
Abstract
Purpose
Meningiomas are the most common primary intracranial tumor in older adults (Ostrom et al. in Neuro Oncol 21(Suppl 5):v1–v100, 2019). Treatment is largely driven by, in addition to patient characteristics and extent of resection/Simpson grade, the World Health Organization (WHO) grading of meningiomas. The current grading scheme, based predominantly on histologic features and only limited molecular characterization of these tumors (WHO Classification of Tumours Editorial Board, in: Central nervous system tumours, International Agency for Research on Cancer, Lyon, 2021), (Mirian et al. in J Neurol Neurosurg Psychiatry 91(4):379–387, 2020), does not consistently reflect the biologic behavior of meningiomas. This leads to both under-treatment and over-treatment of patients, and hence, suboptimal outcomes (Rogers et al. in Neuro Oncol 18(4):565–574). The goal of this review is to synthesize studies to date investigating molecular features of meningiomas as they relate to patient outcomes, in order to clarify best practices in assessing and, therefore, treating meningiomas.
Methods
The available literature of genomic landscape and molecular features of in meningioma was screened using PubMed.
Results
Greater understanding of meningiomas is reached by integrating histopathology, mutational analysis, DNA copy number changes, DNA methylation profiles, and potentially additional modalities to fully capture the clinical and biologic heterogeneity of these tumors.
Conclusion
Diagnosis and classification of meningioma is best accomplished using a combination of histopathology with genomic and epigenomic factors. Future classification schemes may benefit from such an integrated approach.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Meningioma classification with multi-omics data predicts tumor behavior
Currently, the WHO classification of CNS tumors includes the use of two molecular features, for which sufficient evidence of the relationship with patient outcomes exists, in the grading criteria for meningiomas: TERT promoter mutation, and CDKN2A and/or CDKN2B homozygous deletion [1,2,3]. The presence of either one of these features satisfy criteria for a grade 3 meningioma diagnosis. Otherwise, grading is based on histologic criteria, including mitotic activity and brain invasion. However, this is not optimal for risk-stratification in all cases. For example, an important clinical problem relates to risk stratification of grade 2 meningiomas for which treatment decisions depend on extent of surgical resection [4], and an unmet need is standardization of recurrence risk and adjuvant therapy in these patients. To meet this need, evidence has been accumulating in support of epigenetic analysis as a valuable tool in meningioma prognostication [5, 6]. In fact, in clinical practice, some pathologists utilize immunohistochemistry for H3K27 trimethylation to assist in assessing tumor aggressiveness, as loss of trimethylation has been associated with increased risk of recurrence [7]; however, epigenetic assessment is not yet incorporated in the WHO guidelines.
Well-known molecular features that correlate with meningioma aggressiveness are specific copy number variations (CNVs) [5]. Increased risk of recurrence is seen with increasing numbers of CNVs, including losses of 1p, 6q, 14, 10 and 22 [8, 9]. Mitotic index and CNVs have been incorporated with clinical factors to create a nomogram for recurrence risk, yielding an “Integrated Grade” [10]. The Integrated Grade reclassified 32% of the meningiomas in the cohort into higher or lower grades compared to the grades assigned to the tumors by the 2016 WHO based solely on histologic criteria. More recently, genome-wide methylation classification has been shown to be a powerful method for determining meningioma behavior [10,11,12,13,14]. The methylation data builds on earlier studies separating meningiomas into more and less aggressive groups based on subsets of hyper- and hypomethylated loci [15,16,17,18].
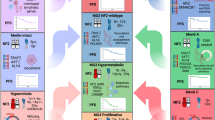
Multiple -omics technologies have been combined to better understand meningioma behavior and biology, and several studies have revealed similar findings (Fig. 1, details in Table 1). In addition to showing that losses of 1p, 6q, 14, 18 and 22q exist in higher-risk molecular groups [13], cluster-of-cluster assignments of DNA methylation, transcriptomic and proteomic data demonstrated that these diverse types of molecular information all contributed to classifying meningiomas, with the combination revealing four stable molecular groups (immunogenic, benign NF2 wild-type, hypermetabolic, proliferative). The molecular groups were also associated with recurrent oncogenic variants and inactivating mutations in tumor suppressors, and the most aggressive molecular groups demonstrated increased genomic instability. A similar study [11] identified three subgroups of meningiomas (immune-rich, NF2/Merlin-intact, hypermitotic), again with distinct clinical outcomes and biology that correlated with response to therapies. CDKN2A/2B homozygous deletion, indicative of grade 3 or anaplastic meningioma, was detected with increasing frequency in the hypermitotic (aggressive) meningiomas [11]. Another study [19] found two predominant methylation clusters, one with dismal outcomes (8/11 patients with poor outcomes), and the other with better outcomes. A larger and more comprehensive study used a 6-class methylation classifier scheme [20] and proposed an integrative model of histologic grade, specific CNVs, and methylation class to create a meningioma grading system [12]. By their model, a subset of WHO grade 1 meningiomas are reclassified as “intermediate” grade, subsets of WHO grade 2 meningiomas are reclassified as “low” and “high” grade, respectively, and rare WHO grade 3 cases are reclassified as “low” or “intermediate” grade. These discrepancies presumably reflect the inadequacies of current WHO grading, with the integrated model classifications better reflecting prognosis, and therefore better guiding patient care, although use of the system and resulting outcomes data are necessary to verify its effectiveness. An advantage of the proposed integrated model is that for most cases, meningiomas could be fully characterized into a risk group solely with the use of methylation profiling, suggesting feasibility in the clinic.
Comparison of classifications made by the various algorithms on a common cohort may be helpful to harmonize these multi-omic classification systems and delineate precise recommendations for clinical implementation. Some work has been performed on this front, with encouraging results [21].

Multiple molecular studies of meningiomas have divided tumors into similar groups, demonstrating that characterizing these tumors by methylation profile, CNVs and gene variants is a replicable method of determining meningioma type, indicative of clinical outcome. Four studies are indicated at the top of the diagram, with the meningioma groups found by the studies depicted vertically from best to worst outcomes. The similar molecular features of the groups are highlighted across studies, with gene mutations in light grey, and CNVs in dark grey
The correlation of mutational profile with both tumor location and prognostic methylation status deserves comment. Non-NF2-mutated tumors in general are enriched in the clinically less aggressive methylation classes. Specifically, mutations in NF2 tend to be mutually exclusive with mutations in TRAF7, AKT1, KLF4 or SMO. Tumors with mutations in TRAF7/AKT1 and SMO are preferentially associated with anterior fossa, median middle fossa, or anterior calvarial locations. Tumors with TRAF7 and KLF4 mutations tend to be located in anterior and medial fossa and may show secretory meningioma histopathology [22]. PIK3CA variants are also seen in lower risk meningiomas, but may be present in rare higher histologic grade meningiomas that are clinically aggressive [23,24,25,26]. Mutations in POLR2A indicate a lower risk subset of genomically stable meningioma, with low grade meningothelial histology, and frequent tuberculum sellae origin [27]. Further, NF2-altered meningiomas may have low- or high-risk behavior [11,12,13, 20]. Overall, while meningiomas can be classified based on genetic mutations to an extent [28], clinical utility of sequencing analysis may be best interpreted in the context of additional -omics technologies, including methylation profiling and DNA copy number aberrations.
Three histologic classes of meningioma deserve mention based on specific correlations of histopathology with genetics. Clear cell meningiomas have intermediate risk (equivalent to WHO grade 2) [29]. In fact, as studied separately, clear cell meningiomas fall in a distinct methylation class, perhaps due to the fact that they almost always have mutations in SMARCE1 and/or loss of the encoded protein, which is a subunit of the SWI/SNF chromatin remodeling complex [29,30,31]. Rhabdoid meningiomas [32, 33] are currently considered grade 3 tumors, regardless of the presence or absence of high-grade histologic features [34]. However, not all meningiomas with rhabdoid features have the equivalent of a grade 3 outcome [35]; specifically those tumors with loss of BAP1 tend to show worse outcomes [36]. Finally, papillary meningiomas, a rare type of grade 3 meningioma, show frequent inactivation (truncating mutation or homozygous deletion) of the PBAF complex gene PBRM1 [37].
Transcriptomics data divides meningiomas into biologic groups and supports CNV- and methylation-based meningioma classification
Meningioma gene expression data has been generated and analyzed to determine molecularly different groups and their clinical correlations. One study identified three groups via transcriptomic analysis, with distinct outcomes [38]. A second study used consensus clustering of gene expression data across all grades to identify four subtypes of meningioma, which also had different DNA methylation patterns [40]. Despite the differences in approach, their groups are again reminiscent of the divisions found in other studies, demonstrating both the strengths and limitations of histologic grading. Mutations in genes such as TRAF7, AKT1 and KLF4 were seen in “subtype 1” meningioma, NF2 mutations with chromosome 22q loss only detected in “subtype 2,” and almost all meningiomas containing chromosome 1p loss with NF2 mutation or chromosome 22q loss falling in “subtype 3.” A scoring system based on up- and down-regulated genes predicted recurrence-free survival, but not overall survival in validation cohorts [40].
Additional approaches, employed by several groups, have focused on outcomes, and characterized the difference between poor and better outcomes within their cohort. For example, Schmidt et al. determined gene expression profiles defining aggressive versus less aggressive meningiomas, which revealed low PTTG1 and high LEPR expression associated with aggressive behavior, both of which were confirmed by immunohistochemical staining [41]. Additional studies have confirmed the potential utility of gene expression profiling to identify prognostic classes [42], and a gene expression profile to predict recurrence from a cohort including all histologic grades of meningiomas has been proposed [43]. In addition, transcriptomic analyses were performed in the multi-omics/methylation studies described above, and the results support the proteomics and methylation profiling data [11, 13, 21]. For example, meningiomas in proposed hypermitotic groups were enriched for cell cycle regulation pathways and proliferation-associated transcription factor networks and protein complexes [11, 13]. Meningiomas in the immunogenic group had enrichment of immune regulation and signaling pathways [13], and immune-enriched meningiomas were enriched for meningeal lymphatic genes [11]. Comparing NF2-mutant meningiomas with the methylation class of clear cell meningiomas demonstrated decreased NF2 expression in the former, and relative decreased SMARCE1 and increased EZH2 expression in the latter [29]. While gene expression profiling is less directly translatable than methylation profiling for clinical diagnostics, it holds promise in the potential identification of markers that could be developed into immunohistochemical tests for clinical use.
Radiation-induced meningiomas
Analyses have been undertaken to understand the differences and similarities between sporadic meningiomas and radiation-induced meningiomas (RIMs), generally post therapeutic radiation for a tumor in childhood [44]. Over 20 years ago, studies of 7 cases [45] and of 25 cases [46] found CNV patterns that characterized these tumors.
More recent studies have revealed that in general, RIMs are more genomically unstable than sporadic meningiomas [47]. However, many molecular similarities are seen between RIMs and sporadic meningiomas. Loss of chromosome 1p, known to be associated with higher-grade meningiomas, is seen in the vast majority of RIMs, in line with the 90% seen by Sahm et al. [48], with, in fact, dual loss of 1p and 22q present in some tumors. RIMs also demonstrate NF2 changes, with several types of alterations seen [48]. One study revealed NF2 inactivation due to structural rearrangements, with 12 of 31 RIMs demonstrated NF2 intronic rearrangements [49].
Molecular characterization of pediatric meningiomas highlights differences with adult meningiomas
Pediatric meningiomas are relatively uncommon [50], and WHO grading has historically been less predictive of clinical outcomes than for adult meningiomas, with an increased proportion showing higher grade by WHO criteria [51,52,53]. In contrast to adults, higher proportions of tumors in children are seen in male versus female patients, and more have a spinal or intraventricular location compared to adult meningiomas [52,53,54,55,56]. These differences have spurred investigation into their molecular features to better understand pediatric meningiomas.
Many pediatric meningiomas are seen in neurofibromatosis type 2 syndrome, and this has been understood for more than 20 years [52, 54, 56, 57]. One cohort of pediatric meningiomas found that close to half of their cases had deleterious variants in NF2, almost always with concomitant loss of chromosome 22 [58]. Rare cases with TERT promoter, SMARCB1, BRAF, FUBP1, SMAD2 or GATA3 pathogenic or likely pathogenic variants were identified, but no mutations were seen in TRAF7, SMO, KLF4, AKT1 and PIK3CA, in contrast to adult meningiomas (Fig. 2). Of note, the TERT promoter mutation was found in a histologic grade 3 case. Regarding CNVs, aside from loss of chromosome 22 (68% of cases), chromosome 1 partial losses were seen frequently (29% of cases), and partial losses in 9, 10 and 14 were occasionally seen [57, 58]. Given the differences between adult and pediatric meningiomas, and the high number of pediatric tumors considered grade 2, it has been suggested than alternative grading schemes may provide a better indicator of recurrence-free survival for pediatric meningioma [58].

Comparison of pediatric, spinal and adult intracranial meningiomas highlights similarities and differences among the tumor types. Except for spinal meningioma methylation group 1 clustering with the adult intracranial meningioma MC benign group with TRAKL genotype mutations (TRAF7, AKT1 and KLF4), and proximity of the few spinal meningiomas with increased CNVs to the malignant methylation group of adult intracranial meningiomas, the methylation groups are different for pediatric, spinal and adult intracranial meningiomas, despite overlap in genotypes, such as similarities across all compartments with NF loss-of-function (NF2 LOF) alterations
Adding to this body of work, another study found that their pediatric cohort also predominantly showed NF2 alterations [55]. The most frequent CNV was chromosome 22 loss; losses of chromosomes 1, 18 and 14 were also found, to a lesser degree. Importantly, DNA methylation profiling suggests that pediatric meningiomas are epigenetically distinct from adult meningiomas. Despite the similarities in NF2 molecular changes, a majority of the pediatric meningiomas cluster separately from adult meningiomas and three methylation groups have been suggested. The first group consisted almost entirely of clear cell meningiomas with SMARCE1 mutations, with frequent losses on chromosomes 22 and 19. Whether these clear cell meningiomas cluster with the adult clear cell meningiomas described above [29], and whether the SMARCE1 variants were germline or somatic in these specific cases remain open questions. The remaining meningiomas could be divided into two subgroups. The first subgroup (“NF2-driven”) consisted mainly of atypical meningiomas with loss of chromosome 22, and most patients carried the clinical diagnosis of NF2. As mentioned, although these young patients carried NF2 alterations, their tumors clustered separately from adult NF2-altered meningiomas. The balance of the tumors clustering in the final subgroup were a mixture, including rhabdoid meningiomas, and tumors with loss of chromosome 11, but not including NF2 patients [55]. The group also performed DNA sequencing, and found, similar to Toland et al., that the mutations seen in adult meningiomas in TRAF7, SMO, KLF4, AKT1 and PIK3CA, and even the TERT promoter (unlike Toland et al.), were not found in their pediatric cohort. Aside from the NF2 alterations and the SMARCE1 changes, only variants of uncertain significance in a few genes were seen. Conclusions regarding any prognostic significance of the molecular findings were not made. An additional and important difference between pediatric and adult meningiomas is the presence to YAP1 fusions in pediatric tumors [59], where 9 out of 102 cases of pediatric meningioma were shown to have YAP1 fusions. The most frequent fusion partner was MAML2, although other partners (LMO1, PYGO1) were also identified. A second study extended these findings, identifying YAP1-FAM118B fusions in 2 cases [60]. A third study [61] identified an additional case with a YAP1-MAML2 fusion and showed on the CNV plot that such a fusion can be suggested by copy number breakpoints on chromosome 11. It has been speculated that fusions involving YAP1 may be a phenocopy to NF2 inactivation, since NF2 alterations have not been identified in the fusion-positive cases.
Similarities and differences between spinal and intracranial meningiomas
Recently, spinal meningiomas have been studied separately, as they have not been well-represented in prior cohorts. Hua et al. performed targeted sequencing on WHO grade 1 spinal meningiomas and found two mutation types [62]. One group with AKT1 p.E17K mutations were slightly older (median age 71 years) and had tumors most often occurring in a ventral/ventrolateral cervical location. The identification of the AKT1 mutation in spinal meningiomas was in line with a prior molecular study [63]. AKT1-mutant tumors in the spine in one study appeared to be genetically distinct from intracranial AKT1-mutant meningiomas, which frequently harbor co-occurring mutations in TRAF7 [64], a combination only exceptionally seen in the spinal meningiomas. In that study, a second group (median age 65.5 years) frequently harbored NF2 mutations, were predominantly females, and more commonly showed thoracic and dorsal/dorsolateral tumor location [62].
Spinal meningiomas have also been studied by DNA methylation profiling [65]. T-distributed Stochastic Neighbor Embedding demonstrated that a majority of the spinal meningiomas clustered in two groups, separate from intracranial meningiomas; in addition, several tumors were found to be separate from the two clusters, close to the intracranial meningiomas [65]. The two clusters were assessed using the framework of six tumor types previously generated for intracranial meningiomas [20]. The mutational profiles of the spinal meningiomas were consistent with the findings of Hua et al. [62]. A majority of tumors in cluster 1 matched to the less aggressive intracranial methylation class whose tumors had mutations in AKT1, SMO, KLF4 and TRAF7. Many tumors in cluster 1 demonstrated AKT1 mutations, but no other mutations seen in intracranial meningiomas. Although only a few tumors in cluster 2 matched to a defined class, a majority showed NF2 mutations and hemizygous chromosome 22q loss. In contrast, cluster 1 included only one NF2-mutated tumor and three tumors with 22q loss [65]. The several spinal meningiomas that did not cluster with the other cases had an increased number of CNVs. Although clear cell meningiomas have been found not uncommonly in the spine [29, 66], only one case in this cohort was a clear cell meningioma. Whether this case would cluster with the intracranial clear cell meningiomas [29] is unclear. The clinical relevance of the different molecular groups in studies of spinal meningiomas requires further study; thus far, outcome data documents an occasional recurrence, but systemic patient outcomes remain to be discerned [62, 65].
Conclusions
Although meningiomas are frequently considered “benign,” the morbidity and the therapeutic challenges encountered in some cases bring this designation into question. As described here, multiple genomic and epigenomic studies have established the existence of distinct biologic groups of meningiomas, which have clinical correlates. Several of these studies have established multi-omic classification schemes that yield robust and coherent groups; harmonization of these schemes may serve to further clarify optimal biological grouping strategies. Improvements in this realm are particularly important for grade 2 meningiomas, as patients with these tumors display a wide variety of clinical courses and circumstances in which the decision between adjuvant therapy versus surveillance can be difficult. Biomarker assessment is also important for the identification of the subset of histologic grade 1 meningiomas that display poor-prognostic markers, for example those that show an unfavorable methylation class, specific CNVs (e.g. loss of 1p, 6q or 14q) or those that have TERT promoter mutations. From a clinical perspective, grading and marker assessment needs to be performed in the context of the factors such as extent of surgical resection, an important predictor of recurrence.
Patient and provider access to the DNA methylation, SNV and CNV profiling necessary for molecular classification for meningiomas can be a challenge. However, access to molecular testing is increasing, with more institutions developing the capacity and infrastructure for molecular testing. In addition, creation of central reference labs and/or utilization of existing reference would allow increased access, although test re-imbursement issues remain a challenge. Data-driven studies that include molecular analyses and reviews of such studies are essential to promote changes in diagnostic practices, which require time for development.
Finally, the molecular characterization of meningiomas may open the door to precision therapy approaches, with the hope of improved patient outcomes. Treatment development and clinical trials are underway to attempt to attack the various molecular targets that have been found in meningiomas. For example, treatment of an AKT1-mutated meningioma by an AKT1 inhibitor has demonstrated long-term control of disease, a dual TORC1-mTORC2 inhibitor is in clinical trials for treating meningiomas with loss of Merlin function in NF2-dependent meningiomas, and a phase II clinical trial with a SMO receptor antagonist is in process for patients with SMO-mutated meningiomas [65]. Multi-omic studies have begun to unravel the biological characteristics of meningiomas and further work in this area will likely yield insights into new therapeutic strategies, which are urgently needed.
The molecular underpinnings of meningioma are only just beginning to be understood. The current WHO classification has begun to incorporate molecular characteristics of meningiomas into their diagnostic criteria and grading. Given the clinical implications of the multi-omics classification schemes, the combination of mutational profiles with DNA copy number changes and methylation subclasses may serve, along with traditional histopathology, as components of a meningioma grading or classification scheme in the future. However, further studies, especially prospective clinical trials, are needed to investigate the value and use of mutational profiles, CNV patterns or methylation classes as relevant clinical markers in meningioma.
References
WHO Classification of Tumours Editorial Board (2021) Central nervous system tumours. International Agency for Research on Cancer, Lyon
Sahm F et al (2016) TERT promoter mutations and risk of recurrence in meningioma. J Natl Cancer Inst 108(5):djv377
Mirian C et al (2020) Poor prognosis associated with TERT gene alterations in meningioma is independent of the WHO classification: an individual patient data meta-analysis. J Neurol Neurosurg Psychiatry 91(4):378–387
Rogers CL et al (2016) Pathology concordance levels for meningioma classification and grading in NRG Oncology RTOG Trial 0539. Neuro Oncol 18(4):565–574
Suppiah S et al (2019) Molecular and translational advances in meningiomas. Neuro Oncol 21(Suppl 1):i4–i17
Galani V et al (2017) Genetic and epigenetic alterations in meningiomas. Clin Neurol Neurosurg 158:119–125
Katz LM et al (2018) Loss of histone H3K27me3 identifies a subset of meningiomas with increased risk of recurrence. Acta Neuropathol 135(6):955–963
Aizer AA et al (2016) A prognostic cytogenetic scoring system to guide the adjuvant management of patients with atypical meningioma. Neuro Oncol 18(2):269–274
McNulty SN et al (2018) Analysis of point mutations and copy number variation in Grade II and III meningioma. Exp Mol Pathol 105(3):328–333
Driver J et al (2022) A molecularly integrated grade for meningioma. Neuro Oncol 24(5):796–808
Choudhury A et al (2022) Meningioma DNA methylation groups identify biological drivers and therapeutic vulnerabilities. Nat Genet 54(5):649–659
Maas SLN et al (2021) Integrated molecular-morphologic meningioma classification: a multicenter retrospective analysis, retrospectively and prospectively validated. J Clin Oncol 39(34):3839–3852
Nassiri F et al (2021) A clinically applicable integrative molecular classification of meningiomas. Nature 597(7874):119–125
Nassiri F et al (2019) DNA methylation profiling to predict recurrence risk in meningioma: development and validation of a nomogram to optimize clinical management. Neuro Oncol 21(7):901–910
Gao F et al (2013) DNA methylation in the malignant transformation of meningiomas. PLoS ONE 8(1):e54114
Kishida Y et al (2012) Epigenetic subclassification of meningiomas based on genome-wide DNA methylation analyses. Carcinogenesis 33(2):436–441
Olar A et al (2017) Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol 133(3):431–444
Vengoechea J et al (2013) Methylation markers of malignant potential in meningiomas. J Neurosurg 119(4):899–906
Millesi M et al (2022) DNA methylation associates with clinical courses of atypical Meningiomas: a matched case-control study. Front Oncol 12:811729
Sahm F et al (2017) DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol 18(5):682–694
Bayley JCt et al (2022) Multiple approaches converge on three biological subtypes of meningioma and extract new insights from published studies. Sci Adv 8(5):eabm6247
Yuzawa S, Nishihara H, Tanaka S (2016) Genetic landscape of meningioma. Brain Tumor Pathol 33(4):237–247
Birzu C, Peyre M, Sahm F (2020) Molecular alterations in meningioma: prognostic and therapeutic perspectives. Curr Opin Oncol 32(6):613–622
Abedalthagafi M et al (2016) Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro Oncol 18(5):649–655
Bujko M et al (2014) EGFR, PIK3CA, KRAS and BRAF mutations in meningiomas. Oncol Lett 7(6):2019–2022
Pang JC et al (2006) Rare mutation of PIK3CA in meningiomas. Acta Neuropathol 111(3):284–285
Clark VE et al (2016) Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat Genet 48(10):1253–1259
Lynes J et al (2022) Molecular determinants of outcomes in meningiomas. Front Oncol 12:962702
Sievers P et al (2021) Clear cell meningiomas are defined by a highly distinct DNA methylation profile and mutations in SMARCE1. Acta Neuropathol 141(2):281–290
Smith MJ et al (2017) SMARCE1 mutation screening in classification of clear cell meningiomas. Histopathology 70(5):814–820
Tauziede-Espariat A et al (2018) Loss of SMARCE1 expression is a specific diagnostic marker of clear cell meningioma: a comprehensive immunophenotypical and molecular analysis. Brain Pathol 28(4):466–474
Kepes JJ et al (1998) Rhabdoid transformation of tumor cells in meningiomas: a histologic indication of increased proliferative activity: report of four cases. Am J Surg Pathol 22(2):231–238
Perry A et al (1998) “Rhabdoid” meningioma: an aggressive variant. Am J Surg Pathol 22(12):1482–1490
Louis D, Scheithaur B, Budka H (2000) Meningiomas. WHO classification of tumour: pathology and genetics of tumoursof the nervous system. IARC Press, Lyon
Vaubel RA et al (2016) Meningiomas with rhabdoid features lacking other histologic features of malignancy: a study of 44 cases and review of the literature. J Neuropathol Exp Neurol 75(1):44–52
Shankar GM et al (2017) Germline and somatic BAP1 mutations in high-grade rhabdoid meningiomas. Neuro Oncol 19(4):535–545
Williams EA et al (2020) Frequent inactivating mutations of the PBAF complex gene PBRM1 in meningioma with papillary features. Acta Neuropathol 140(1):89–93
Patel AJ et al (2019) Molecular profiling predicts meningioma recurrence and reveals loss of DREAM complex repression in aggressive tumors. Proc Natl Acad Sci U S A 116(43):21715–21726
Harmanci AS et al (2017) Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat Commun 8:14433
Liu F, Qian J, Ma C (2021) MPscore: a novel predictive and prognostic scoring for progressive meningioma. Cancers (Basel) 13(5):1113
Schmidt M et al (2016) Transcriptomic analysis of aggressive meningiomas identifies PTTG1 and LEPR as prognostic biomarkers independent of WHO grade. Oncotarget 7(12):14551–14568
Chen WC et al (2020) A prognostic gene-expression signature and risk score for meningioma recurrence after resection. Neurosurgery 88(1):202–210
Olar A et al (2018) A gene expression signature predicts recurrence-free survival in meningioma. Oncotarget 9(22):16087–16098
Lee JY et al (2004) Loss of heterozygosity analysis of benign, atypical, and anaplastic meningiomas. Neurosurgery 55(5):1163–1173
Shoshan Y et al (2000) Radiation-induced meningioma: a distinct molecular genetic pattern? J Neuropathol Exp Neurol 59(7):614–620
Joachim T et al (2001) Comparative analysis of the NF2, TP53, PTEN, KRAS, NRAS and HRAS genes in sporadic and radiation-induced human meningiomas. Int J Cancer 94(2):218–221
Brastianos PK et al (2013) Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet 45(3):285–289
Sahm F et al (2017) Meningiomas induced by low-dose radiation carry structural variants of NF2 and a distinct mutational signature. Acta Neuropathol 134(1):155–158
Agnihotri S et al (2017) Therapeutic radiation for childhood cancer drives structural aberrations of NF2 in meningiomas. Nat Commun 8(1):186
Ostrom QT et al (2016) American brain tumor association adolescent and young adult primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro Oncol 18(Suppl 1):i1–i50
Perry A, Dehner LP (2003) Meningeal tumors of childhood and infancy. An update and literature review. Brain Pathol 13(3):386–408
Rushing EJ et al (2005) Central nervous system meningiomas in the first two decades of life: a clinicopathological analysis of 87 patients. J Neurosurg 103(6 Suppl):489–495
Caroli E, Russillo M, Ferrante L (2006) Intracranial meningiomas in children: report of 27 new cases and critical analysis of 440 cases reported in the literature. J Child Neurol 21(1):31–36
Perry A et al (2001) Aggressive phenotypic and genotypic features in pediatric and NF2-associated meningiomas: a clinicopathologic study of 53 cases. J Neuropathol Exp Neurol 60(10):994–1003
Kirches E et al (2021) Molecular profiling of pediatric meningiomas shows tumor characteristics distinct from adult meningiomas. Acta Neuropathol 142(5):873–886
Erdincler P et al (1998) Intracranial meningiomas in children: review of 29 cases. Surg Neurol 49(2):136–40 (discussion 140-1)
Battu S et al (2018) Clinicopathological and molecular characteristics of pediatric meningiomas. Neuropathology 38(1):22–33
Toland A et al (2020) Pediatric meningioma: a clinicopathologic and molecular study with potential grading implications. Brain Pathol 30(6):1134–1143
Sievers P et al (2020) YAP1-fusions in pediatric NF2-wildtype meningioma. Acta Neuropathol 139(1):215–218
Schieffer KM et al (2021) YAP1-FAM118B fusion defines a rare subset of childhood and young adulthood meningiomas. Am J Surg Pathol 45(3):329–340
Esposito S et al (2022) Interhemispheric pediatric meningioma, YAP1 fusion-positive. Diagnostics (Basel) 12(10):2367
Hua L et al (2022) Two predominant molecular subtypes of spinal meningioma: thoracic NF2-mutant tumors strongly associated with female sex, and cervical AKT1-mutant tumors originating ventral to the spinal cord. Acta Neuropathol. https://doi.org/10.1007/s00401-022-02474-9
Sahm F et al (2013) AKT1E17K mutations cluster with meningothelial and transitional meningiomas and can be detected by SFRP1 immunohistochemistry. Acta Neuropathol 126(5):757–762
Clark VE et al (2013) Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 339(6123):1077–80
Ricklefs FL et al (2022) Genetic and epigenetic profiling identifies two distinct classes of spinal meningiomas. Acta Neuropathol. https://doi.org/10.1007/s00401-022-02504-6
Smith MJ et al (2014) Germline SMARCE1 mutations predispose to both spinal and cranial clear cell meningiomas. J Pathol 234(4):436–440
Okano A et al (2022) Advances in Molecular biological and translational studies in World Health Organization grades 2 and 3 meningiomas: a literature review. Neurol Med Chir (Tokyo) 62(8):347–360
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
MPN and KDA: wrote the main manuscript text, and prepared the figures and table.
Corresponding author
Ethics declarations
Conflict of interest
MPN and KDA each have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nasrallah, M.P., Aldape, K.D. Molecular classification and grading of meningioma. J Neurooncol 161, 373–381 (2023). https://doi.org/10.1007/s11060-022-04228-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-022-04228-9