
Abstract
Background
Glioblastoma (GBM) is the most common and malignant gliomas of adults and recur, resulting in death, despite surgery, radiotherapy, and temozolomide-based chemotherapy. There are a few reports on immunotherapy for the mismatch repair (MMR)-deficient GBMs with high tumor mutational burden (TMB). However, the clinicopathological and genetic features of the MMR genes altered in GBMs have not been elucidated yet.
Methods
The authors analyzed targeted next-generation sequencing (NGS) data from 282 (276 primary and 6 recurrent) glioblastomas to evaluate the mutational status of six DNA repair-related genes: MLH1, MSH2, MSH6, PMS2, POLE, and POLD1. Tumors harboring somatic or germline mutations in one or more of these six genes were classified as an MMR gene-altered GBM. The clinicopathologic and molecular characteristics of MMR gene-altered GBMs were compared to those of tumors without MMR gene alterations.
Results
Sixty germline or somatic mutations were identified in 37 cases (35 primary and two recurrent) of GBM. The most frequently mutated genes were MSH6 and POLE. Single nucleotide variants were the most common, followed by frameshift deletions or insertions and approximately 60% of the mutations were germline mutations. Two patients who showed MSH2 (c.2038C > T) and MSH6 (c.1082G > A) mutations had familial colon cancer. The clinical findings were not different between the two groups. However, the presence of MGMT promoter methylation and high tumor mutation burden (TMB) values (> 20) were correlated with MMR gene alterations.
Conclusion
Since MMR-related genes can be found even in primary glioblastoma and are correlated with high TMB and MGMT promoter methylation, MMR genes should be carefully analyzed in NGS study on glioblastomas.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM) is the most frequent malignant brain tumor in adults. This tumor is invariably fatal, with less than 5% of the patients alive after five years [1]. In addition, it tends to recur after surgery, radiotherapy, and temozolomide-based chemotherapy.
Recently, immune therapy has improved the overall survival rates of patients with various cancer types such as advanced melanoma, non-small cell lung cancer, urothelial carcinoma, and renal cell carcinoma [2]. However, GBM is an immunologically quiet tumor compared to other tumors that remain obstacles to successful immunotherapy [3, 4]. Thus, there have been a few reports on immunotherapy for DNA mismatch repair (MMR)-deficient GBMs [5, 6].
Deficiency of MMR is caused by germline and somatic mutation of MMR related genes, such as MutL homo- logue 1 (MLH1), MutS protein homologue 2 (MSH2), MutS homologue 6 (MSH6) and PMS1 homologue 2 (PMS2) [7]. Germline mutation of these MMR genes cause autosomal dominant inherited disorder, Lynch syndrome (LS) [8]. However, about 35% of suspected LS patients show negative germline MMR gene alteration and showed pathogenic somatic mutations and loss of heterozygosity [8]. Lynch syndrome is the most common cause of hereditary colorectal cancer, and increases the risk of colorectal cancer as well as cancers in other organs including the endometrium, stomach, small intestine, ovary, pancreas, upper urinary tract, biliary tract, brain, skin, and prostate [9].
The response to immunotherapy is closely related to the hypermutation of the tumor [10]. Hypermutation of the tumor can be caused by environmental factors, such as UV radiation, cigarette smoking, or MMR gene mutations such as in Lynch syndrome [10]. Besides widely known MMR genes such as MLH1, MSH2, MSH6, and PMS2, mutations in polymerase delta 1 (POLD1) and DNA polymerase epsilon, catalytic subunit (POLE) genes are also associated with MMR deficiency [11,12,13,14]. In addition to the germline mutations or MLH1 promoter hypermethylation, acquired somatic mutations of MMR genes can lead to the development of hypermutated tumor phenotype and affect patients’ survival [13, 15]. Hypermutated tumors typically had mutations in at least one of the MMR genes MLH1, MSH2, MSH6, and PMS2 [14, 16, 17]. In addition, mutations in POLE and POLD1 led to hypermutation and neo-antigen production [14, 18]. MMR deficiency is known to be related to negative chemotherapy responses and favorable responses to anti-PD-1/PD-L1 therapy in other cancers such as colorectal cancer, non-small cell lung cancer, and bladder cancer [19,20,21]. Several studies demonstrated a higher rate of acquired MMR deficiency in treated or recurrent GBMs [22,23,24]. The Cancer Genome Atlas (TCGA) demonstrated that approximately 26% of the recurrent tumors in patients who received alkylating agents acquired mutations in MSH6 and demonstrated increased mutational rates [17, 23,24,25].
MMR deficiency can be detected by various methods. Immunohistochemical staining (IHC) for MMR protein is widely used [26] and shows high sensitivity to detect germline mutation of MMR genes in CRC patients [27]. However, there is possibility of discordance between MMR gene status and IHC results, due to technical factors (such as false negative staining and aberrant staining) or other biological factors [26, 28, 29]. Polymerase chain reaction (PCR), such as Bethesda and pentaplex PCR tests, is gold standard for MSI detection in colorectal carcinomas [30, 31]. This method shows high concordance with IHC for MMR proteins in CRC patients [32]. In GBM, the loss of MSH6 expression in IHC correlated with germline or somatic mutation of MSH6 [33]. However, the low correlation between MSI data with MSH6 protein expression is concerning [34]. In addition, no gold standard method has been established for the detection of MSI or MMR deficiency in GBM. Recently, next-generation sequencing (NGS) with computational tools has emerged for the detection of MMR-deficiency [30, 31, 35,36,37]. NGS can be used for various tumor types and reduces the need for separate tests such as IHC or MSI by PCR [38] in cases with limited tissue samples.
According to the fourth revised edition of the World Health Organization’s (WHO) classification of central nervous system (CNS) [39] tumors, the integration of genetic profile is important in the diagnosis of CNS tumors. NGS testing is widely used for defining tumor mutation profiles, integration of diagnosis and determining treatment approaches. However, whether to integrate the MMR gene status in GBM is not mentioned and histologic differences based on the genetic status have not been elucidated yet. To identify the molecular and clinicopathologic characteristics of MMR-altered GBMs, we analyzed targeted NGS data of 282 glioblastomas.
Materials and methods
Case selection and slide review
We retrospectively reviewed NGS data of 282 glioblastomas from the pathology file of the Samsung Medical Center from April 2017 to April 2019. All 282 cases were studied by targeted NGS for 232 brain cancer-related genes using paraffin-embedded tumor tissues. The tumor tissues were obtained from biopsies (n = 10) or surgical resections (n = 272). All the cases were diagnosed as glioblastomas, and classified as IDH-mutant and IDH-wildtype, based on the 2016 WHO classification of the central nervous system tumors. This cohort consisted of 276 cases of newly diagnosed glioblastomas and 6 recurrent tumors. There were each case of giant cell glioblastoma and epithelioid glioblastoma. Diffuse midline glioma with H3K27M-mutant or H3G34M-mutant was already excluded. Two cases of BRAF V600E mutated glioblastoma were included. The Institutional Review Board of Samsung Medical Center approved this study and waived the requirement for informed consent (IRB number: 2020–04-011).
The targeted NGS gene panel included six MMR related genes: MLH1, MLH6, PMS2, MSH2, POLE, and POLD1. Of 282 glioblastomas, 46 cases showed somatic or germline alterations in at least one of the six genes [5, 11, 14]. Among 46 MMR altered cases, nine were reported as benign or likely benign in ClinVar and reclassified into the MMR non-altered group. Finally, a total of 37 cases were categorized as MMR-altered GBMs. Clinicopathological characteristics of MMR altered group (n = 37 cases) were compared to those of MMR non-altered group that were 39 age- and sex-matched patients selected from 282 cases.
Various clinicopathologic factors, including age at operation, sex, tumor location, familial history, and clinical follow-up data were obtained by medical record review. All H&E stained slides of the selected 76 cases (37 MMR altered and 39 MMR non-altered tumors) were reviewed by two independent pathologists (Y.L. Suh and Y.A. Cho), and quantified histological features including necrosis, microvascular proliferation, multinucleated giant cells, small cell change, and perivascular lymphocytic infiltration. Presence of multinucleated giant cells were assessed as scores 0 (absent), 1 (less than 50% of tumor cells) and 2 (more than 50% of the tumor cells. Small cell change was defined as high N/C ratio with minimal or absence of cytoplasm and hyperchromatic nuclei. When small cells were observed more than 5% of the total tumor area, it was defined as the presence of small cell change. The representative histologic features are presented in Fig. 1.

Representative figures of histologic features of glioblastomas. a multinucleated giant cells, b small cell changes, c perivascular lymphocytic infiltration
Acquisition and analysis of targeted next-generation sequencing study
A panel containing 232 genes with potential clinical relevance was used to capture the target regions. The targeted genes are listed in Supplementary Table S1. DNA was fragmented using a Covaris S2 (Covaris, Woburn, MA, USA) to generate DNA fragments, and a library was prepared using the Sureselect XT Automation Reagent Kit according to the manufacturer’s protocol. The median size of the DNA was measured with an Agilent 4200 Tape Station (Agilent, Santa Clara, CA, USA) to confirm that the median size was between 300 and 400 bp. The DNA concentration was measured with the Qubit™ dsDNA High Sensitivity Assay Kit (Thermo Fisher Scientific) and sequencing was conducted using the TG NextSeq® 500/550 High Output Kit v2 and the TG NextSeq® 500/550 Mid Output Kit v2 (NextSeq 550 Dx, Illumina). Data analysis and interpretation were performed by the BrainTumorSCAN V2.0 pipeline, which is the same as the CancerSCAN V2.0 pipeline [40] and differs only in the target gene list.
The sequence data were analyzed for clinically relevant classes of genomic alterations, including single nucleotide variants (SNVs), small insertions/deletions (indels), copy number alterations (CNAs), and rearrangements/fusions. The SNVs and indels with a variant allele frequency (VAF) of less than 1% were excluded. An average copy number of more than four was considered a gain and less than one was called a loss. A translocation with supporting reads ranged from four to twelve was called a translocation, depending upon the quality of the sample. The NGS study results were reviewed by two pathologists (Y.L. Suh and Y.A. Cho) and three bioinformatics experts (D.G. Kim, B.R. Lee, and J.H. Shim). MMR gene variants were searched through ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and excluded if they were reported as benign. The visualization of genomic data analysis was done via cBioPortal Oncoprinter [41].
MGMT methylation
Five hundred nanograms of DNA was modified by sodium bisulfite, which converts unmethylated cytosine to uracil, following the instructions of the MGMT Plus Kit (Diatech Pharmacogenetics, Jesi, Italy) manufacturer. Modified DNA was subjected to PCR using the Rotor-Gene Q, and during the amplification, thymine was incorporated at the uracil position. The PCR products were then sequenced, and the extent of methylation at each of the 10 analyzed CpG sites (CpG 74–83; chr 10:131,265,507– 131,265,556) was evaluated by pyrosequencing using the PyroMark Q96 ID system (Qiagen). The assay evaluates a cytosine (position chr10: 131,265,528) not followed by guanine, which is, therefore, unmethylated, to test the incomplete conversion by sodium bisulfite in each sample. The extent of methylation was obtained by calculating the average methylation of the 10 CpG sites using PyroMark CpG software (Qiagen).
An analytical cutoff of 5% was considered to discriminate methylated from unmethylated samples as proposed by the MGMT Plus Kit.
Estimation of TMB
The tumor mutation burden (TMB) was estimated as the somatic SNVs per Mb that were (1) non-synonymous mutations, (2) with a mutant allele frequency of > 5% (to avoid counting the variants caused by FFPE artifacts), and (3) a minor allele frequency of less than 0.0001 in gnomAD r2.0.2. and less than 0.001 in the Korean Reference Genome Database (http://152.99.75.168/KRGDB/) and Korean Variant Archive (to exclude germline mutations) [42]. Using these criteria, a previous study revealed a strong correlation with the TMB calculated from whole-exome sequencing [40]. The TMB was categorized into three tiers of low, < 6; intermediate, 6–19; and high, ≥ 20 mutations per Mb, as previously described [14, 43].
Statistical analysis
The T-test or the Wilcoxon rank-sum test was used to compare continuous variables. Fisher’s exact test or the Chi-squared test was used to compare categorical variables. All statistical analyses were performed with SPSS for Windows version 21.0 (SPSS Inc., Chicago, IL, USA). Statistical significance was defined as a two-sided P-value of < 0.05. In addition, to identify the genes associated with MMR alterations, we applied the Benjamini and Hochberg method for false discovery rate (FDR) correction, and significant changes were considered when the adjusted P-value was < 0.05.
Results
Incidence, clinicopathological and molecular features of MMR gene-altered glioblastomas
Out of a total of 282 GBMs, 46 cases (16.3%) showed somatic or germline alterations involving at least one of the six MMR-related genes. Among 46 MMR-altered GBMs, nine were reported as benign or likely benign in ClinVar and reclassified into the non-MMR altered group. Finally, a total of 37 cases (37/282, 13.1%) were categorized as MMR-altered GBMs. The clinicopathological and molecular features of MMR altered group (n = 37) were compared to those of non-MMR altered group (n = 39) (Table 1). The median age at diagnosis was not different between the patients with or without MMR gene alterations (60.5 years vs. 59.1 years). Most of the tumors were unilateral in the location, which was not associated with MMR alterations. The tumors with MMR alterations were associated with the presence of MGMT methylation (P = 0.039, Table 1). The follow-up period was one month to 12 months and neither tumor progression nor tumor recurrence was significantly associated with MMR gene alterations (Table 1). When comparing the histopathological findings between MMR altered and non-altered GBMs, MMR altered tumors showed tendency to have small cell change (P = 0.077) but there were no significant differences between two groups, as shown in Table 1.
Among the 282 patients, 13 patients had a family history of cancers, of which six had MMR gene alterations (Table 1). Of these six patients, two had familial colorectal cancers. The first patient was a 47-year-old woman with IDH-wildtype glioblastoma, who had been diagnosed with MSI-high, ascending colon cancer at the age of 43. She had familial colorectal cancer (her mother, older brother, and uncle had the disease) and biliary cancer (her younger brother), which met Amsterdam II criteria and the revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (HNPCC). She showed a germline mutation in MSH6 (c.1082G > A), which was reported in the HNPCC, although of uncertain significance in the ClinVar report https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000089168.7).
The second patient was a 53-year-old man with glioblastoma, not elsewhere classified (NEC), who had a family history of colon cancer and breast cancer. He showed a germline mutation in MSH2 (c.2038C > T) that was reported to be pathogenic for Lynch Syndrome, HNPCC, or hereditary cancer-predisposing syndrome in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000036572.5). In the remaining four patients with MMR gene alterations, prostate or gastric cancer, lymphoma, or glioblastoma were present in their family members. A mutation in MSH2 (c.1342 T > C) detected in a patient with a familial history of GBM was not reported in ClinVar.
This study cohort included 64 and 8 cases of IDH-wildtype and IDH-mutant GBM, respectively, and four cases of GBM, NEC. The MMR gene-altered GBMs consisted of IDH-wildtype (33), IDH-mutant (3), and NEC-type (1) tumors. The IDH1 alteration c.781 T > A was classified as NEC-type GBM. The classification of tumors by the IDH gene alteration status was not correlated with alterations in the MMR-related genes (P = 0.470). Our study included six cases of recurrent glioblastomas, of which two showed alterations in MMR-related genes (Supplementary Table S2).
Incidence and type of MMR gene alterations
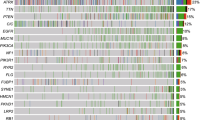
The median tumor volume submitted for NGS was 80.0% (range 5 – 95%). The overall sequencing quality of the NGS study was tolerable, with an average on-target rate of 94.07% (range 92.19 – 96.08%), a mapping quality (Q30%) of 96.98 (range 92.6 – 98), a mean depth of 1032.50 (range 514.5 – 1418.47), and uniformity of 77.40 (range 72 – 85) (Supplementary Table S3). The average total depth of the MMR genes was MLH1: 1271.9 (range 245.2 – 3701.2), MSH2: 1003.6 (range 506.8 – 1562.4), MSH6: 697.3 (range 384.0 – 1066.3), PMS2: 1253.0 (range 738.7 – 4468.2), POLD1: 626.8 (range: 326.5 – 991.1), and POLE: 984.1 (range 671.3 – 1539.0) (Supplementary Table S4). Sixty somatic or germline mutations (43 nonsynonymous, 10 frameshift deletions, one non-frameshift deletion, three frameshift insertions, two stop-gains, and one stop-loss) were identified in 37 glioblastomas and the most frequently mutated genes were MSH 6 (15/37, 41%) and POLE (15/37, 41%), followed by MLH1 (8/37, 22%), MSH2 (7/37, 19%), POLD1 (7/37, 19%), and PMS2 (8/37, 19%) (Fig. 2). Twenty-one patients had mutations in two or more genes, including three patients with mutations in five genes. The major alteration was SNVs, and a few amplifications or deletions were found in MLH1 and PMS2. In 58% of the SNVs observed in the MMR genes, the VAF was around 40 to 60, indicating a suspected germline mutation (Supplementary Table S5). Pathogenic mutations were identified in only one patient and the remaining variants were not reported or reported as conflicting or of uncertain significance (Supplementary Table S5).

Alterations in MMR genes in the 37 cases of glioblastoma. Oncotype printer reveals that most of the alterations were SNVs and amplifications or deletions in MLH1 or PMS2 genes. Gene alterations are the most frequently detected mutations in POLE and MSH6. Each type of genetic alteration is designated in colors under the figure
Molecular features and TMB according to MMR gene alteration
All selected cases showed molecular features of glioblastoma. CDKN2A/2B deletions (51/76, 67.1%), EGFR amplifications (24/76, 31.6%), and PTEN alterations (17/76, 22.4%) were found regardless of the MMR gene alteration status. Nine cases showed IDH1 mutations; seven cases showed c.395G > A (R132H), one case showed c.395C > A (R132S), and one case showed c.781 T > A (S261T), but there was no statistically significant difference according to the MMR gene alteration status. MMR gene-altered GBMs demonstrated mutations in the following genes: ARID1B (P = 0.04), ARID2 (P = 0.04), BICRA (P = 0.04), CCND1 (P = 0.018), CHEK1 (P = 0.035), FGFR1 (P = 0.035), FIZ1 (P = 0.035), FLT1 (P = 0.04), GLI2 (P = 0.035), GLTSCR1 (P = 0.009), HNF1A (P = 0.018), KMT2B (P = 0.033), PIK2R2 (P = 0.02), PTCH2 (P = 0.04), TSC1 (P = 0.009), WWTR1 (P = 0.013), and ZNF865 (P = 0.035). Most of the alterations in these genes were SNVs, especially missense mutations but there were also truncations or in-frame shift mutations in the ARID1B and TSC1 genes. However, there was no significant difference in the genetic alterations between the two groups, when an FDR adjusted P-value of < 0.05 was applied. The genetic alteration profiles are described in Fig. 3.

Genetic characteristics of the MMR gene alterations. Comparison of genetic alterations, MGMT methylation status, and tumor mutation burden (TMB) values. Each alteration status is indicated in color in the figure. MMR mismatch repair, TMB tumor mutation burden
The TMB of the entire case ranged from 1.5 to 143.7 with an average of 13.6. There was a statistically significant association between TMB and MMR gene alterations when the patients were divided into three (P = 0.006) or two groups (0.009) according to the TMB values (Table 1). There were six cases of GBMs with high TMB (more than 20/Mb). All high TMB tumors showed alterations in MMR-related genes but were not significantly associated with MGMT promoter methylation. The mean age of the patients with high TMB tumors was younger than in the total patient cohort (50.8 vs. 60 years). The highest TMB (143.7 and 132) was found in two tumors with mutations in MLH1, POLD1, and POLE. Additionally, the highest TMB tumor showed MSH2 c.2038C > T (R680*), MLH1 c.649C > T (R217C), and PMS2 c.2519C > A (P840H). The second highest TMB tumor showed MLH1 c.498delA (L166fs) and MSH6 c.1082G > A (R361H) (Table 2). These two tumors showed familial colorectal cancers. Another high TMB tumor (37.8) was in a patient with a family history of prostate cancer. Histologically, high TMB tumors showed considerable numbers of tumor giant cells (P < 0.001), small cell change (P < 0.001), and dense perivascular lymphocytic cuffing (P < 0.001), which were statistically significant compared with low TMB tumors (Supplementary Table S6).
Discussion
Our study demonstrated 60 somatic or germline mutations in at least one of six MMR related genes in 37 (13.1%) of 282 patients with GBM, using targeted NGS. The most frequently mutated genes were MSH 6 and POLE, followed by MLH1, MSH2, POLD1, and PMS2. Most cases showed SNVs of the MMR-related genes, with amplifications or deletions in MLH1 and PMS2 genes. This rate was much higher than that (0.25%) reported by The Cancer Genome Atlas (TCGA) using computational software with NGS data [44]. This might be due to differences in the analyses the studies used for the somatic mutations in repair genes, including MSH2, MSH6, MLH1, PMS2, EXO1, POLD1, and POLE. Whether the incidence of MMR gene alterations differs between races should be investigated through large cohort data in a multi-institute study. Since we reviewed consecutive cases, selection bias in the data collection may be excluded. However, GBM patients with high risk tend to undergo surgery in a referral center, which could explain the relatively high incidence of MMR gene alterations in our study. About half of the MMR gene alterations had a VAF of around 40 to 60, indicating the possibility of a germline mutation but we could not find a statistically significant correlation between familial history and MMR gene alteration status. However, one patient with a family history of colon cancer and breast cancer demonstrated a mutation in MSH2 (c.2038C > T), which was reported to be related to Lynch Syndrome or hereditary non-polyposis colon cancer (HNPCC) in ClinVar. In addition, although of uncertain significance in the ClinVar report, a patient with a mutation in MSH6 (c.1082G > A) had MSI-high colon cancer and a family history of hepatobiliary and colorectal cancer, which met Amsterdam II criteria and the revised Bethesda guidelines for HNPCC.
Previous studies revealed that germline mutations in MMR genes might lead to high susceptibility to glioma or an association with tumor progression [10, 24]. However, our study demonstrated a significant association of MMR gene-altered GBMs with MGMT methylation, which might indicate a favorable outcome. Unfortunately, this study did not investigate the prognosis of the patients with MMR due to the short follow-up period.
Furthermore, previous studies demonstrated that the loss of function of MMR genes contributed to resistance to TMZ by defective programmed cell death in the tumor cells [22, 23, 45]. A higher MMR deficiency rate was reported to be acquired during the treatment and recurrence of GBM. Approximately 26% of the recurrent tumors have been reported to acquire mutations in MSH6 and demonstrate increased mutation rates. All of these patients received alkylating agents (most commonly temozolomide) as their initial treatment, and the resulting mutation pattern was indicative of alkylator-induced mutations in the setting of MMR defects [17, 23, 25]
The present study demonstrated germline or somatic mutations in MMR-related genes in 35 (12.7%) of 272 cases of primary glioblastomas, and in two (33.3%) of six recurrent GBMs. The higher rate of MMR gene alterations in our recurrent GBM patients seems to be from selection bias due to the small numbers of recurrent cases analyzed in this study. In our recurrent cases, mutations in MMR-related genes were detected in MSH2 (c.14C > A) and MSH6 (c.128C > T). Mutations in MMR-related genes such as MSH2, MSH3, MSH6, and MLH3 in primary glioblastomas were also reported in a previous study [46].
Unlike previous studies using IHC or an in vitro study [17, 23], not all MMR gene alterations detected in targeted NGS might lead to the loss of protein function. MMR alteration status and IHC status were highly concordant in colorectal cancer and endometrial cancer [47, 48]. However, concordance is not guaranteed in some tumor types, such as ovarian cancer [49], and not fully studied in GBM.
There are previous reports that MSI and mutations in POLE and POLD1, which are related to DNA repair, resulted in high TMB [50,51,52]. In our cases, the TMB of the entire cohort ranged from 1.5 to 143.7 and the MMR gene-altered group showed a statistically significant correlation with high TMB (more than 20), consistent with previous results [14, 43]. High TMB seems to be related to the immunotherapy response in various tumors [53]. In other tumors, such as melanoma and non-small cell lung cancer, high TMB predicts favorable outcomes after anti-PD-1/PD-L1 immunotherapy [53]. Brain tumors are known to be immunologically quiet and have low TMB, which is barrier for efficient immunotherapy response [54]. Temozolomide treatment can result in mutations in MMR genes and lead to high TMB, which can be more immunogenic compared to other GBM [55, 56]. Acquired mutation of MMR genes can lead to high mutational load of tumor [46]. TBM can be increased in recurrent GBM and high TMB favors the response to checkpoint inhibition [3]. There are previous studies of GBM with high TMB showed favorable response to anti PD-1 therapy [14, 57, 58]. However, there is a report about the failure of checkpoint inhibitor therapy in hypermutated GBMs [59]. Careful patient selection for immunotherapy may be necessary.
In summary, the targeted NGS of 282 glioblastomas revealed that about 13.1% harbored germline or somatic alterations in at least one of six MMR-related genes. Genetic alterations were most frequent in POLE and MSH6. Alterations in MMR-related genes were not associated with the WHO classification of GBM or histologic features. MMR gene-altered GBMs did not show different molecular profiles but were significantly associated with high TMB. Since MMR-related genes can be found even in primary glioblastoma and are correlated with high TMB and MGMT promoter methylation, a careful analysis of MMR genes is necessary in NGS studies of glioblastomas.
Data availability
The data generated and analyzed in this study are provided in the text and the figures, tables, and supplementary tables. Any additional data needed may be requested from the corresponding author.
References
Dolecek TA, Propp JM, Stroup NE, Kruchko C (2012) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol 14(Suppl 5):v1-49. https://doi.org/10.1093/neuonc/nos218
Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, Peters S (2019) Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol 30:44–56. https://doi.org/10.1093/annonc/mdy495
McGranahan T, Therkelsen KE, Ahmad S, Nagpal S (2019) Current state of immunotherapy for treatment of glioblastoma. Curr Treat Options Oncol 20:24. https://doi.org/10.1007/s11864-019-0619-4
Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, Ziv E, Culhane AC, Paull EO, Sivakumar IKA, Gentles AJ, Malhotra R, Farshidfar F, Colaprico A, Parker JS, Mose LE, Vo NS, Liu J, Liu Y, Rader J, Dhankani V, Reynolds SM, Bowlby R, Califano A, Cherniack AD, Anastassiou D, Bedognetti D, Mokrab Y, Newman AM, Rao A, Chen K, Krasnitz A, Hu H, Malta TM, Noushmehr H, Pedamallu CS, Bullman S, Ojesina AI, Lamb A, Zhou W, Shen H, Choueiri TK, Weinstein JN, Guinney J, Saltz J, Holt RA, Rabkin CS, Lazar AJ, Serody JS, Demicco EG, Disis ML, Vincent BG, Shmulevich I (2018) The immune landscape of cancer. Immunity 48:812-830.e814. https://doi.org/10.1016/j.immuni.2018.03.023
Indraccolo S, Lombardi G, Fassan M, Pasqualini L, Giunco S, Marcato R, Gasparini A, Candiotto C, Nalio S, Fiduccia P, Fanelli GN, Pambuku A, Della Puppa A, D’Avella D, Bonaldi L, Gardiman MP, Bertorelle R, De Rossi A, Zagonel V (2019) Genetic, epigenetic, and immunologic profiling of mmr-deficient relapsed glioblastoma. Clin Cancer Res 25:1828–1837. https://doi.org/10.1158/1078-0432.ccr-18-1892
Martinez R, Schackert HK, Appelt H, Plaschke J, Baretton G, Schackert G (2005) Low-level microsatellite instability phenotype in sporadic glioblastoma multiforme. J Cancer Res Clin Oncol 131:87–93. https://doi.org/10.1007/s00432-004-0592-5
Mills AM, Liou S, Ford JM, Berek JS, Pai RK, Longacre TA (2014) Lynch syndrome screening should be considered for all patients with newly diagnosed endometrial cancer. Am J Surg Pathol 38:1501–1509. https://doi.org/10.1097/pas.0000000000000321
Geurts-Giele WR, Leenen CH, Dubbink HJ, Meijssen IC, Post E, Sleddens HF, Kuipers EJ, Goverde A, van den Ouweland AM, van Lier MG, Steyerberg EW, van Leerdam ME, Wagner A, Dinjens WN (2014) Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J Pathol 234:548–559. https://doi.org/10.1002/path.4419
Lin KM, Shashidharan M, Thorson AG, Ternent CA, Blatchford GJ, Christensen MA, Watson P, Lemon SJ, Franklin B, Karr B, Lynch J, Lynch HT (1998) Cumulative incidence of colorectal and extracolonic cancers in MLH1 and MSH2 mutation carriers of hereditary nonpolyposis colorectal cancer. J Gastrointest Surg 2:67–71. https://doi.org/10.1016/s1091-255x(98)80105-4
Finocchiaro G, Langella T, Corbetta C, Pellegatta S (2017) Hypermutations in gliomas: a potential immunotherapy target. Discov Med 23:113–120
Chang SC, Lan YT, Lin PC, Yang SH, Lin CH, Liang WY, Chen WS, Jiang JK, Lin JK (2020) Patterns of germline and somatic mutations in 16 genes associated with mismatch repair function or containing tandem repeat sequences. Cancer Med 9:476–486. https://doi.org/10.1002/cam4.2702
Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, Sherborne A, Chubb D, Carvajal-Carmona LG, Ma Y, Kaur K, Dobbins S, Barclay E, Gorman M, Martin L, Kovac MB, Humphray S, Lucassen A, Holmes CC, Bentley D, Donnelly P, Taylor J, Petridis C, Roylance R, Sawyer EJ, Kerr DJ, Clark S, Grimes J, Kearsey SE, Thomas HJ, McVean G, Houlston RS, Tomlinson I (2013) Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 45:136–144. https://doi.org/10.1038/ng.2503
Haraldsdottir S, Hampel H, Tomsic J, Frankel WL, Pearlman R, de la Chapelle A, Pritchard CC (2014) Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 147:1308-1316.e1301. https://doi.org/10.1053/j.gastro.2014.08.041
Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, Huse JT, de Groot J, Li S, Overwijk WW, Spetzler D, Heimberger AB (2017) Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol 19:1047–1057. https://doi.org/10.1093/neuonc/nox026
Liu Y, Chen L, Zhang S, Shu Y, Qi Q, Zhu M, Peng Y, Ling Y (2020) Somatic mutations in genes associated with mismatch repair predict survival in patients with metastatic cancer receiving immune checkpoint inhibitors. Oncol Lett 20:27. https://doi.org/10.3892/ol.2020.11888
Comprehensive genomic characterization defines human glioblastoma genes and core pathways (2008). Nature 455:1061–1068. https://doi.org/https://doi.org/10.1038/nature07385
Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, Batchelor TT, Futreal PA, Stratton MR, Curry WT, Iafrate AJ, Louis DN (2007) Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res 13:2038–2045. https://doi.org/10.1158/1078-0432.Ccr-06-2149
Yao J, Gong Y, Zhao W, Han Z, Guo S, Liu H, Peng X, Xiao W, Li Y, Dang S, Liu G, Li L, Huang T, Chen S, Song L (2019) Comprehensive analysis of POLE and POLD1 Gene Variations identifies cancer patients potentially benefit from immunotherapy in Chinese population. Scientific Reports 9:15767. https://doi.org/10.1038/s41598-019-52414-z
Kim JE, Chun SM, Hong YS, Kim KP, Kim SY, Kim J, Sung CO, Cho EJ, Kim TW, Jang SJ (2019) Mutation Burden and I Index for Detection of Microsatellite Instability in Colorectal Cancer by Targeted Next-Generation Sequencing. J Mol Diagn 21:241–250. https://doi.org/10.1016/j.jmoldx.2018.09.005
Lommatzsch M, Bratke K, Stoll P (2018) Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N Engl J Med 379:e14. https://doi.org/10.1056/NEJMc1808251
Necchi A, Anichini A, Raggi D, Briganti A, Massa S, Luciano R, Colecchia M, Giannatempo P, Mortarini R, Bianchi M, Fare E, Monopoli F, Colombo R, Gallina A, Salonia A, Messina A, Ali SM, Madison R, Ross JS, Chung JH, Salvioni R, Mariani L, Montorsi F (2018) Pembrolizumab as Neoadjuvant Therapy Before Radical Cystectomy in Patients With Muscle-Invasive Urothelial Bladder Carcinoma (PURE-01) An Open-Label Single-Arm. J Clin Oncol, Phase II Study. https://doi.org/10.1200/jco.18.01148
McCord M, Steffens A, Javier R, Kam KL, McCortney K, Horbinski C (2020) The efficacy of DNA mismatch repair enzyme immunohistochemistry as a screening test for hypermutated gliomas. Acta Neuropathol Commun 8:15. https://doi.org/10.1186/s40478-020-0892-2
Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, Louis DN (2009) MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res 15:4622–4629. https://doi.org/10.1158/1078-0432.Ccr-08-3012
Xie C, Sheng H, Zhang N, Li S, Wei X, Zheng X (2016) Association of MSH6 mutation with glioma susceptibility, drug resistance and progression. Mol Clin Oncol 5:236–240. https://doi.org/10.3892/mco.2016.907
Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, Greenman C, Edkins S, Bignell G, Davies H, O’Meara S, Parker A, Avis T, Barthorpe S, Brackenbury L, Buck G, Butler A, Clements J, Cole J, Dicks E, Forbes S, Gorton M, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Kosmidou V, Laman R, Lugg R, Menzies A, Perry J, Petty R, Raine K, Richardson D, Shepherd R, Small A, Solomon H, Tofts C, Varian J, West S, Widaa S, Yates A, Easton DF, Riggins G, Roy JE, Levine KK, Mueller W, Batchelor TT, Louis DN, Stratton MR, Futreal PA, Wooster R (2006) A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 66:3987–3991. https://doi.org/10.1158/0008-5472.Can-06-0127
Luchini C, Bibeau F, Ligtenberg MJL, Singh N, Nottegar A, Bosse T, Miller R, Riaz N, Douillard JY, Andre F, Scarpa A (2019) ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: a systematic review-based approach. Ann Oncol 30:1232–1243. https://doi.org/10.1093/annonc/mdz116
Southey MC, Jenkins MA, Mead L, Whitty J, Trivett M, Tesoriero AA, Smith LD, Jennings K, Grubb G, Royce SG, Walsh MD, Barker MA, Young JP, Jass JR, St John DJB, Macrae FA, Giles GG, Hopper JL (2005) Use of Molecular Tumor Characteristics to Prioritize Mismatch Repair Gene Testing in Early-Onset Colorectal Cancer. J Clin Oncol 23:6524–6532. https://doi.org/10.1200/JCO.2005.04.671
Engel KB, Moore HM (2011) Effects of preanalytical variables on the detection of proteins by immunohistochemistry in formalin-fixed, paraffin-embedded tissue. Arch Pathol Lab Med 135:537–543. https://doi.org/10.1043/2010-0702-rair.1
Shia J (2008) Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn 10:293–300. https://doi.org/10.2353/jmoldx.2008.080031
Baudrin LG, Deleuze JF, How-Kit A (2018) Molecular and Computational Methods for the Detection of Microsatellite Instability in Cancer. Front Oncol 8:621. https://doi.org/10.3389/fonc.2018.00621
Waalkes A, Smith N, Penewit K, Hempelmann J, Konnick EQ, Hause RJ, Pritchard CC, Salipante SJ (2018) Accurate Pan-Cancer Molecular Diagnosis of Microsatellite Instability by Single-Molecule Molecular Inversion Probe Capture and High-Throughput Sequencing. Clin Chem 64:950–958. https://doi.org/10.1373/clinchem.2017.285981
Bartley AN, Luthra R, Saraiya DS, Urbauer DL, Broaddus RR (2012) Identification of Cancer Patients with Lynch Syndrome: Clinically Significant Discordances and Problems in Tissue-Based Mismatch Repair Testing. Cancer Prevention Research 5:320. https://doi.org/10.1158/1940-6207.CAPR-11-0288
Forsström LM, Sumi K, Mäkinen MJ, Oh JE, Herva R, Kleihues P, Ohgaki H, Aaltonen LA (2017) Germline MSH6 Mutation in a Patient With Two Independent Primary Glioblastomas. J Neuropathol Exp Neurol 76:848–853. https://doi.org/10.1093/jnen/nlx066
Maxwell JA, Johnson SP, McLendon RE, Lister DW, Horne KS, Rasheed A, Quinn JA, Ali-Osman F, Friedman AH, Modrich PL, Bigner DD, Friedman HS (2008) Mismatch repair deficiency does not mediate clinical resistance to temozolomide in malignant glioma. Clinical cancer research : an official journal of the American Association for Cancer Research 14:4859–4868. https://doi.org/10.1158/1078-0432.CCR-07-4807
Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC (2014) Microsatellite instability detection by next generation sequencing. Clin Chem 60:1192–1199. https://doi.org/10.1373/clinchem.2014.223677
Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, Wendl MC, Ding L (2013) MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 30:1015–1016. https://doi.org/10.1093/bioinformatics/btt755
Kautto EA, Bonneville R, Miya J, Yu L, Krook MA, Reeser JW, Roychowdhury S (2016) Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 8(5):7452–7463
Haraldsdottir S (2017) Microsatellite Instability Testing Using Next-Generation Sequencing Data and Therapy Implications. JCO Precision Oncology 1:1–4. https://doi.org/10.1200/PO.17.00189
Louis DN, Ohgaki, H., Wiestler, O.D., Cavenee, W.K. (2016) WHO Classification of Tumours of the Central Nervous System, Revised. Fourth Edition.
Park S, Lee H, Lee B, Lee SH, Sun JM, Park WY, Ahn JS, Ahn MJ, Park K (2019) DNA Damage Response and Repair Pathway Alteration and Its Association With Tumor Mutation Burden and Platinum-Based Chemotherapy in SCLC. J Thorac Oncol 14:1640–1650. https://doi.org/10.1016/j.jtho.2019.05.014
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. https://doi.org/10.1126/scisignal.2004088
Lee S, Seo J, Park J, Nam JY, Choi A, Ignatius JS, Bjornson RD, Chae JH, Jang IJ, Lee S, Park WY, Baek D, Choi M (2017) Korean Variant Archive (KOVA): a reference database of genetic variations in the Korean population. Sci Rep 7:4287. https://doi.org/10.1038/s41598-017-04642-4
Johnson A, Severson E, Gay L, Vergilio JA, Elvin J, Suh J, Daniel S, Covert M, Frampton GM, Hsu S, Lesser GJ, Stogner-Underwood K, Mott RT, Rush SZ, Stanke JJ, Dahiya S, Sun J, Reddy P, Chalmers ZR, Erlich R, Chudnovsky Y, Fabrizio D, Schrock AB, Ali S, Miller V, Stephens PJ, Ross J, Crawford JR, Ramkissoon SH (2017) Comprehensive Genomic Profiling of 282 Pediatric Low- and High-Grade Gliomas Reveals Genomic Drivers, Tumor Mutational Burden, and Hypermutation Signatures. Oncologist 22:1478–1490. https://doi.org/10.1634/theoncologist.2017-0242
Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen H-Z, Reeser JW, Yu L, Roychowdhury S (2017) Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precision Oncology. https://doi.org/10.1200/po.17.00073
Daniel P, Sabri S, Chaddad A, Meehan B, Jean-Claude B, Rak J, Abdulkarim BS (2019) Temozolomide Induced Hypermutation in Glioma: Evolutionary Mechanisms and Therapeutic Opportunities. Front Oncol 9:41. https://doi.org/10.3389/fonc.2019.00041
Sa JK, Choi SW, Zhao J, Lee Y, Zhang J, Kong DS, Choi JW, Seol HJ, Lee JI, Iavarone A, Rabadan R, Nam DH (2019) Hypermutagenesis in untreated adult gliomas due to inherited mismatch mutations. Int J Cancer 144:3023–3030. https://doi.org/10.1002/ijc.32054
Stelloo E, Jansen AML, Osse EM, Nout RA, Creutzberg CL, Ruano D, Church DN, Morreau H, Smit V, van Wezel T, Bosse T (2017) Practical guidance for mismatch repair-deficiency testing in endometrial cancer. Ann Oncol 28:96–102. https://doi.org/10.1093/annonc/mdw542
Yuan L, Chi Y, Chen W, Chen X, Wei P, Sheng W, Zhou X, Shi D (2015) Immunohistochemistry and microsatellite instability analysis in molecular subtyping of colorectal carcinoma based on mismatch repair competency. Int J Clin Exp Med 8:20988–21000
Lee JH, Cragun D, Thompson Z, Coppola D, Nicosia SV, Akbari M, Zhang S, McLaughlin J, Narod S, Schildkraut J, Sellers TA, Pal T (2014) Association between IHC and MSI testing to identify mismatch repair-deficient patients with ovarian cancer. Genet Test Mol Biomarkers 18:229–235. https://doi.org/10.1089/gtmb.2013.0393
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, Huang F, He Y, Sun J, Tabori U, Kennedy M, Lieber DS, Roels S, White J, Otto GA, Ross JS, Garraway L, Miller VA, Stephens PJ, Frampton GM (2017) Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9:34. https://doi.org/10.1186/s13073-017-0424-2
Kroeze LI, de Voer RM, Kamping EJ, von Rhein D, Jansen EAM, Hermsen MJW, Barberis MCP, Botling J, Garrido-Martin EM, Haller F, Lacroix L, Maes B, Merkelbach-Bruse S, Pestinger V, Pfarr N, Stenzinger A, van den Heuvel MM, Grunberg K, Ligtenberg MJL (2020) Evaluation of a Hybrid Capture-Based Pan-Cancer Panel for Analysis of Treatment Stratifying Oncogenic Aberrations and Processes. J Mol Diagn. https://doi.org/10.1016/j.jmoldx.2020.02.009
Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J (2018) Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med 7:746–756. https://doi.org/10.1002/cam4.1372
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R (2017) Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 16:2598–2608. https://doi.org/10.1158/1535-7163.mct-17-0386
Medikonda R, Dunn G, Rahman M, Fecci P, Lim M (2020) A review of glioblastoma immunotherapy. J Neurooncol. https://doi.org/10.1007/s11060-020-03448-1
Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, Greenman C, Edkins S, Bignell G, Davies H, O’Meara S, Parker A, Avis T, Barthorpe S, Brackenbury L, Buck G, Butler A, Clements J, Cole J, Dicks E, Forbes S, Gorton M, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Kosmidou V, Laman R, Lugg R, Menzies A, Perry J, Petty R, Raine K, Richardson D, Shepherd R, Small A, Solomon H, Tofts C, Varian J, West S, Widaa S, Yates A, Easton DF, Riggins G, Roy JE, Levine KK, Mueller W, Batchelor TT, Louis DN, Stratton MR, Futreal PA, Wooster R (2006) A Hypermutation Phenotype and Somatic <em>MSH6</em> mutations in recurrent human malignant gliomas after alkylator chemotherapy. Can Res 66:3987–3991. https://doi.org/10.1158/0008-5472.Can-06-0127
Choi S, Yu Y, Grimmer MR, Wahl M, Chang SM, Costello JF (2018) Temozolomide-associated hypermutation in gliomas. Neuro-Oncology 20:1300–1309. https://doi.org/10.1093/neuonc/noy016
Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, Zhukova N, Mason G, Farah R, Afzal S, Yalon M, Rechavi G, Magimairajan V, Walsh MF, Constantini S, Dvir R, Elhasid R, Reddy A, Osborn M, Sullivan M, Hansford J, Dodgshun A, Klauber-Demore N, Peterson L, Patel S, Lindhorst S, Atkinson J, Cohen Z, Laframboise R, Dirks P, Taylor M, Malkin D, Albrecht S, Dudley RW, Jabado N, Hawkins CE, Shlien A, Tabori U (2016) Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 34:2206–2211. https://doi.org/10.1200/jco.2016.66.6552
Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry A, Uppaluri R, Ferguson C, Schmidt RE, Dahiya S, Ansstas G, Mardis ER, Dunn GP (2016) Immunogenomics of hypermutated glioblastoma: a patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov 6:1230–1236. https://doi.org/10.1158/2159-8290.Cd-16-0575
Ahmad H, Fadul CE, Schiff D, Purow B (2019) Checkpoint inhibitor failure in hypermutated and mismatch repair-mutated recurrent high-grade gliomas. Neuro-oncology practice 6:424–427. https://doi.org/10.1093/nop/npz016
Funding
None.
Author information
Authors and Affiliations
Contributions
Contribution to design: YLS, YAC. Contribution to analysis and interpretation of the data: YAC, DGK, BL, JHS. YAC and YLS wrote the manuscript. All authors were involved in writing this manuscript and read and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflicts of interest.
Ethical approval
The Institutional Review Board of Samsung Medical Center approved this study and waived the requirement for informed consent (IRB Number: 2020–04-011).
Consent for publication
No individual patient data were reported in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cho, Y.A., Kim, D., Lee, B. et al. Incidence, clinicopathologic, and genetic characteristics of mismatch repair gene-mutated glioblastomas. J Neurooncol 153, 43–53 (2021). https://doi.org/10.1007/s11060-021-03710-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-021-03710-0