
Abstract
Antiangiogenic therapy can rapidly reduce vascular permeability and cerebral edema but high doses of bevacizumab may induce selective pressure to promote resistance. This trial evaluated the efficacy of low dose bevacizumab in combination with lomustine (CCNU) compared to standard dose bevacizumab in patients with recurrent glioblastoma. Patients (N = 71) with recurrent glioblastoma who previously received radiation and temozolomide were randomly assigned 1:1 to receive bevacizumab monotherapy (10 mg/kg) or low dose bevacizumab (5 mg/kg) in combination with lomustine (90 mg/m2). The primary end point was progression-free survival (PFS) based on a blinded, independent radiographic assessment of post-contrast T1-weighted and non-contrast T2/FLAIR weighted magnetic resonance imaging (MRI) using RANO criteria. For 69 evaluable patients, median PFS was not significantly longer in the low dose bevacizumab + lomustine arm (4.34 months, CI 2.96–8.34) compared to the bevacizumab alone arm (4.11 months, CI 2.69–5.55, p = 0.19). In patients with first recurrence, there was a trend towards longer median PFS time in the low dose bevacizumab + lomustine arm (4.96 months, CI 4.17–13.44) compared to the bevacizumab alone arm (3.22 months CI 2.5–6.01, p = 0.08). The combination of low dose bevacizumab plus lomustine was not superior to standard dose bevacizumab in patients with recurrent glioblastoma. Although the study was not designed to exclusively evaluate patients at first recurrence, a strong trend towards improved PFS was seen in that subgroup for the combination of low dose bevacizumab plus lomustine. Further studies are needed to better identify such subgroups that may most benefit from the combination treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma is the most common malignant primary brain tumor in adults and invariably carries a poor prognosis despite maximal safe surgical resection, radiotherapy with concurrent temozolomide followed by cycles of adjuvant temozolomide [1]. Despite optimal multimodality treatment, recent clinical trials have reported a median survival of only 14–16 months with a 26−33 % 2-year survival rate [1, 2]. At tumor progression, treatment options are limited, and response to single or combinational chemotherapy is poor, with an objective response rate of 6 %, 6-month progression free survival (PFS-6) of 15 % and median overall survival of 6 months [3]. Nitrosoureas such as lomustine continue to be used commonly as salvage therapy. In a phase III study comparing enzastaurin to the lomustine (CCNU), lomustine was found to be superior suggesting that nitrosoureas still have a role in the treatment of glioblastoma in the recurrent setting [4].
Angiogenesis is a pathologic hallmark of glioblastoma with the expression of vascular endothelial growth factor (VEGF) among other pro-angiogenic cytokines as one of the most important regulators of angiogenesis [5]. The expression of VEGF and other proangiogenic cytokines in glioblastoma results in the development of abnormal tumor vasculature. This aberrant tumor vasculature is believed to enhance tumor hypoxia and impair the delivery of cytotoxic chemotherapy [6].
Bevacizumab (Avastin, Genentech/Roche) is a humanized monoclonal antibody that binds to VEGF preventing its interaction with VEGFRs resulting in suppression of VEGF signaling. Bevacizumab was approved by the FDA in 2009 for treatment of recurrent glioblastoma [7]. Multiple clinical trials in patients with recurrent glioblastoma targeting the VGEF pathway alone or in combination with cytotoxic chemotherapy have shown promise for a meaningful prolongation of progression free survival [8−10]. Unfortunately despite impressive radiographic responses, bevacizumab has not resulted in a durable survival benefit either in the recurrent or the upfront setting [11, 12].
Antiangiogenic therapy has been shown to prune abnormal vessels and “normalize” existing vasculature which may paradoxically improve drug and oxygen delivery to the tumor for a period of time following drug administration [13]. Ideally, this window of “normalization” leads to a temporary improvement in tumor oxygenation and blood flow, which enhances radiation [14] and chemotherapy effectiveness [6]. In an orthotopic murine model of glioma, vascular normalization was induced by both low and high doses of bevazicumab [15]. The potential consequence of higher doses of bevacizumab has been associated with the promotion of tumor hypoxia, a well-known mediator of treatment resistance, and promotion of an aggressive phenotype of glioblastoma [16]. Thus, lower doses of antiangiogenic therapy may potentially improve chemotherapy delivery and ultimately patient outcome. In one retrospective analysis, low dose intensity bevacizumab (<5 mg/kg/week) was associated with improved PFS and OS with an inverse relationship seen between dose-intensity and overall survival when compared to normal dose intensity bevacizumab (r = −0.48, p > 0.00001) [17].
The current standard of care for bevacizumab dosing for recurrent glioblastoma is 10 mg/kg IV every 2 weeks (the t1/2 of bevacizumab is approximately 3 weeks). This dose and frequency was based on the schedule used in other cancer types and a small series of patients with glioblastoma [18]. Although this dose has been found to be safe and efficacious in most patients, there may be detrimental effects of giving chronic, high-dose anti-VEGF therapy to patients with glioblastoma. The normalization window may be narrowed by excessive vasculature suppression in the presence of high-dose, continuous antiangiogenic therapy. Over-suppression of the tumor vasculature may prematurely or permanently close the normalization window and consequently reduce tumor exposure and uptake of anti-cancer agents [19]. Since no other combination regimens have been shown to improve outcomes compared to bevacizumab alone, we sought to overcome antiangiogenic therapy resistance by adopting a low-dose bevacizumab regimen which we hypothesized may enhance lomustine delivery, improve its therapeutic efficacy, and reduce tumor resistance compared to the standard of care—standard dose bevacizumab monotherapy [20]. We additionally hypothesized that a low dose bevacizumab regimen in combination with lomustine would result in prolonged PFS and OS when compared to standard dose bevacizumab alone.
Materials and methods
Patients
Patients with recurrent glioblastoma were included in this study. Inclusion criteria included age ≥18 years, histologically confirmed glioblastoma in first, second, or third relapse, prior standard radiation for glioblastoma, prior treatment with temozolomide chemotherapy, Karnofsky performance status (KPS) ≥60, and adequate hematologic, renal, and hepatic function. Exclusion criteria included prior treatment with an antiangiogenic agent or a nitrosurea. All patients were required to sign an informed consent form approved by the institutional review board.
Trial design and treatment arms
The study was a phase II comparative, randomized, single center trial with patients randomly assigned [stratified by first, second, or third recurrence, age (≤50 versus >50 years), KPS low (60–80) versus high (90–100), and scheduled surgery (yes or no)] to receive treatment with bevacizumab [10 mg/kg every 2 weeks (5 mg/kg/week)] or low dose bevacizumab [5 mg/kg every 3 weeks (1.66 mg/kg/week)] plus lomustine (90 mg/m2) each 6 week cycle. The primary end point of the study was to compare the efficacy between the two arms by independent, blinded, radiographic assessment of PFS. Secondary objectives included the proportion of patients progression-free at 6 months after randomization, OS, radiographic response rate, time to progression, and safety. Exploratory end points, to be reported elsewhere, included determination of baseline plasma myeloid chemokines or circulating myeloid cells and the association with response or resistance to bevacizumab as measured by radiographic response and PFS.
Treatment plan
Single agent bevacizumab was given intravenously at a dose of 10 mg/kg every 2 weeks until disease progression or unacceptable toxicity. In the combination group, bevacizumab was given intravenously at a dose of 5 mg/kg every 3 weeks. Lomustine was initially given at 90 mg/m2 every 6 weeks but was later reduced to 75 mg/m2 following the occurrence of 17 grade 3 and 7 grade 4 hematologic adverse events observed in 12 patients and 27 cycles of treatment. For those patients randomized to the combination group, lomustine was given on day 3 of each 6-week cycle. After every 6-week cycle, patients underwent clinical evaluation and radiographic tumor assessment with MRI. Lomustine was given up to a maximum of six cycles. In the setting of hematologic toxicity from lomustine, the lomustine dose could be reduced a maximum of two times. Further reduction in dose was not permitted, and the patient was removed from the protocol.
Evaluations
The primary PFS end points were determined in patients based on gadolinium enhanced, T1 weighted and T2/FLAIR MRI scans assessed separately by treating physicians and by an independent, treatment-arm blinded, radiographic review by a neuro-radiologist based on published RANO criteria [21]. For patients with measurable disease at study entry (defined as bi-dimensionally measurable disease with a minimum measurement of 1 cm on MRI), progression was defined as either (1) 25 % increase in the sum of products of all measurable lesions over smallest sum observed (over baseline if no decrease) using the same techniques as baseline; (2) clear worsening of any evaluable disease; (3) appearance of any new lesion/site; (4) clear clinical worsening or failure to return for evaluation due to death or deteriorating condition (unless clearly unrelated to this cancer). The neurologic status and KPS were determined based on physical examination by the treating investigator before and during treatment. Adverse events (AEs) were recorded and graded according to the Common Terminology Criteria for Adverse Events (CTCAE) version 3.0.
Statistical analysis
This was a randomized, two-arm, comparative, single-center, phase II trial with patients randomized to either treatment using a 1:1 randomization scheme. The primary measure of efficacy was PFS. The method advocated by Rubenstein et al. was used for this randomized phase II screening design where a direct, but non-definitive, “screening” comparison of the experimental versus standard treatments occurred [22]. Within this framework the one-sided type I error rate was set to 0.10 and the power to 0.90. Differences in PFS was monitored at three time points and took place: (1) after a total of 28 events occur (to monitor futility), (2) after 55 events occur and (3) after at least 82 events occur. The test statistic used was based on the log-rank test. An early stopping rule for futility served as guidance for early termination of patient accrual. The interim stopping rule consisted of a group sequential test based on a Gamma family Type I error spending function. Results from the interim analysis were reported to an independent Data Monitoring Committee (DMC) convened at MD Anderson Cancer Center. The DMC assessed the data along with supportive data including other efficacy outcomes, and safety data. All analyses were by intention to treat. Time-to-event endpoints are descriptively summarized by Kaplan–Meier curves. Point and interval estimates of treatment effects are based on maximum likelihood methods. For binomially distributed variables, proportions are reported with their 95 % confidence intervals, differences in proportions and 95 % confidence intervals for the difference in proportions. Confidence intervals were constructed using two-sided 95 % and were based on the normal approximation.
Results
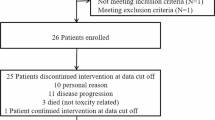
Between January 2010 and December 2014, 83 patients were enrolled. Twelve patients were judged ineligible and were excluded from all analyses. After these exclusions, 71 patients were randomly assigned in a 1:1 ratio to receive bevacizumab monotherapy (n = 36) or low dose bevacizumab plus lomustine (n = 35) (Fig. 1). The demographic and baseline clinical characteristics were well-balanced in terms of major prognostic factors (age, KPS, and number of prior recurrences) across the two treatment arms (Table 1).

Trial profile
For the 12 patients treated with lomustine dosed at 90 mg/m2 in combination with low dose bevacizumab, there were 7 grade 4 (leukopenia n = 1, neutropenia n = 1, thrombocytopenia n = 2, lymphopenia n = 3) and 17 grade 3 (leukopenia n = 4, neutropenia n = 3, thrombocytopenia n = 4, lymphopenia n = 6) hematologic adverse events prompting revision of the protocol to lower the lomustine dose to 75 mg/m2. There were no unexpected AEs or treatment related deaths observed in any arm during the course of the study. The reported toxicities are in accordance with the known toxicities of both drugs (Table 2).
At the time of analysis, 62 patients had progressed or died among 69 radiographically evaluable patients, and the median PFS was not significantly longer in the low dose bevacizumab plus lomustine arm (4.3 months, CI 2.96, 8.34) compared to the bevacizumab alone arm (4.1 months, CI 2.69, 5.55, p = 0.19) (Fig. 2). In 47 patients treated at first recurrence, there was a trend towards statistical significance with longer median PFS time per blinded radiology evaluation in the low dose bevacizumab plus lomustine arm (5.0 months, CI 4.17–13.44) compared to the bevacizumab alone arm (3.2 months CI 2.5–6.01, p = 0.08) (Fig. 3). Median OS in all patients treated with low dose bevacizumab in combination with lomustine was 9.6 months (95 % CI 6.26–16.73) and was not significantly longer than those treated with bevacizumab alone (8.3 months, CI 6.42–11.58, p = 0.75). Median OS in patients with first recurrence on low dose bevacizumab plus lomustine was 13.05 months (95 % CI 7.08–17.82), not significantly longer than those treated with bevacizumab alone (8.8 months, CI 6.42–20.22, p = 0.98) (Fig. 4). The trial was closed early due to futility for the primary end point of PFS.

Median PFS per blinded radiology evaluation by treatment arms

Median PFS per blinded radiology evaluation by treatment arms in patients with first recurrence

Median OS in patients with first recurrence
Six-month PFS was 36.4 % (95 % CI 23.3 %, 57.1 %) in the low dose bevacizumab plus lomustine group and 23.6 % (95 % CI 12.9 %, 43.3 %) in the bevacizumab group. In patients treated at first recurrence, 6-month PFS was 45.8 % (95 % CI 29.7 %, 70.8 %) in the low dose bevacizumab plus lomustine group and 27.6 % (95 % CI 14 %, 54.5 %) in the bevacizumab group. The objective response rate based on blinded radiographic review was 2/12 (17 %) in the low dose bevazicumab group plus lomustine 90 mg/m2, 8/21 (38 %) in the low dose bevazicumab group plus lomustine 75 mg/m2, and 7/36 (19 %) in the bevacizumab alone group.
Discussion
This single center study failed to meet its primary end point of demonstrating a benefit in PFS for the combination of low dose bevacizumab plus lomustine versus bevacizumab alone. The trial was closed early due to futility for its primary end point. Median PFS and OS were both longer in the combination treatment arm particularly in glioblastoma patients with first recurrence suggesting the possibility of clinical activity in this subgroup. However, these findings did not reach statistical significance possibly due to the small numbers of patients treated on this trial. The trend in PFS favoring the combination supports the findings of the published BELOB study [23]. In the randomized phase III follow-up study to BELOB, EORTC 26101, PFS was longer in the combination of bevazicumab and lomustine (4.2 months) compared to lomustine alone (1.5 months), but no overall survival benefit was seen in the combination arm (9.1 months) compared to the lomustine arm alone (8.6 months) [24].
The combination of bevazicumab and lomustine has also been evaluated in a number of other studies in an attempt to address questions regarding the optimal dosing and timing of initiation of bevacizumab [25−27]. In one of these studies, bevacizumab was intentionally reduced to 5 mg/kg every 2 weeks in combination with lomustine 90 mg/m2 based on preclinical work suggesting that even subclinical doses of bevacizumab had an equal effect on regression on tumor vasculature in vivo [15]. The reduced dose of bevacizumab in combination with lomustine led to an extension of PFS and OS of 2 months compared to bevacizumab 10 mg/kg every 2 weeks [25].
An unexpectedly large number of patients on the combination arm treated with lomustine developed grade 3 or 4 myelotoxicity requiring a protocol amendment. Following dose reduction of lomustine from 90 mg/m2 to 75 mg/m2, fewer grade 3 and 4 hematologic adverse events were noted, and the treatment was overall well tolerated. In the BELOB trial, lomustine was initially dosed at 110 mg/m2 with subsequent lowering of the dose to 90 mg/m2 due to hematologic side effects noted at the time of a preplanned safety review. At our institution, patients are typically treated to a goal of 12 cycles of adjuvant temozolomide as tolerated. The median number of temozolomide cycles for patients entered in our trial was six. The greater exposure of our patients to temozolomide prior to the introduction of lomustine may potentially explain why the patients on this trial were less able to tolerate the 90 mg/m2 dose of lomustine compared to the patients on the BELOB trial.
Low dose single agent bevacizumab dosed 5 mg/kg every 3 weeks has been evaluated in a retrospective review of recurrent glioblastoma patients with substantial activity noted (median PFS of 3.6 months and median OS of 6.4 months) comparable to other studies evaluating single agent bevacizumab at higher doses [28]. This comparable efficacy suggests that low dose bevacizumab has activity in recurrent glioblastoma and is a reasonable treatment regimen to investigate. In addition, no grade III-IV toxicities were observed at this low dose [28]. It is possible that low dose bevacizumab may be associated with less toxicity when compared to standard dose bevacizumab.
To the best of our knowledge, this is the first prospective study incorporating low dose bevacizumab in combination with lomustine. Lomustine was administered on day 3 of the 6-week cycle during the hypothesized normalization window in the effort to optimize the time when drug delivery is proposed to be most effective. Numerous studies combining bevacizumab with other therapies have been ineffective [29−32]. Although the reasons for this are unknown, there are no studies that have been performed to determine the optimal biologic dose of bevacizumab or other non-cytotoxic, biologic therapies alone or in combination with other therapeutics. We chose the lower dose bevacizumab schedule based on the 3 week half-life, but the actual dose of 5 mg/kg may still have been too high. If so, lowering the dose further may potentiate the efficacy of bevacizumab combinations. Likewise, the normalization window was assumed to be ‘open’ on day three but this may not have been the optimal time of cytotoxic drug delivery. Although it is not known if optimal biologic dosing will translate into enhanced therapeutic efficacy in the clinic, it is important for investigators to recognize the potential importance of optimal biologic dose and the potential for antagonistic effects of one drug on the other. Preclinical studies to optimize therapies should be attempted prior to combining drugs in the clinical setting.
Limitations of this study included the small number of patients evaluated and the trial being completed at a single institution. The trial design also did not sufficiently answer the question whether low dose bevazicumab delayed or prevented resistance to antiangiogenic therapy. Without a third treatment arm of standard dose bevacizumab in combination with lomustine, this trial could not appropriately answer the question. Although this study was not designed to exclusively evaluate patients at first recurrence, a strong trend towards improved PFS was seen in that subgroup for the combination of low dose bevacizumab plus lomustine. The optimal dosing schema of bevazicumab remains unknown, and low dose bevacizumab may still be an appealing therapeutic option that could be further investigated in a trial powered to directly compare the efficacy on survival between chemotherapy combined with either low dose bevacizumab or standard dose bevacizumab. The future challenge is to better identify the subgroup of patients that may most benefit from the combination treatment. Determining the optimal biologic dosing of non-cytotoxic agents should be an important consideration in the design of agents targeting the tumor microenvironment.
References
Stupp R, Mason WP, van den Bent MJ et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352(10):987–996
Gilbert MR, Wang M, Aldape KD et al (2013) Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol 31(32):4085–4091
Wong ET, Hess KR, Gleason MJ et al (1999) Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol 17(8):2572–2578
Wick W, Puduvalli VK, Chamberlain MC et al (2010) Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol 28(7):1168–1174
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438(7070):932–936
Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307(5706):58–62
Cohen MH, Shen YL, Keegan P, Pazdur R (2009) FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 14(11):1131–1138
Vredenburgh JJ, Desjardins A, Herndon JE 2nd et al (2007) Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol 25(30):4722–4729
Vredenburgh JJ, Desjardins A, Herndon JE 2nd et al (2007) Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res 13(4):1253–1259
Friedman HS, Prados MD, Wen PY et al (2009) Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27(28):4733–4740
Chinot OL, Wick W, Cloughesy T (2014) Bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370(21):2049
Gilbert MR, Dignam JJ, Armstrong TS et al (2014) A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370(8):699–708
Jain RK (2001) Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med 7(9):987–989
Winkler F, Kozin SV, Tong RT et al (2004) Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell 6(6):553–563
von Baumgarten L, Brucker D, Tirniceru A et al (2011) Bevacizumab has differential and dose-dependent effects on glioma blood vessels and tumor cells. Clin Cancer Res 17(19):6192–6205
de Groot JF (2011) High-dose antiangiogenic therapy for glioblastoma: less may be more? Clin Cancer Res 17(19):6109–6111
Lorgis V, Maura G, Coppa G et al (2012) Relation between bevacizumab dose intensity and high-grade glioma survival: a retrospective study in two large cohorts. J Neurooncol 107(2):351–358
Stark-Vance V (2005) Bevacizumab and CPT-11 in the treatment of relapsed malignant glioma. Paper presented at World Federation of Neuro-Oncology, Edinburgh
Ma J, Waxman DJ (2008) Combination of antiangiogenesis with chemotherapy for more effective cancer treatment. Mol Cancer Ther 7(12):3670–3684
Field KM, Jordan JT, Wen PY, Rosenthal MA, Reardon DA (2015) Bevacizumab and glioblastoma: scientific review, newly reported updates, and ongoing controversies. Cancer 121(7):997–1007
Wen PY, Macdonald DR, Reardon DA et al (2010) Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 28(11):1963–1972
Rubinstein LV, Korn EL, Freidlin B, Hunsberger S, Ivy SP, Smith MA (2005) Design issues of randomized phase II trials and a proposal for phase II screening trials. J Clin Oncol 23(28):7199–7206
Taal W, Oosterkamp HM, Walenkamp AM et al (2014) Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol 15(9):943–953
Wick W, Brandes A, Gorlia T et al (2015) LB-05PHASE III trial exploring the combination of bevacizumab and lomustine in patients with first recurrence of a glioblastoma: the EORTC 26101 trial. Neurooncol 17(suppl 5):v1
Heiland DH, Masalha W, Franco P, Machein MR, Weyerbrock A (2016) Progression-free and overall survival in patients with recurrent glioblastoma multiforme treated with last-line bevacizumab versus bevacizumab/lomustine. J Neurooncol 126(3):567–575
Tonder M, Eisele G, Weiss T et al (2014) Addition of lomustine for bevacizumab-refractory recurrent glioblastoma. Acta Oncol 53(10):1436–1440
Wiestler B, Radbruch A, Osswald M et al (2014) Towards optimizing the sequence of bevacizumab and nitrosoureas in recurrent malignant glioma. J Neurooncol 117(1):85–92
Kaloshi G, Brace G, Rroji A et al (2013) Bevacizumab alone at mg/kg in an every-3-week schedule for patients with recurrent glioblastomas: a single center experience. Tumori 99(5):601–603
Desjardins A, Reardon DA, Coan A et al (2012) Bevacizumab and daily temozolomide for recurrent glioblastoma. Cancer 118(5):1302–1312
Reardon DA, Desjardins A, Peters K et al (2011) Phase II study of metronomic chemotherapy with bevacizumab for recurrent glioblastoma after progression on bevacizumab therapy. J Neurooncol 103(2):371–379
Reardon DA, Desjardins A, Peters KB et al (2012) Phase II study of carboplatin, irinotecan, and bevacizumab for bevacizumab naive, recurrent glioblastoma. J Neurooncol 107(1):155–164
Lu-Emerson C, Norden AD, Drappatz J et al (2011) Retrospective study of dasatinib for recurrent glioblastoma after bevacizumab failure. J Neurooncol 104(1):287–291
Funding
National Institutes of Health [1R21CA152024-01] to J. D. National Institutes of Health [CCSG-P30 CA016672] to R. D.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
S. W. serves on the advisory board for Actelion. X. H. has no disclosures. D. L. has no disclosures. C. C. has consultant relationships with Actelion, DNAtrix, Reata Pharma, Newlink Genetics and Cytrx Corp. M. G. has no disclosures. M. L. has no disclosures. B. O. has no disclosures. M. P-P. has no disclosures. V. P. is a consultant for Orbus Therapeutics, Foundation Medicine, Celgene, Genetech, and Merck. I. T-L. has no disclosures. R. C. has no disclosures. W. Y. is a consultant and serves on the advisory board for Actelion, DNATrix, Merck, and Novartis. J. D. serves on the advisory board for Genentech, Inc., Novartis, Celldex Therapeutics, and Foundation Medicine, Inc. J. D. serves on the DSMB for VBL Therapeutics and is a consultant for Celldex Therapeutics, OXiGENE, Omniox, Inc. and Deciphera Pharmaceuticals. J. D. receives research support from Sanofi-Aventis, AstraZeneca, EMD-Serono, Eli Lilly, Novartis, and Deciphera Pharmaceuticals.
Rights and permissions
About this article

Cite this article
Weathers, SP., Han, X., Liu, D.D. et al. A randomized phase II trial of standard dose bevacizumab versus low dose bevacizumab plus lomustine (CCNU) in adults with recurrent glioblastoma. J Neurooncol 129, 487–494 (2016). https://doi.org/10.1007/s11060-016-2195-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-016-2195-9