
Abstract
We investigated the pharmacological actions of a slow-releasing H2S donor, GYY 4137; a substrate for the biosynthesis of H2S, l-cysteine and its precursor, N-acetylcysteine on potassium (K+; 50 mM)-evoked [3H]D-aspartate release from bovine isolated retinae using the Superfusion Method. GYY 4137 (10 nM–10 µM), l-cysteine (100 nM–10 µM) and N-acetylcysteine (10 µM–1 mM) elicited a concentration-dependent decrease in K+-evoked [3H]D-aspartate release from isolated bovine retinae without affecting basal tritium efflux. At equimolar concentration of 10 µM, the rank order of activity was as follows: l-cysteine > GYY 4137 > N-acetylcysteine. A dual inhibitor of the biosynthetic enzymes for H2S, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE), amino-oxyacetic acid (AOA; 3 mM) reversed the inhibitory responses caused by GYY 4137, l-cysteine and N-acetylcysteine on K+-evoked [3H]D-aspartate release. Glibenclamide (300 µM), an inhibitor of KATP channels blocked the inhibitory action of GYY 4137 and l-cysteine but not that elicited by N-acetylcysteine on K+-induced [3H]D-aspartate release. The inhibitory effect of GYY 4137 and l-cysteine on K+-evoked [3H]D-aspartate release was reversed by the non-specific inhibitor of nitric oxide synthase (NOS), l-NAME (300 µM). Furthermore, a specific inhibitor of inducible NOS (iNOS), aminoguanidine (10 µM) blocked the inhibitory action of l-cysteine on K+-evoked [3H]D-aspartate release. We conclude that both donors and substrates for H2S production can inhibit amino acid neurotransmission in bovine isolated retinae, an effect that is dependent, at least in part, upon the intramural biosynthesis of this gas, and on the activity of KATP channels and NO synthase.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although hydrogen sulfide (H2S) was previously known as a toxic gas, it is now widely accepted as a signaling molecule that has—physiological roles in the central nervous, cardiovascular and the immune systems [1]. H2S is endogenously biosynthesized from l-cysteine and d-cysteine by cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and cysteine aminotransferase (CAT) and d-aminotransferase in combination with 3-mercaptosulfurtransferase (3MST). In the central nervous system, H2S has been shown to facilitate the induction of hippocampal long-term potentiation by enhancing the activity of N-methyl-d-aspartate (NMDA) receptors in neurons and by the stimulation of calcium waves in astrocytes [2, 3]. H2S can protect neurons from the deleterious action of oxygen-derived free radicals by enhancing the activity of glutathione synthase, scavenging reactive oxygen species and preventing the excessive increase in intracellular calcium concentration [4,5,6]. H2S has also been reported to exhibit neurotransmitter-like activities because of its ability to regulate synaptic activities induced by steroid hormones and other neurotransmitters [7, 8]. Evidence of a pathophysiological role for H2S has been demonstrated in neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, vascular dementia, Huntington’s disease and amyotrophic lateral sclerosis [9,10,11]. In the eye, the presence of enzymes of the trans-sulfuration pathway (CBS, CSE and 3MST) have been localized in tissues of both anterior and posterior segments [12,13,14]. There is evidence that deficiency of CBS due to a mutation in the gene encoding the enzyme can lead to several eye disorders such as glaucoma and retinal detachment [15]. High levels of homocysteine, a substrate for H2S biosynthesis have been observed in the aqueous humor, tear fluid and plasma of patients with primary open-angle glaucoma [16, 17]. Furthermore, elevated concentrations of H2S have been demonstrated in the vitreous body and plasma of patients with proliferative diabetic retinopathy [18]. The ability of H2S to exert pharmacological actions on mammalian ocular tissues from both anterior and posterior segments has been reported by us and other investigators [6, 19,20,21,22,23,24,25,26,27]. For instance, we have evidence that H2S donors such as sodium hydrosulfide (NaHS) can relax both isolated mammalian irides and long posterior ciliary arteries [19,20,21,22]. Moreover, H2S donors enhanced cyclic AMP production in bovine and porcine isolated neural retinae and retinal pigment epithelial cells, an effect that was dependent upon the biosynthesis of H2S by CSE and CBS and, on the activity of KATP channels [23, 24]. In other studies, we demonstrated that H2S donors can inhibit both electrically-evoked [3H]-norepinephrine release and endogenous catecholamine concentrations from isolated porcine iris-ciliary bodies in a concentration-related manner [25]. Interestingly, the inhibitory action of H2S donors on norepinephrine release was antagonized by inhibitors of CSE and CBS suggesting that the observed response was due, at least in part, on the intramural biosynthesis of this gas [25]. In bovine and porcine isolated retinae, the H2S donors, NaHS and Na2S inhibited K+-evoked [3H]D-aspartate release by a mechanism that was dependent upon the intramural biosynthesis of the gas [26]. Since an increase in retinal glutamate concentrations have been linked to excitotoxicity, the ability of H2S to inhibit glutamate release suggests a potential neuroprotective action of this gas in the retinae. Indeed, evidence for a neuroprotective effect of H2S in the retinae has been reported by other investigators [6, 27]. Thus, we sought to delineate the role of other H2S producing compounds on excitatory neurotransmission. The aim of the present study was twofold: (a) to investigate the pharmacological actions of a slow-releasing H2S donor, GYY 4137 and l-cysteine, a substrate for H2S biosynthesis on K+-evoked [3H]D-aspartate release, and (b) to examine the role of KATP channels and NO in the responses elicited by these compounds on neurotransmitter release.
Methods
Tissue Preparation
Freshly harvested bovine eyeballs were obtained from a local slaughterhouse (Greater Omaha Packing or J. F. O’Neil packing company) within 3 h after enucleation and delivered to the laboratory in an ice bucket. The anterior segment tissues and the vitreous humor were carefully removed and the eyecup everted to isolate the retinae.
[3H]D-Aspartate Release Studies
The method used for assessment of [3H]D-aspartate release studies was similar to the one previously described by us [26, 28,29,30] and others [31, 32]. Briefly, bovine retinae were incubated in oxygenated (95% O2/5% CO2) Krebs buffer solution containing 200 nM [3H]D-aspartate at 37 °C for 1 h. The Krebs buffer solution was prepared as follows (mM): NaCl 118; KCl 4.8; CaCl2 2.5; KH2PO4 1.2; NaHCO3 25; MgSO4 2.0; dextrose 10 (pH 7.4). The cyclooxygenase inhibitor, flurbiprofen (3 µM) was added to the Krebs buffer solution to inhibit the endogenous production of prostanoids [30]. Following the incubation period, the radiolabeled retinae were rinsed three times with 25 mL of Krebs buffer solution for 5 min each. The radiolabeled tissues were then mounted between nylon mesh cloths and placed in thermostatically-controlled Superfusion Chambers. In order to establish a stable baseline of spontaneous tritium efflux, retinal tissues were superfused with oxygenated Krebs buffer solution at the rate of 1.5 mL/min for 60 min. Fractional collection was commenced at the same rate (1.5 mL/min) and effluent collected every six min. Neurotransmitter ([3H]D-aspartate) release was evoked by the application of iso-osmotic concentrations of KCl (50 mM) stimuli applied between 84 and 96 min (S1) and 132–144 min (S2) after onset of superfusion. Superfusates were assessed for radioactivity by liquid scintillation spectrometry (LS 6500 Multipurpose Scintillation Counter, Beckman Coulter) and [3H]D-aspartate release was estimated by subtraction of the extrapolated basal tritium efflux from total tritium released during the 20 min period after the onset of stimulation. The basal tritium efflux declined linearly between pre-stimulation and post-stimulation fractions.
To determine the effects of a H2S-releasing compound on K+-evoked [3H]D-aspartate release, the compound was applied 12 min before S2 (120 min after start of superfusion). To examine the effect of enzyme inhibitors and ion channel blockers on responses to H2S-releasing compounds, the inhibitors/blockers were added 40 min from onset of Superfusion and were present during both S1 and S2 stimulation periods.
Data Analysis
Results are expressed as absolute S2/S1 ratios of the K+-evoked [3H]D-aspartate release in control and treated preparations. Values are given as the arithmetic means ± standard error of the mean (SEM). Significance of differences between control and test values was tested using the analysis software by analysis of variance (ANOVA) test followed by Dunnett’s test (Graph Pad Prism software San Diego, CA). The level of significance selected was at least P < 0.05.
Results
Effects of Compounds on [3H]D-Aspartate Release
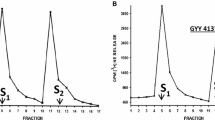
Application of an iso-osmotic high potassium chloride (K+; 50 mM) stimuli to isolated retinae loaded with [3H]D-aspartate and mounted in Superfusion Chambers elicited an overflow of tritium over basal levels, a response that can be repeated more than twice (S1, S2, S3, etc). In control experiments in which no compound was present, application of two subsequent K+ stimuli yielded two peaks of [3H]D-aspartate overflow, as depicted in Fig. 1 (Panel a). The area under curve (AUC) calculated for both the peaks (S1 and S2) yielded S2/S1 ratios of 1.033 ± 0.03 (n = 12). As illustrated in Fig. 1 (Panel b), application of GYY 4137 (10 µM) 12 min before the second train of field stimulation (S2) caused a marked inhibition of K+-evoked [3H]D-aspartate release from bovine retinae. A similar profile of inhibitory response was observed with l-cysteine and N-acetylcysteine on stimulated [3H]D-aspartate release from isolated bovine retinae (data not shown).

Effect of slow hydrogen sulfide (H2S) releasing compound, GYY 4137 (10 µM) on KCl (K+, 50 mM)-induced release of [3H]D-aspartate from isolated, superfused bovine retinae. K+ stimuli were applied at fractions 5/6 (S1) and 13/14 (S2). a control (no compound present); b GYY 4137 (10 µM) applied 12 min before S2. Fractions of the superfusate were collected at 6 min. intervals and analyzed for radioactivity as described under Methods
Since there is evidence that fast-releasing H2S donors (such as NaHS and Na2S) can alter the release of norepinephrine from sympathetic nerves in the anterior uvea and excitatory amino acids from iris-ciliary bodies and mammalian retinae, in vitro [25, 26], we investigated the effect of the slow-releasing H2S donor, GYY 4137 (0.01–10 µM) on K+-induced [3H]D-aspartate release from isolated bovine retinae (Fig. 2, Panel a). GYY 4137 exhibited a concentration-dependent inhibition of [3H]D-aspartate release yielding a maximum inhibition of 21.48 ± 2.53% (n = 5; p < 0.05) at a concentration of 10 µM.

Inhibitory effect of hydrogen sulfide (H2S) releasing compounds on K+-evoked [3H]D-aspartate release from isolated, bovine retinae. a the slow H2S donor, GYY 4137 (0.01–10 µM); b the endogenous substrate for H2S synthesis, l-cysteine (0.1–10 µM); c l-cysteine precursor, N-acetylcysteine (0.01–1 mM). Vertical bars represent means ± SEM. Number of observations is in parenthesis. *P < 0.05; ***P < 0.001 significantly different from the control
We next examined the effect of l-cysteine, a substrate for endogenous biosynthesis of H2S via the enzymes, CSE and CBS, on [3H]D-aspartate release. In the concentration range 0.1–10 µM, l-cysteine inhibited K+-induced [3H]D-aspartate release in a concentration-dependent manner, achieving a maximum inhibition of 54.28 ± 3.61% (n = 4; p < 0.001) at 10 µM and an IC50 value of 9.2 µM (Fig. 2, Panel b).
To further delineate the role of endogenous production of H2S on the inhibitory effect on neurotransmission, we examined the effect of N-acetylcysteine, an acetylated derivative of the amino acid l-cysteine and a precursor to l-cysteine on K+-induced [3H]D-aspartate release. N-acetylcysteine (10 µM–1 mM) also elicited a concentration-dependent inhibition of [3H]D-aspartate release from bovine isolated retinae yielding a maximum inhibition of 29.63 ± 14.7% (n = 5; p < 0.05) at the 1 mM concentration and an IC25 value of 843.5 µM (Fig. 2, Panel c). At an equimolar concentration of 10 µM, the rank order of inhibitory activity elicited by the H2S producing compounds was as follows: l-cysteine > GYY 4137 > N-Acetylcysteine.
Effect of an Inhibitor of H2S-Biosynthesis
There is evidence that amino-oxyacetic acid (AOA) can inhibit both CBS and CSE, enzymes involved in the biosynthesis of H2S [33]. We examined the effect of AOA on the inhibitory action of the H2S producing compounds, l-cysteine, N-acetylcysteine and GYY 4137. Whereas AOA (3 mM) had no effect on K+-induced [3H]D-aspartate release, it reversed the inhibitory action of GYY 4137, l-cysteine and N-acetylcysteine on neurotransmitter release (Fig. 3, Panels a–c).

Effect of the cystathionine β-synthase inhibitor, amino-oxyacetic acid (AOA; 3 mM) on the inhibitory effect of hydrogen sulfide (H2S) releasing compounds on K+-evoked [3H]D-aspartate release from isolated, bovine retinae. a control or GYY 4137 (10− 6 and 10− 5 M) in presence or absence of AOA (3 mM); b control or l-cysteine (10− 6 and 10− 5 M) in presence or absence of AOA (3 mM); c control or N-acetylcysteine (10− 4 and 10− 3 M) in presence or absence of AOA (3 mM). Vertical bars represent means ± SEM. Number of observations is in parenthesis. *P < 0.05; ***P < 0.001 significantly different from the control; ^P < 0.05; ^^^P < 0.001 significantly from H2S releasing compound in presence of AOA (3 mM)
Effect of a Potassium Channel (KATP) Blocker
In the next series of experiments, we examined the potential involvement of KATP channels on the inhibitory action of the H2S donors on K+-evoked [3H]D-aspartate release. There is evidence that glibenclamide, a KATP channel inhibitor can antagonize the pharmacological actions of H2S producing compounds in several tissues [34]. Consequently, we investigated the effect of glibenclamide (300 µM) on the inhibitory responses elicited by the H2S producing compounds. Although glibenclamide (300 µM) had no effect on K+-induced [3H]D-aspartate release, it reversed the inhibitory action of l-cysteine and GYY 4137 on neurotransmitter release (Fig. 4, Panels a, b). Interestingly, glibenclamide had no significant (P > 0.05) effect on the inhibitory action of N-acetylcysteine on neurotransmitter release (data not shown).

Effect of the KATP channel blocker, glibenclamide (0.3 mM) on the inhibitory effect of hydrogen sulfide (H2S) releasing compounds on K+-evoked [3H]D-aspartate release from isolated, bovine retinae. a control or GYY 4137 (10− 6 and 10− 5 M) in presence or absence of glibenclamide (0.3 mM); b control or l-cysteine (10− 6 and 10− 5 M) in presence or absence of glibenclamide (0.3 mM). Vertical bars represent means ± SEM. Number of observations is in parenthesis. *P < 0.05; ***P < 0.001 significantly different from the control; ^P < 0.05; ^^^P < 0.001 significantly different from H2S releasing compound in presence of glibenclamide (0.3 mM)
Effect of Inhibitors of Nitric Oxide Synthase
The gaseous transmitter, nitric oxide (NO) has been reported to interact with H2S in various mammalian tissues and organs as reviewed by [35]. Therefore, in a series of experiments, we examined the effect of a non-specific inhibitor of nitric oxide synthase (NOS), l-NAME on the inhibitory responses elicited by l-cysteine and GYY 4137 on K+-evoked [3H]D-aspartate release. On its own, l-NAME (300 µM) had no effect on K+-induced [3H]D-aspartate release. However, l-NAME (300 µM) completely reversed the inhibitory effects of l-cysteine and GYY 4137 on [3H]D-aspartate release (Fig. 5, Panels a, b) .

Effect of the non-specific nitric oxide synthase (NOS) inhibitor, l-NAME (0.3 mM) and the inducible NOS (iNOS) inhibitor, aminoguanidine (10 µM) on the inhibitory effect of hydrogen sulfide (H2S) releasing compounds on K+-evoked [3H]D-aspartate release from isolated, bovine retinae. a control or GYY 4137 (10− 6 and 10− 5 M) in presence or absence of l-NAME (0.3 mM). b control or l-cysteine (10− 6 and 10− 5 M) in presence or absence of l-NAME (0.3 mM). c control or l-cysteine (10− 7 to 10− 5 M) in presence or absence of aminoguanidine (10 µM). Vertical bars represent means ± SEM. Number of observations is in parenthesis. *P < 0.05; **P < 0.001 significantly different from the control; ^P < 0.05; ^^^P < 0.001 significantly different from H2S releasing compound in presence of NOS inhibitor
To further elucidate the role of NO in the inhibitory effect of H2S-releasing compounds, we investigated the effect of the specific inhibitor of inducible NOS (iNOS), aminoguanidine on the inhibitory response elicited by l-cysteine [36, 37]. Although aminoguanidine (10 µM) had no effect on the neurotransmitter release, it reversed the inhibitory effects of l-cysteine (1 and 10 µM) on [3H]D-aspartate release (Fig. 6).

Schematic representation of putative mechanisms by which hydrogen sulfide (H2S) regulates excitatory neurotransmitter release in isolated bovine retina. The slow releasing H2S donor, GYY 4137, and the substrate for endogenous production of H2S, l-cysteine and its prodrug, N-acetylcysteine produce the gasotransmitter to attenuate K+-induced [3H]D-aspartate release in bovine retina. This effect is facilitated by mechanisms that involve either (1) the H2S biosynthetic enzymes, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) enzymes; (2) endogenous generation of nitric oxide (NO) gasotransmiter; or (3) opening of potassium (K)ATP (KATP) channels. N-Acetylcysteine is presumed to release l-cysteine, in situ, which then attenuates neurotransmitter release. However, the KATP antagonist did not reverse its effect, suggesting that other mechanisms may be involved in the effect elicited by N-acetylcysteine (A). The interaction between NO and CSE/CBS (B) and/or KATP channels (C) has not been completely elucidated in these tissues
Discussion
In the central nervous system, H2S has been reported to play neuro-modulatory and neuroprotective roles by virtue of its effect on synaptic transmission [2, 3] and its preventive action against toxic insults such as oxidative stress, excitotoxicity and neuronal injury [4,5,6, 38]. H2S has also been shown to play a role in the pathophysiology of neurodegenerative disease such as Alzheimer’s disease, Parkinson’s disease, vascular dementia, Huntington’s disease and amyotrophic lateral sclerosis [9,10,11]. The three enzymes responsible for the biosynthesis of H2S from amino acids, l-cysteine and d-cysteine (i.e., CBS, CSE and 3MST) have been localized in tissues of the eye [12,13,14]. A deficiency in the activity of CBS has been implicated in some eye diseases such as glaucoma and retinal detachment [15]. On the other hand, high concentrations of H2S have been reported in the vitreous body and plasma of patients with diabetic retinopathy [18]. Taken together, these observations support a pathophysiological role for H2S in the eye. Studies to determine the pharmacological actions of H2S in biological systems have utilized sulfide salts such as NaHS and Na2S as gas donors because of their ability to generate H2S in vivo [2]. These sulfide salts have been shown to release large amounts of H2S in a short duration of time. In contrast, the release of endogenous H2S from cells may occur at a much slower rate than that from sulfide salts [39, 40]. Indeed, compounds have been synthesized (e.g., GYY 4137, AP67, AP72) that can release H2S slowly in biological systems [40, 41]. Using NaHS and Na2S as gas donors, we have evidence that H2S can relax both mammalian irides and long posterior ciliary arteries [19,20,21,22] and can increase cyclic AMP production in bovine and porcine isolated retinae and retinal pigment epithelial cells [23, 24]. Both NaHS and Na2S inhibited electrically-induced [3H]-norepinephrine release and reduced basal catecholamine levels in porcine isolated irides [25] and K+-evoked [3H]D-aspartate overflow from bovine and porcine retinae, in vitro [26]. In the present study, we examined the effects of three compounds on K+-depolarization induced release of [3H]D-aspartate from bovine isolated retinae: a slow-releasing H2S donor, GYY 4137; a substrate for H2S biosynthesis, l-cysteine; and a precursor of l-cysteine, N-acetylcysteine. We employed [3H]D-aspartate as a marker for glutamate because this amino acid has been reported to utilize the same transport system as l-glutamate and l-aspartate and, therefore, it has been presumed to be an appropriate marker for neurons that employ glutamate and/or aspartate as neurotransmitters in the retinae [31, 32, 42].
In the present study, all three compounds tested (GYY 4137, l-cysteine and N-acetylcysteine) inhibited K+-evoked [3H]D-aspartate release from bovine isolated retinae without affecting basal tritium efflux. At an equimolar concentration of 10 µM, the rank order of inhibitory activity on K+-evoked [3H]D-aspartate release was: l-cysteine > GYY 4137 > N-acetylcysteine. The ability of the slow-releasing H2S compound, GYY 4137 to attenuate [3H]D-aspartate release is consistent with earlier observations made using fast-releasing gas compounds such as NaHS and Na2S in bovine and porcine isolated neural retinae [26]. It is pertinent to note that H2S-releasing compounds have also been shown to inhibit [3H]-norepinephrine from porcine isolated iris-ciliary bodies [25]. It is, however, unclear whether the mechanisms involved in the inhibition of neurotransmitter release by H2S-releasing compounds in noradrenergic and glutamatergic neurons are identical. Be that as it may, the finding that both the slow-releasing H2S compounds and substrates of the H2S biosynthetic pathways (l-cysteine and N-acetylcysteine) inhibited K+-depolarization evoked [3H]D-aspartate overflow suggest that this gas exerts a negative regulatory role on glutamatergic transmission in the retinae. Interestingly, the ability of H2S to regulate neurotransmission has been described in various biological systems such as the cat carotid body, pig intravesical ureter, brain and endocrine system [7, 8, 43,44,45].
It has been well established that H2S can be produced by three enzymes, CBS, CSE and 3MST, along with cysteine aminotransferase (CAT), which is identical to aspartate aminotransferase (as reviewed by [46]). In 2013, Asimakopoulou et al. showed evidence that, amino-oxyacetic acid (AOA) can inhibit both CBS and CSE activities [33]. In the present study, we found that while AOA had no effect on basal tritium efflux, it blocked the inhibitory action of GYY 4137, l-cysteine and N-acetylcysteine on K+-induced [3H]D-aspartate release without affecting basal tritium efflux. A blockade of NaHS-induced attenuation of K+-evoked [3H]D-aspartate release in isolated mammalian retinae and field-stimulated [3H]-norepinephrine overflow in isolated porcine irides by the CSE inhibitor, propargylglycine has also been reported by Opere et al. [26] and Kulkarni et al. [25]. It is noteworthy that AOA blocked responses elicited by both the H2S-releasing compound and substrates in the present study indicating that endogenously produced H2S could account, at least in part, for the observed inhibitory action of these compounds on glutamatergic neurotransmission in the bovine retinae.
A potential mechanism of action of H2S in mammalian tissues and organs is via its action on ion channels such potassium or calcium channels [47, 48]. In the vasculature, ATP-sensitive K+ channels or voltage-dependent K+ channels have been implicated in the relaxations induced by H2S donors such as NaHS [21, 22, 49]. In the present study, the KATP channel inhibitor, glibenclamide blocked the inhibitory effect of GYY 4137 and l-cysteine on K+-evoked [3H]D-aspartate release from bovine isolated retinae without affecting basal tritium efflux. Glibenclamide has also been reported to block NaHS-induced decrease in insulin release from Syrian hamster pancreatic β-cells (HIT-T15 cells) [50]. These authors found that NaHS, by increasing K+ efflux will lead to hyperpolarization which then prevents the opening of voltage-gated calcium channels. The subsequent prevention of calcium influx by NaHS leads to a decrease in the release of insulin from the HIT-T15 cells [50]. The observation in the present study that the inhibitory action of a H2S-releasing compound and its substrate on glutamatergic neurotransmission can be antagonized by glibenclamide supports the data reported by Ali et al. [50] that KATP channels are involved in the effects of H2S on neurotransmission. Glibenclamide has also been shown by us and other investigators to block the pharmacological actions of H2S-releasing compound in ocular and non-ocular tissues [21, 22, 49,50,51,52]. Surprisingly, we found that the inhibition of K+-induced [3H]D-aspartate overflow by the l-cysteine precursor, N-acetylcysteine was not blocked by glibenclamide. It is unclear why glibenclamide had no effect on the inhibition of evoked excitatory amino acid transmission caused by N-acetylcysteine. It may well be that at the concentration of N-acetylcysteine employed in the present study, this compound may involve other non-specific actions such as those affecting the cystine-glutamate transporter [53].
It is well known that H2S can interact with other gaseous molecules such as NO and CO to produce physiological and pharmacological actions in biological tissues and organs [34]. In addition to interaction at the level of enzyme activity between gaseous molecules such as H2S and NO, Whiteman et al. proposed that a chemical reaction between these gases can occur leading to the formation of nitrosothiols [54]. In the eye, an interaction between H2S and NO has been reported in rabbit ophthalmic artery [55] and bovine isolated posterior ciliary arteries [26]. In the present study, the NOS inhibitor, l-NAME blocked the inhibitory actions of GYY 4137 and l-cysteine on K+-evoked [3H]D-aspartate release without affecting basal tritium efflux. Furthermore, the specific inhibitor of iNOS, aminoguanidine antagonized the inhibitory effect of l-cysteine on K+-depolarization-induced [3H]D-aspartate release. A similar observation was made by Moustafa and Habara in rat pancreatic acini where NaHS induced an increase in intracellular calcium release was inhibited by l-NAME [56]. These authors also showed evidence that H2S can directly stimulate NO production in a dose-dependent manner [56]. Taken together, our findings support the view that H2S and NO can act synergistically to inhibit glutamatergic neurotransmission in the bovine isolated retinae. Data obtained in experiments with aminoguanidine suggests that the iNOS could be involved in the response elicited by l-cysteine in the bovine isolated retinae. Since excessive glutamate release has been implicated in neurotoxic processes leading to neurodegeneration, it is conceivable that H2S and NO donors could find therapeutic application in ocular neuropathies such as glaucoma and age-related macular degeneration. Figure 6 provides a schematic representation of the possible H2S interactions that lead to attenuation of [3H]D-aspartate release in isolated bovine retinae.
There is evidence that K+-stimulated release of [3H]D-aspartate in mammalian retinae dependent on calcium homeostasis [31, 57]. In a previous study, we reported that the fast releasing H2S donor, NaHS inhibited glutamatergic neurotransmission in bovine and porcine isolated retinae, a response that was dependent, at least in part, on the intramural biosynthesis of H2S [26]. The present observation that both a slow-releasing H2S donor and l-cysteine (a substrate for H2S biosynthesis) can inhibit K+-depolarization induced [3H]D-aspartate release suggests that this gas may serve a protective role in preventing damage to retinal neurons under conditions that involve excessive glutamate production leading to calcium overload.
We conclude that both the slow-releasing H2S donor, GYY 4137 and a substrate for the production of this gas, l-cysteine can inhibit K+-depolarization-induced release of [3H]D-aspartate from the bovine isolated retinae. The pharmacological actions elicited by these compounds on glutamatergic neurotransmission is dependent, at least in part, upon the intramural biosynthesis of this gas, and on the activity of KATP channels and NOS activity. The exact mechanism/s that underlie the effect of H2S on the pathway leading to the release of glutamate from retinal neurons merits further investigation.
References
Kimura H (2014) Hydrogen sulfide and polysulfides as biological mediators. Molecules 19:16146–16157. https://doi.org/10.3390/molecules191016146
Abe K, Kimura H (1996) The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 16:1066–1071
Nagai Y, Tsugane M, Oka J, Kimura H (2004) Hydrogen sulfide induces calcium waves in astrocytes. FASEB J 18:557–559. https://doi.org/10.1096/fj.03-1052fje
Kimura Y, Kimura H (2004) Hydrogen sulfide protects neurons from oxidative stress. FASEB J 18:1165–1167. https://doi.org/10.1096/fj.04-1815fje
Kimura Y, Goto Y, Kimura H (2010) Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal 12(1):1–13. https://doi.org/10.1089/ars.2008.2282
Mikami Y, Shibuya N, Kimura Y, Nagahara N, Yamada M, Kimura H (2011) Hydrogen sulfide protects the retina from light-induced degeneration by the modulation of Ca2+ influx. J Biol Chem 286:39379–39386. https://doi.org/10.1074/jbc.M111.298208
Gerasimova E, Lebedeva J, Yakovlev A, Zefirov A, Giniatullin R, Sitdikova G (2015) Mechanisms of hydrogen sulfide (H2S) action on synaptic transmission at the mouse neuromuscular junction. Neuroscience 303:577–585. https://doi.org/10.1016/j.neuroscience.2015.07.036
Kimura H (2002) Hydrogen sulfide as a neuromodulator. Mol Neurobiol 26:13–19. https://doi.org/10.1385/MN:26:1:013
Gong QH, Shi XR, Hong ZY, Pan LL, Liu XH, Zhu YZ. (2011) A new hope for neurodegeneration: possible role of hydrogen sulfide. J Alzheimers Dis 24(Suppl 2):173–182. https://doi.org/10.3233/JAD-2011-110128
Wei HJ, Li X, Tang XQ (2014) Therapeutic benefits of H2S in Alzheimer’s disease. J Clin Neurosci 21:1665–1669. https://doi.org/10.1016/j.jocn.2014.01.006
Davoli A, Greco V, Spalloni A, Guatteo E, Neri C, Rizzo GR, Cordella A, Romigi A, Cortese C, Bernardini S, Sarchielli P, Cardaioli G, Calabresi P, Mercuri NB, Urbani A, Longone P (2015) Evidence of hydrogen sulfide involvement in amyotrophic lateral sclerosis. Ann Neurol 77:697–709. https://doi.org/10.1002/ana.24372
Persa C, Osmotherly K, Chao-Wei CK, Moon S, Lou MF (2006) The distribution of cystathionine beta-synthase (CBS) in the eye: implication of the presence of a trans-sulfuration pathway for oxidative stress defense. Exp Eye Res 83:817–823. https://doi.org/10.1016/j.exer.2006.04.001
Pong WW, Stouracova R, Frank N, Kraus JP, Eldred WD (2007) Comparative localization of cystathionine beta-synthase and cystathionine gamma-lyase in retina: differences between amphibians and mammals. J Comp Neurol 505:158–165. https://doi.org/10.1002/cne.21468
Kulkarni M, Njie-Mbye YF, Okpobiri I, Zhao M, Opere CA, Ohia SE (2011) Endogenous production of hydrogen sulfide in isolated bovine eye. Neurochem Res 36:1540–1545. https://doi.org/10.1007/s11064-011-0482-6
Kraus JP, Kozich V (2001) Cystathionine B synthase and its deficiency. In: Carmel R, Jacobsen DW (eds) Homocysteine in health and disease. Cambridge University Press, Cambridge, pp 223–243
Roedl JB, Bleich S, Reulbach U, von Ahsen N, Schlötzer-Schrehardt U, Rejdak R, Naumann GO, Kruse FE, Kornhuber J, Jünemann AG (2007) Homocysteine levels in aqueous humor and plasma of patients with primary open-angle glaucoma. J Neural Transm (Vienna) 114:445–450. https://doi.org/10.1007/s00702-006-0556-9
Roedl JB, Bleich S, Schlötzer-Schrehardt U, von Ahsen N, Kornhuber J, Naumann GO, Kruse FE, Jünemann AG (2008) Increased homocysteine levels in tear fluid of patients with primary open-angle glaucoma. Ophthalmic Res 40:249–256. https://doi.org/10.1159/000127832
Ran R, Du L, Zhang X, Chen X, Fang Y, Li Y, Tian H (2014) Elevated hydrogen sulfide levels in vitreous body and plasma in patients with proliferative diabetic retinopathy. Retina 34:2003–2009. https://doi.org/10.1097/IAE.0000000000000184
Monjok EM, Kulkarni KH, Kouamou G, McKoy M, Opere CA, Bongmba ON, Njie YF, Ohia SE (2008) Inhibitory action of hydrogen sulfide on muscarinic receptor-induced contraction of isolated porcine irides. Exp Eye Res 87:612–616. https://doi.org/10.1016/j.exer.2008.09.011
Ohia SE, Opere CA, Monjok EM, Kouamou G, LeDay AM, Njie-Mbye YF (2010) Role of hydrogen sulfide production in inhibitory action of L-cysteine on isolated porcine irides. Curr Eye Res 35:402–407. https://doi.org/10.3109/02713680903576716
Chitnis MK, Njie-Mbye YF, Opere CA, Wood ME, Whiteman M, Ohia SE (2013) Pharmacological actions of the slow release hydrogen sulfide donor GYY4137 on phenylephrine-induced tone in isolated bovine ciliary artery. Exp Eye Res 116C:350–354. https://doi.org/10.1016/j.exer.2013.10.004
Kulkarni-Chitnis M, Njie-Mbye YF, Mitchell L, Robinson J, Whiteman M, Wood ME, Opere CA, Ohia SE (2015) Inhibitory action of novel hydrogen sulfide donors on bovine isolated posterior ciliary arteries. Exp Eye Res 134:73–79. https://doi.org/10.1016/j.exer.2015.04.001
Njie-Mbye YF, Bongmba OY, Onyema CC, Chitnis A, Kulkarni M, Opere CA, LeDay AM, Ohia SE (2010) Effect of hydrogen sulfide on cyclic AMP production in isolated bovine and porcine neural retinae. Neurochem Res 35:487–494. https://doi.org/10.1007/s11064-009-0085-7
Njie-Mbye YF, Kulkarni M, Opere CA, Ohia SE (2012) Mechanism of action of hydrogen sulfide on cyclic AMP formation in rat retinal pigment epithelial cells. Exp Eye Res 98:16–22. https://doi.org/10.1016/j.exer.2012.03.001
Kulkarni KH, Monjok EM, Zeyssig R, Kouamou G, Bongmba ON, Opere CA, Njie YF, Ohia SE (2009) Effect of hydrogen sulfide on sympathetic neurotransmission and catecholamine levels in isolated porcine iris-ciliary body. Neurochem Res 34:400–406. https://doi.org/10.1007/s11064-008-9793-7
Opere CA, Monjok EM, Kulkarni KH, Njie YF, Ohia SE (2009) Regulation of [3H]D-aspartate release from mammalian isolated retinae by hydrogen sulfide. Neurochem Res 34:1962–1968. https://doi.org/10.1007/s11064-009-9984-x
Biermann J, Lagreze WA, Schallner N, Schwer CI, Goebel U (2011) Inhalative preconditioning with hydrogen sulfide attenuated apoptosis after retinal ischemia/reperfusion injury. Mol Vis 17:1275–1286. (PMID: 21633713; PMCID: PMC3103742)
Opere CA, Zheng WD, Huang J, Adewale A, Kruglet M, Ohia SE (2005) Dual effects of isoprostanes on the release of [3H]D-aspartate from isolated bovine retinae: Role of arachidonic acid metabolites. Neurochem Res 30:129–137. (PMID: 15756941)
Jamil JM, Bankhele P, Salvi A, Mannix JE, Oger C, Guy A, Galano JM, Durand T, Njie-Mbye YF, Ohia SE, Opere CA (2014) Role of the non-enzymatic metabolite of eicosapentaenoic acid, 5-epi-5-F3t-isoprostane in the regulation of [3H]D-aspartate release in isolated bovine retina. Neurochem Res 39:2360–2369. https://doi.org/10.1007/s11064-014-1436-6
LeDay AM, Kulkarni KH, Opere CA, Ohia SE (2004) Arachidonic acid metabolites and peroxide-induced inhibition of [3H]D-aspartate release from bovine isolated retinae. Curr Eye Res 28:367–372. (PMID: 15287374).
Lopez-Colome AM, Roberts PJ (1987) Effect of excitatory amino acid analogue on release of D-3H-Aspartate from chick retina. Eur J Pharmacol 142:409–417. (PMID: 3428354)
Santos PF, Duarte CB, Carvalho AP (1996) Glutamate receptor agonists evoked Ca(2+)-dependent and Ca(2+)-independent release of [3H]D-aspartate from cultured chick retina cells. Neurochem Res 21:361–368. (PMID: 9139243)
Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, Giannis A, Szabo C, Spyroulias GA, Papapetropoulos A (2013) Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br J Pharmacol 169:922–932. https://doi.org/10.1111/bph.12171
Wang R (2012) Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92:791–896. https://doi.org/10.1152/physrev.00017.2011
Kajimura M, Fukuda R, Bateman RM, Yamamoto T, Suematsu M (2010) Interactions of multiple gas-transducing systems: hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxid Redox Signal 13:157–192. https://doi.org/10.1089/ars.2009.2657
Corbett JA, McDaniel ML (1996) The use of aminoguanidine, a selective iNOS inhibitor, to evaluate the role of nitric oxide in the development of autoimmune diabetes. Methods 10:21–30. (PMID: 8812641)
Chen C, Yun XJ, Liu LZ, Guo H, Liu LF, Chen XL. (2017) Exogenous nitric oxide enhances the prophylactic effect of aminoguanidine, a preferred iNOS inhibitor, on bleomycin-induced fibrosis in the lung: Implications for the direct roles of the NO molecule in vivo. Nitric Oxide. https://doi.org/10.1016/j.niox.2017.07.005
Wang JF, Li Y, Song JN, Pang HG (2014) Role of hydrogen sulfide in secondary neuronal injury. Neurochem Int 64:37–47. https://doi.org/10.1016/j.neuint.2013.11.002
Li L, Salto-Tellez M, Tan CH, Whiteman M, Moore PK (2009) GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radic Biol Med 47(1):103–113. https://doi.org/10.1016/j.freeradbiomed.2009.04.014
Li L, Whiteman M, Guan YY, Neo KL, Cheng Y, Lee SW, Zhao Y, Baskar R, Tan CH, Moore PK (2008) Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation 117:2351–2360. https://doi.org/10.1161/CIRCULATIONAHA.107.753467
Whiteman M, Perry A, Zhou Z, Bucci M, Papapetropoulos A, Cirino G, Wood ME. (2015). Phosphinodithioate and Phosphoramidodithioate. Hydrogen Sulfide Donors. Handb Exp Pharmacol, 337–363. https://doi.org/10.1007/978-3-319-18144-8_17
de Mello MC, Klein WL, de Mello FG (1988) L-glutamate evoked release of GABA from cultured avian retina cells does not require glutamate receptor activation. Brain Res 443:166–172. (PMID: 2896053)
Fitzgerald RS, Shirahata M, Chang I, Kostuk E, Kiihl S (2011) The impact of hydrogen sulfide (H2S) on neurotransmitter release from the cat carotid body. Respir Physiol Neurobiol 176:80–89. https://doi.org/10.1016/j.resp.2011.01.010
Fernandes VS, Ribeiro ASF, Martínez P, López-Oliva ME, Barahona MV, Orensanz LM et al (2014) Hydrogen sulfide plays a key role in the inhibitory neurotransmission to the pig intravesical ureter. PLoS ONE 9(11):e113580. https://doi.org/10.1371/journal.pone.0113580
Wang M, Zhu J, Pan Y, Dong J, Zhang L, Zhang X, Zhang L (2015) Hydrogen sulfide functions as a neuromodulator to regulate striatal neurotransmission in a mouse model of Parkinson’s disease. J Neurosci Res 93:487–494. https://doi.org/10.1002/jnr.23504
Kimura H, Shibuya N, Kimura Y (2012) Hydrogen sulfide is a signaling molecule and a cytoprotectant. Antioxid Redox Signal 17:45–57. https://doi.org/10.1089/ars.2011.4345
Peers C, Bauer CC, Boyle JP, Scragg JL, Dallas ML (2012) Modulation of ion channels by hydrogen sulfide. Antioxid Redox Signal 17:95–105. https://doi.org/10.1089/ars.2011.4359
Munaron L, Avanzato D, Moccia F, Mancardi D (2013) Hydrogen sulfide as a regulator of calcium channels. Cell Calcium 53:77–84. https://doi.org/10.1016/j.ceca.2012.07.001
Cheng Y, Ndisang JF, Tang G, Cao K, Wang R (2004) Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol 287:H2316-2323. https://doi.org/10.1152/ajpheart.00331.2004
Ali MY, Whiteman M, Low CM, Moore PK (2007) Hydrogen sulphide reduces insulin secretion from HIT-T15 cells by a KATP channel-dependent pathway. J Endocrinol 195:105–112. https://doi.org/10.1677/JOE-07-0184
Distrutti E, Sediari L, Mencarelli A, Renga B, Orland Si, Antonelli E, Roviezzo F, Morelli A, Cirino G, Wallace JL, Fiorucci S. Evidence that hydrogen sulfide exerts antinociceptive effects in the gastrointestinal tract by activating KATP Channels. J Pharmacol Exp Ther 316:325–335; https://doi.org/10.1124/jpet.105.091595
Salvi A, Bankhele P, Jamil JM, Kulkarni-Chitnis M, Njie-Mbye Y, Ohia SE, Opere CA (2016) Pharmacological actions of hydrogen sulfide donors on sympathetic neurotransmission in the bovine anterior uvea, in vitro. Neurochem Res 41:1020–1028. https://doi.org/10.1007/s11064-015-1784-x
Bauzo RM, Kimmel HL, Howell LL (2011) The cystine-glutamate transporter enhancer N-acetyl-L-cysteine attenuates cocaine-induced changes in striatal dopamine but not self-administration in squirrel monkeys. Pharmacol Biochem Behav 101:288–296. https://doi.org/10.1016/j.pbb.2011.12.018
Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M, Moore PK (2006) Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem Biophys Res Commun 343:303–310. https://doi.org/10.1016/j.bbrc.2006.02.154
Salomone S, Foresti R, Villari A, Giurdanella G, Drago F, Bucolo C (2014) Regulation of vascular tone in rabbit ophthalmic artery: cross talk of endogenous and exogenous gas mediators. Biochem Pharmacol 92:661–668. https://doi.org/10.1016/j.bcp.2014.10.011
Moustafa A, Habara Y (2014) Hydrogen sulfide regulates Ca(2+) homeostasis mediated by concomitantly produced nitric oxide via a novel synergistic pathway in exocrine pancreas. Antioxid Redox Signal 20:747–758. https://doi.org/10.1089/ars.2012.5108
Ohia SE, Opere CA, Awe SO, Adams L, Sharif NA (2000) Human, bovine, and rabbit retinal glutamate-induced [3H]D-aspartate release: role in excitotoxicity. Neurochem Res 25:853–860. (PMID: 10944004).
Acknowledgements
We thank J.F. O’Neill Packing Co., Omaha, NE for their generous donation of bovine eyeballs and the Creighton University School of Pharmacy and Health Professions, Department of Pharmacy Sciences for Graduate Students support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bankhele, P., Salvi, A., Jamil, J. et al. Comparative Effects of Hydrogen Sulfide-Releasing Compounds on [3H]D-Aspartate Release from Bovine Isolated Retinae. Neurochem Res 43, 692–701 (2018). https://doi.org/10.1007/s11064-018-2471-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-018-2471-5