplementary Fig. 2).
VGSCs are composed of α and β subunits, with α subunit alone being sufficient to conduct sodium and β subunit modulating the channel activity. Four α subunits of VGSCs, Nav1.1, Nav1.2, Nav1.3, and Nav1.6, are abundant in the CNS. Among them, Nav1.3 is highly expressed in rodent fetal nervous tissues, whereas Nav1.1, Nav1.2, and Nav1.6 are abundant in the juvenile and adult CNS [10, 11]. Unlike the acute inhibition of I Na, the present study showed that the protein levels of Nav1.1, Nav1.2, and Nav1.6 increased in GSK-injected mice, whereas those of Nav1.3 and Navβ1 remained unchanged (Fig. 5). We hypothesize that the increased expression of α subunit upon chronic TRPV4 activation may compensate for the functional inhibition of I Na by acute TRPV4 activation. Additionally, the increase in α subunit protein expression indicates that an increase of I Na is possible when TRPV4 activation is sustained or it is over-activated. Previous studies report that activating TRPV1 inhibits TTX-R current in primary sensory neurons, which may account for the analgesia of capsaicin [29, 30]. However, the increased TRPV1 and TTX-R current is required for developing chronic hyperalgesia [31]. In the future study, more experiments are required to clarify whether the increased α-subunit expression upon the chronic TRPV4 activation will lead to an increased of I Na.
Under normal conditions, there are two forms of firing in hippocampal pyramidal neurons, single AP or bursts of multiple APs, with the latter being more important in both the processing and storage of neuronal information [32]. As a calcium-permeable channel, activation of TRPV4 induces an inward current, helping depolarize the cell membrane, and thus TRPV4 is reported to facilitate spike induction in hippocampal neurons [1, 3, 8]. The spontaneous firing rates of retinal ganglion cells are increased by treatment with TRPV4 agonists, which is accompanied with an increase of [Ca2+] i [6]. VGSCs play an important role in the depolarization of AP, but the contribution of other factors to the generation or bursts of AP cannot be neglected. For example, in the region of axon initial segment of hippocampal pyramidal neurons, KCNQ potassium channels determine the threshold for firing an AP [33]. Somatodendritic A-type voltage-gated potassium channels are key regulators of neuronal excitability, contributing to the resting membrane potential and AP repolarization and functioning to modulate the frequency of repetitive firing, the thresholds for AP generation, and the back-propagation of AP into dendrites [33–36]. In hippocampal pyramidal neurons, the generation of an afterdepolarization (ADP) is known to prolong the somatic depolarization required for the initiation of bursts of APs [37]. Multiple ionic factors, including a persistent sodium current, calcium current, and KCNQ potassium current, are involved in ADP [38–40]. Meanwhile, the neural circuitry in the hippocampus is crucial for modulating neuronal excitability. The activation of TRPV4 has been shown to facilitate glutamatergic transmission in the hippocampus and promote glutamate receptor activation (including N-methyl-d-aspartate receptors (NMDARs) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs)) in hippocampal pyramidal neurons [9, 20, 41]. In TG neurons, more evoked APs are produced by activating TRPV4, although TTX-R current is inhibited [7, 15]. Additionally, blocking PKA attenuates TRPV4-inhibited TTX-R current markedly, but does not affect TRPV4-increased AP generation [7, 15]. Therefore, upon acute TRPV4 activation, the hyperexcitability of hippocampal neurons may result from the activation of TRPV4 per se, from the modulations of ion channels/receptors and from synaptic integration. As discussed above, the protein levels of VGSCs were increased by chronic treatment with a TRPV4 agonist. Therefore, when TRPV4 activation is sustained or TRPV4 is hyper-activated, changes in the protein levels of ion channels may also be responsible for TRPV4-induced hyperexcitability.
Recently, it has been reported that hyperthermia-induced seizures recorded in the larval zebrafish forebrain are blocked by the application of TRPV4 and NMDAR antagonists, revealing a possible role for TRPV4 in seizure [42]. Concerning the characteristics of multipolar activation, TRPV4-induced regulation of ion channels or receptors may play an important role in pathological conditions, such as seizure and cerebral ischemia [20, 42], thus making TRPV4 a promising target for the development of new drugs.
References
Nilius B, Szallasi A (2014) Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacol Rev 66(3):676–814
Vincent F, Duncton MA (2011) TRPV4 agonists and antagonists. Curr Top Med Chem 11(17):2216–2226
Nilius B, Voets T (2013) The puzzle of TRPV4 channelopathies. EMBO Rep 14(2):152–163
Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, Altier C, Cenac N, Zamponi GW, Bautista-Cruz F, Lopez CB, Joseph EK, Levine JD, Liedtke W, Vanner S, Vergnolle N, Geppetti P, Bunnett NW (2007) Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol 578(Pt 3):715–733
Egbuniwe O, Grover S, Duggal AK, Mavroudis A, Yazdi M, Renton T, Di Silvio L, Grant AD (2014) TRPA1 and TRPV4 activation in human odontoblasts stimulates ATP release. J Dent Res 93(9):911–917
Ryskamp DA, Witkovsky P, Barabas P, Huang W, Koehler C, Akimov NP, Lee SH, Chauhan S, Xing W, Rentería RC, Liedtke W, Krizaj D (2011) The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J Neurosci 31(19):7089–7101
Chen L, Liu C, Liu L (2009) Osmolality-induced tuning of action potentials in trigeminal ganglion neurons. Neurosci Lett 452(1):79–83
Shibasaki K, Suzuki M, Mizuno A, Tominaga M (2007) Effects of body temperature on neural activity in the hippocampus: regulation of resting membrane potentials by transient receptor potential vanilloid 4. J Neurosci 27(7):1566–1575
Li L, Yin J, Jie PH, Lu ZH, Zhou LB, Chen L, Chen L (2013) Transient receptor potential vanilloid 4 mediates hypotonicity-induced enhancement of synaptic transmission in hippocampal slices. CNS Neurosci Ther 19(11):854–862
Yu FH, Catterall WA (2003) Overview of the voltage-gated sodium channel family. Genome Biol 4(3):207
Catterall WA (2000) From ionic currents to molecular mechanisms: the structure and function of voltagegated sodium channels. Neuron 26(1):13–25
Scheuer T (2011) Regulation of sodium channel activity by phosphorylation. Semin Cell Dev Biol 22(2):160–165
Gasser A, Cheng X, Gilmore ES, Tyrrell L, Waxman SG, Dib-Hajj SD (2010) Two Nedd4-binding motifs underlie modulation of sodium channel Nav1.6 by p38 MAPK. J Biol Chem 285(34):26149–26161
Wittmack EK, Rush AM, Hudmon A, Waxman SG, Dib-Hajj SD (2005) Voltage-gated sodium channel Nav1.6 is modulated by p38 mitogen-activated protein kinase. J Neurosci 25(28):6621–6630
Chen L, Liu C, Liu L, Cao X (2009) Changes in osmolality modulate voltage-gated sodium channels in trigeminal ganglion neurons. Neurosci Res 64:199–207
Lin L, Liu C, Chen L, Chen L (2011) Hypotonicity modulates tetrodotoxin-sensitive sodium current in trigeminal ganglion neurons. Mol Pain 7:27–32
Chen L, Liu C, Liu L (2008) The modulation of voltage-gated potassium channels by anisotonicity in trigeminal ganglion neurons. Neuroscience 154:482–495
Chen L, Liu C, Liu L (2008) Changes in osmolality modulate voltage-gated calcium channels in trigeminal ganglion neurons. Brain Res 1208:56–66
Liu L, Chen L, Liedtke W, Simon SA (2007) Changes in osmolality sensitize the response to capsaicin in trigeminal sensory neurons. J Neurophysiol 97:2001–2015
Li L, Qu W, Zhou L, Lu Z, Jie P, Chen L, Chen L (2013) Activation of transient receptor potential vanilloid 4 increases NMDA-activated current in hippocampal pyramidal neurons. Front Cell Neurosci 7:17–26
Cantrell AR, Smith RD, Goldin AL, Scheuer T, Catterall WA (1997) Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel αsubunit. J Neurosci 17:7330–7338
Cantrell AR, Ma JY, Scheuer T, Catterall WA (1996) Muscarinic modulation of sodium current by activation of protein kinase C in rat hippocampal neurons. Neuron 16:1019–1026
Lidtke W, Tobin DM, Bargmann CI, Friedman JM (2003) Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorbditis elegans. Proc Natl Acad Sci U S A 100(Suppl 2):14531–14536
Borodinsky LN, Spitzer NC (2006) Second messenger pas de deux: the coordinated dance between calcium and cAMP. Sci STKE (336):pe22
Fridlyand LE, Harbeck MC, Roe MW, Philipson LH (2007) Regulation of cAMP dynamics by Ca2+ and G protein-coupled receptors in the pancreatic beta-cell: a computational approach. Am J Physiol Cell Physiol 293(6):C1924–C1933
Willoughby D, Cooper DM (2006) Ca2+ stimulation of adenylyl cyclase generates dynamic oscillations in cyclic AMP. J Cell Sci 119(Pt 5):828–836
Landa LR Jr, Harbeck M, Kaihara K, Chepurny O, Kitiphongspattana K, Graf O, Nikolaev VO, Lohse MJ, Holz GG, Roe MW (2005) Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 beta-cell line. J Biol Chem 280(35):31294–31302
Seale AP, Watanabe S, Grau EG (2012) Osmoreception: perspectives on signal transduction and environmental modulation. Gen Comp Endocrinol 176(3):354–360
Liu L, Oortgiesen M, Li L, Simon SA (2001) Capsaicin inhibits activation of voltage-gated sodium currents in capsaicin-sensitive trigeminal ganglion neurons. J Neurophysiol 85(2):745–758
Onizuka S, Yonaha T, Tamura R, Hosokawa N, Kawasaki Y, Kashiwada M, Shirasaka T, Tsuneyoshi I (2011) Capsaicin indirectly suppresses voltage-gated Na+ currents through TRPV1 in rat dorsal root ganglion neurons. Anesth Analg 112(3):703–709
Chen WN, Lee CH, Lin SH, Wong CW, Sun WH, Wood JN, Chen CC (2014) Roles of ASIC3, TRPV1, and NAv1.8 in the transition from acute to chronic pain in a mouse model of fibromyalgia. Mol Pain 10:40–54
Lisman JE (1997) Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci 20(1):38–43
Bean BP (2007) The action potential in mammalian central neurons. Nat Rev Neurosci 8(6):451–465
Johnston D, Hoffman DA, Colbert CM, Magee JC (1999) Regulation of back-propagating action potentials in hippocampal neurons. Curr Opin Neurobiol 9(3):288–292
Johnston J, Forsythe ID, Kopp-Scheinpflug C (2010) Going native: voltage-gated potassium channels controlling neuronal excitability. J Physiol 588(Pt 17):3187–3200
Vacher H, Mohapatra DP, Trimmer JS (2008) Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev 88(4):1407–1447
Jensen MS, Azouz R, Yaari Y (1996) Spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol 492(Pt 1):199–210
Magee JC, Carruth M (1999) Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. J Neurophysiol 82(4):1895–1901
Azouz R, Jensen MS, Yaari Y (1996) Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol 492(Pt1):211–223
Yue C, Yaari Y (2004) KCNQ/M channels control spike afterdepolarization in hippocampal neurons. J Neurosci 24(19):4614–4624
Cao DS, Yu SQ, Premkumar LS (2009) Modulation of transient receptor potential vanilloid 4-mediated membrane currents and synaptic transmission by protein kinase C. Mol Pain 5:5–17
Hunt RF, Hortopan GA, Gillespie A, Baraban SC (2012) A novel zebrafish model of hyperthermia-induced seizures reveals a role for TRPV4 channels and NMDA-type glutamate receptors. Exp Neurol 237(1):199–206
Acknowledgments
This work was supported by National Natural Science Foundation of China (31271206), Research Award Fund for Outstanding Young Teachers in Nanjing Medical University (JX2161015033) and Qing Lan Project of Jiangsu province (2014–2017).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
I Na and GSK1016790A-evoked current recorded in hippocampal CA1 pyramidal neurons. When recording TRPV4-mediated current, 500 nM GSK1016790A and 0.3 μM TTX were added to mASCF and a ramp protocol depolarizing from −80 to +80 mV over 700 ms was used. Typical recordings show that I Na (a) and GSK1016790A-induced current (b) were recorded in the same neuron. (GIF 5 kb)
Supplementary Fig. 2
Effect of BAPTA-AM on GSK1016790A-induced inhibition of I Na in hippocampal CA1 pyramidal neurons. GAK1016790A-induced inhibition of I Na was partially blocked by pre-application of BAPTA-AM (10 μM). The dose of BATPA-AM was used as previously reported [1] *p<0.05 vs. GSK1016790A 1. Aflaki M, Qi XY, Xiao L, Ordog B, Tadevosyan A, Luo X, Maguy A, Shi Y, Tardif JC, Nattel S (2014) Exchange protein directly activated by cAMP mediates slow delayed-rectifier current remodeling by sustained β-adrenergic activation in guinea pig hearts. Circ Res 114(6):993–1003. (GIF 18 kb)
Rights and permissions
About this article

Cite this article
Hong, Z., Jie, P., Tian, Y. et al. Transient Receptor Potential Vanilloid 4-Induced Modulation of Voltage-Gated Sodium Channels in Hippocampal Neurons. Mol Neurobiol 53, 759–768 (2016). https://doi.org/10.1007/s12035-014-9038-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-9038-5