
Abstract
There is no effective treatment for recurrent glioblastoma (GBM) after bevacizumab failure. Putative mechanisms of resistance to bevacizumab include increased pericyte coverage, mediated partly by platelet-derived growth factor receptor (PDGFR) signaling, and an infiltrative tumor growth pattern potentially dependent on SRC. We explored the efficacy of dasatinib, a SRC, BCR-ABL, c-KIT, EPHA2, and PDGFRβ inhibitor, in patients with recurrent GBM after bevacizumab failure. Adult patients with histologically confirmed GBM who failed bevacizumab therapy were treated with dasatinib 70–100 mg twice daily in combination with bevacizumab (n = 14), until tumor progression or unacceptable toxicity. Fourteen patients were treated. Median age was 55 years (range 32–66) and median KPS was 80 (range 50–90). All patients (100%) had glioblastomas. The median number of prior regimens was 4 (range from 2 to 6). Of the thirteen evaluable patients, none had a complete or partial response. Only one patient had stable disease after an 8 week interval. Median progression-free survival (PFS) was 28 days (95% confidence interval [CI] 26–35 days). Six month progression-free survival (PFS6) was 0%. Median overall survival (OS) was 78 days (95% CI 41–137 days). Treatment was moderately well-tolerated, although one patient sustained a grade 4 intracerebral hemorrhage. Dasatinib in conjunction with bevacizumab does not appear to have activity in patients with recurrent, heavily pretreated GBM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Current treatment of high-grade glioma (HGG) consists of a multi-modality approach including surgery, radiotherapy, and chemotherapy [1]. Despite optimal therapy, the prognosis remains poor with a median overall survival (OS) for newly diagnosed glioblastoma (GBM) of 14.6 months and for anaplastic glioma (AG) of 2–5 years [2–5]. Survival outcomes for recurrent HGG remain grim with 6 month progression-free survival (PFS6) of 9–16% for GBM and 28–31% for AG [6–8]. These dismal outcomes have prompted the search for more effective forms of therapy.
Advances in molecular biology have uncovered clues to glioma pathogenesis. Common aberrations in cellular signaling involved in growth, survival, angiogenesis, and invasion have been identified, and new treatments are aimed at targeting specific molecules in these pathways. Much interest has surrounded the use of anti-angiogenic agents as a novel anticancer therapy. The Food and Drug Administration (FDA) recently granted accelerated approval for bevacizumab, a humanized monoclonal antibody to vascular endothelial growth factor (VEGF), for the treatment of recurrent GBM [9–11]. Unfortunately, the effect on survival is modest with eventual progression of tumors.
As recently reviewed, mechanisms of resistance to anti-angiogenic therapies include upregulation of alternative pro-angiogenic signaling pathways; recruitment of pro-angiogenic bone marrow derived endothelial progenitor cells and monocytes; increase of protective pericyte coverage along tumor vasculature; and activation of invasion [12, 13]. There is increasing evidence that endothelial cells can induce pericyte recruitment for protection when survival signals, normally conveyed by VEGF, are inhibited [12, 14–18]. Platelet derived growth factor-B (PDGF-B) and platelet derived growth factor receptor beta (PDGF-Rβ) have been implicated in pericyte recruitment during developmental vasculogenesis, and PDGF-B expression has been linked to pericyte recruitment in gliomas and other solid tumors [19–22].
Another proposed mechanism of resistance of tumors to anti-angiogenic agents is activation of a more invasive phenotype that is mediated in part by the SRC pathway [12, 23–26]. Dysregulation of the SRC pathway is implicated in tumorigenesis, with Src proteins functioning as a central regulator between extracellular signaling and intracellular activation [27]. Recent work has shown that the Src family kinases, including FYN and SRC, are effectors of oncogenic EGFR signaling, promoting invasion and tumor cell survival [27, 28]. Pharmacologic inhibition of FYN and SRC result in decreased tumor invasion, tumor regression, and tumor cell apoptosis in cell culture [28].
Dasatinib is an oral small molecule inhibitor of SRC, BCR-ABL, c-KIT, EPHA2, and PDGFRβ [29]. It has demonstrated efficacy in malignancies with mutant BCR-ABL, KIT, and PDGFR [30, 31]. Dasatinib is FDA-approved for treatment of chronic myelogenous leukemia (CML) that is resistant or intolerant to other therapies and Philadelphia chromosome positive acute lymphoblastic leukemia (ALL). The role of this drug in solid tumors has yet to be identified, though data from phase I and II studies in advanced melanoma, gastrointestinal stromal tumors (GIST), and prostate cancer have been encouraging [32, 33]. In this report, we review our experience with dasatinib in conjunction with bevacizumab in a heavily pretreated group of patients with recurrent GBM who have failed prior therapy with bevacizumab.
Methods
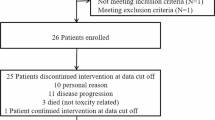
A retrospective review of 14 adult patients with histologically confirmed GBM and radiographically documented recurrent disease was conducted. All patients had prior treatment with radiotherapy, age 18 or older, Karnofsky Performance Status (KPS) of 50 or greater, absolute neutrophil count (ANC) greater than or equal to 1500/Ul, normal renal function, and prior failure of bevacizumab therapy. There was no limit on the number of previous therapies. Endpoints included response rate, progression free survival (PFS), six-month progression free survival (PFS6), overall survival (OS), and toxicities.
Between February 2007 and August 2009, patients were treated with dasatinib 70–100 mg twice daily, in combination with bevacizumab until tumor progression or unacceptable toxicity. In general, patients with a history of myelosuppression received dasatinib 70 mg twice daily, while patients without such a history received 100 mg twice daily. Brain MRI scans were obtained every 8 weeks, or more often if clinically indicated. Response was determined using Macdonald criteria [34]. Toxicities were graded according to the National Cancer Institute Common Toxicity Criteria (CTCAE 3.0). The retrospective analysis was approved by the Institutional Review Board (IRB) of the Dana Farber Cancer Institute. The Kaplan–Meier method was used to estimate survival function.
Results
Patient characteristics are presented in Table 1. Fourteen patients were treated (13 males and 1 female). The median age was 55 years (range 32–66) and median KPS was 80 (range 50–90). All patients (100%) had GBM. The median number of prior regimens was 4 (range from 2 to 6) and included prior treatment with bevacizumab, either alone or in combination with another agent. These fourteen patients received dasatinib in combination with bevacizumab 10 mg/kg every 2 weeks.
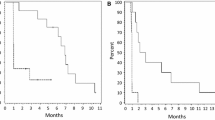
Thirteen patients were evaluable. One patient was censored for enrollment in an impending vaccine trial. Of the thirteen evaluable patients, there were no radiographic responses. One patient (7%) had stable disease after an 8 week interval. Median PFS was 28 days (95% CI 26–35 days), and median OS was 78 days (95% CI 41–137 days).
The treatment was fairly well tolerated (Table 2). The most common toxicities included fatigue, diarrhea, and thrombocytopenia. Two patients (14%) stopped dasatinib therapy because of toxicity. These toxicities included grade 1 fever and rigor (1) and grade 4 hemorrhage (1). Overall, most toxicities were graded 1. The patient who sustained a grade 4 cerebral hemorrhage was not on anticoagulation at the time hemorrhage.
Discussion
This retrospective study showed that dasatinib, a SRC, BCR-ABL, c-KIT, EPHA2, and PDGFRβ inhibitor, in conjunction with bevacizumab, does not have significant activity in patients with recurrent, heavily pretreated GBM following failure of bevacizumab therapy. Median PFS was only 28 days, and PFS6 was 0%. Median OS was 78 days. The treatment was moderately well tolerated with only two patients experiencing adverse effects requiring discontinuation of the drug. The main toxicities included fatigue, diarrhea, rash, fever, thrombocytopenia, and hemorrhage. The observation of one grade 4 cerebral hemorrhage in this small cohort is concerning, and further evaluation of dasatinib in this population warrants caution.
Several reasons could account for these dismal outcomes. The cohort consisted of heavily pre-treated patients (median of 4 prior therapies) who had failed bevacizumab. Clinical deterioration has been reported after disease progression on bevacizumab, secondary to aggressive tumor growth and rebound edema [35]. Attempts to add a chemotherapeutic agent to bevacizumab in these patients have been generally ineffective [36–38]. One study reported median PFS of 2 months (95% CI 1.2–3.3 months), PFS6 of 0%, and median OS of 5.2 months (95% CI 3.3–8.4 months) with salvage chemotherapy after bevacizumab failure [38]. Another recent study reported median PFS of 37.5 days (95% CI 34–42 days) and PFS6 of 2% with a second bevacizumab regime [37]. Our results were slightly worse with a median PFS of 1 month, PFS6 of 0%, and median OS slightly greater than 2 months.
There is increasing animal and human data suggesting that treatment with bevacizumab induces a particularly invasive tumor phenotype in a subset of patients [36, 39]. Several reviews on glioma invasion and migration have recently been published implicating SRC [27, 40, 41]. Although dasatinib inhibits SRC, it is possible that other signaling pathways play a greater role in tumor invasion.
Another potential reason for the lack of response to dasatinib is the inability of the drug to penetrate the blood–brain barrier and achieve adequate concentrations in the tumor. Some murine and human studies suggest that dasatinib’s efficacy against intracranial leukemia is secondary to improved central nervous system (CNS) penetration over imatinib [42]. Unfortunately, pharmacokinetic analysis shows that its CNS penetration, which ranges from 5 to 28% of plasma levels, is still relatively low when compared to other agents known to have good penetration. CNS penetration of this drug remains a real challenge, potentially preventing its effective use in gliomas. Multidrug efflux transporters such as P-glycoprotein (P-gp; ABCB1), breast cancer resistance protein (ABCG2), and multidrug resistance protein 2 (ABCC2) mediate drug resistance to a variety of drugs [43, 44]. Recent animal studies have found that brain accumulation of dasatinib is restricted by P-gp and ABCG2 at the blood brain barrier; inhibition of these transporters increased the brain uptake of this drug [44]. It is possible that the lack of response to dasatinib is partly a consequence of ineffective drug delivery to its molecular target. In addition, the concomitant administration of bevacizumab may reduce disruption of the blood–brain barrier and further decrease CNS penetration of dasatinib. Co-administration of the drug with an inhibitor of P-gp and ABCG2 could theoretically overcome this issue. Preclinical studies demonstrate that P-gp inhibitors, such as elacridar, can increase the concentrations of drugs in brain tissue, although its efficacy in humans is unknown [45, 46]. Despite these arguments, it is well known that the blood–brain barrier is disrupted in the center of high-grade gliomas, which may indicate that tumor penetration is not the major obstacle to dasatinib’s efficacy in this setting.
The activity of single agent dasatinib in recurrent GBM is currently being evaluated by the Radiation Therapy Oncology Group (RTOG 0627). It remains possible that dasatinib could be effective in a particular GBM subgroup defined by molecular genetics. For instance, there is report of increased sensitivity to dasatinib in glioma cells with functional PTEN [47]. However, it must be noted that the applicability of these in vitro data to glioma patients is uncertain. The same study also found significant augmentation of autophagy when dasatinib was combined with temozolomide [47]. Thus, if confirmed in other preclinical models, combination therapy with dasatinib may also be worth exploring. Because of the small number of patients evaluated in this retrospective study and the lack of a uniform treatment approach, subgroup analysis was not pursued in the current dataset.
In summary, this retrospective analysis did not find any significant activity of dasatinib, in combination with bevacizumab, in our cohort of heavily pretreated patients who had failed prior bevacizumab therapy. Whether there may be a role for this drug in the therapy of a selected group of less heavily pretreated patients with recurrent GBM is unclear and will require larger prospective studies. Any future clinical studies regarding this agent should include correlative biomarkers to explore the biological basis of success or failure.
References
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359:492–507
Prados MD, Gutin PH, Phillips TL, Wara WM, Larson DA, Sneed PK, Davis RL, Ahn DK, Lamborn K, Wilson CB (1992) Highly anaplastic astrocytoma: a review of 357 patients treated between 1977 and 1989. Int J Radiat Oncol Biol Phys 23:3–8
Prados MD, Seiferheld W, Sandler HM, Buckner JC, Phillips T, Schultz C, Urtasun R, Davis R, Gutin P, Cascino TL, Greenberg HS, Curran WJ Jr (2004) Phase III randomized study of radiotherapy plus procarbazine, lomustine, and vincristine with or without BUdR for treatment of anaplastic astrocytoma: final report of RTOG 9404. Int J Radiat Oncol Biol Phys 58:1147–1152
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
van den Bent MJ, Carpentier AF, Brandes AA, Sanson M, Taphoorn MJ, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Haaxma-Reiche H, Kros JM, van Kouwenhoven MC, Vecht CJ, Allgeier A, Lacombe D, Gorlia T (2006) Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol 24:2715–2722
Ballman KV, Buckner JC, Brown PD, Giannini C, Flynn PJ, LaPlant BR, Jaeckle KA (2007) The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro Oncol 9:29–38
Lamborn KR, Yung WK, Chang SM, Wen PY, Cloughesy TF, DeAngelis LM, Robins HI, Lieberman FS, Fine HA, Fink KL, Junck L, Abrey L, Gilbert MR, Mehta M, Kuhn JG, Aldape KD, Hibberts J, Peterson PM, Prados MD (2008) Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol 10:162–170
Wong ET, Hess KR, Gleason MJ, Jaeckle KA, Kyritsis AP, Prados MD, Levin VA, Yung WK (1999) Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol 17:2572–2578
Ferrara N, Kerbel RS (2005) Angiogenesis as a therapeutic target. Nature 438:967–974
Gasparini G, Longo R, Toi M, Ferrara N (2005) Angiogenic inhibitors: a new therapeutic strategy in oncology. Nat Clin Pract Oncol 2:562–577
Vredenburgh JJ, Desjardins A, Herndon JE II, Dowell JM, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Wagner M, Bigner DD, Friedman AH, Friedman HS (2007) Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res 13:1253–1259
Bergers G, Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8:592–603
Ebos JM, Lee CR, Kerbel RS (2009) Tumor and host-mediated pathways of resistance and disease progression in response to antiangiogenic therapy. Clin Cancer Res 15:5020–5025
Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D (2003) Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest 111:1287–1295
Jain RK, Booth MF (2003) What brings pericytes to tumor vessels? J Clin Invest 112:1134–1136
Mancuso MR, Davis R, Norberg SM, O’Brien S, Sennino B, Nakahara T, Yao VJ, Inai T, Brooks P, Freimark B, Shalinsky DR, Hu-Lowe DD, McDonald DM (2006) Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest 116:2610–2621
Baluk P, Hashizume H, McDonald DM (2005) Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev 15:102–111
Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307:58–62
Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C (1999) Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 126:3047–3055
Lindahl P, Johansson BR, Leveen P, Betsholtz C (1997) Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277:242–245
Guo P, Hu B, Gu W, Xu L, Wang D, Huang HJ, Cavenee WK, Cheng SY (2003) Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am J Pathol 162:1083–1093
Abramsson A, Lindblom P, Betsholtz C (2003) Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest 112:1142–1151
Casanovas O, Hicklin DJ, Bergers G, Hanahan D (2005) Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8:299–309
Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G (2008) HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 13:206–220
Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O (2009) Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15:220–231
Angers-Loustau A, Hering R, Werbowetski TE, Kaplan DR, Del Maestro RF (2004) SRC regulates actin dynamics and invasion of malignant glial cells in three dimensions. Mol Cancer Res 2:595–605
de Groot J, Milano V (2009) Improving the prognosis for patients with glioblastoma: the rationale for targeting Src. J Neurooncol 95:151–163
Lu KV, Zhu S, Cvrljevic A, Huang TT, Sarkaria S, Ahkavan D, Dang J, Dinca EB, Plaisier SB, Oderberg I, Lee Y, Chen Z, Caldwell JS, Xie Y, Loo JA, Seligson D, Chakravari A, Lee FY, Weinmann R, Cloughesy TF, Nelson SF, Bergers G, Graeber T, Furnari FB, James CD, Cavenee WK, Johns TG, Mischel PS (2009) Fyn and SRC are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res 69:6889–6898
Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius LA, Das J, Doweyko AM, Fairchild C, Hunt JT, Inigo I, Johnston K, Kamath A, Kan D, Klei H, Marathe P, Pang S, Peterson R, Pitt S, Schieven GL, Schmidt RJ, Tokarski J, Wen ML, Wityak J, Borzilleri RM (2004) Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem 47:6658–6661
Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH III, Edeen PT, Floyd M, Ford JM, Grotzfeld RM, Herrgard S, Insko DE, Mehta SA, Patel HK, Pao W, Sawyers CL, Varmus H, Zarrinkar PP, Lockhart DJ (2005) Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci USA 102:11011–11016
Doggrell SA (2005) BMS-354825: a novel drug with potential for the treatment of imatinib-resistant chronic myeloid leukaemia. Expert Opin Investig Drugs 14:89–91
Demetri GD, Lo Russo P, MacPherson IR, Wang D, Morgan JA, Brunton VG, Paliwal P, Agrawal S, Voi M, Evans TR (2009) Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin Cancer Res 15:6232–6240
Yu EY, Wilding G, Posadas E, Gross M, Culine S, Massard C, Morris MJ, Hudes G, Calabro F, Cheng S, Trudel GC, Paliwal P, Sternberg CN (2009) Phase II study of dasatinib in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res 15:7421–7428
Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG (1990) Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol 8:1277–1280
Ananthnarayan S, Bahng J, Roring J, Nghiemphu P, Lai A, Cloughesy T, Pope WB (2008) Time course of imaging changes of GBM during extended bevacizumab treatment. J Neurooncol 88:339–347
Norden AD, Young GS, Setayesh K, Muzikansky A, Klufas R, Ross GL, Ciampa AS, Ebbeling LG, Levy B, Drappatz J, Kesari S, Wen PY (2008) Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology 70:779–787
Quant EC, Norden AD, Drappatz J, Muzikansky A, Doherty L, Lafrankie D, Ciampa A, Kesari S, Wen PY (2009) Role of a second chemotherapy in recurrent malignant glioma patients who progress on bevacizumab. Neuro Oncol 11:550–555
Iwamoto FM, Abrey LE, Beal K, Gutin PH, Rosenblum MK, Reuter VE, DeAngelis LM, Lassman AB (2009) Patterns of relapse and prognosis after bevacizumab failure in recurrent glioblastoma. Neurology 73:1200–1206
de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, Conrad CA (2010) Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol 12:233–242
Chi A, Norden AD, Wen PY (2007) Inhibition of angiogenesis and invasion in malignant gliomas. Expert Rev Anticancer Ther 7:1537–1560
Drappatz J, Norden AD, Wen PY (2009) Therapeutic strategies for inhibiting invasion in glioblastoma. Expert Rev Neurother 9:519–534
Porkka K, Koskenvesa P, Lundan T, Rimpilainen J, Mustjoki S, Smykla R, Wild R, Luo R, Arnan M, Brethon B, Eccersley L, Hjorth-Hansen H, Hoglund M, Klamova H, Knutsen H, Parikh S, Raffoux E, Gruber F, Brito-Babapulle F, Dombret H, Duarte RF, Elonen E, Paquette R, Zwaan CM, Lee FY (2008) Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood 112:1005–1012
Borst P, Elferink RO (2002) Mammalian ABC transporters in health and disease. Annu Rev Biochem 71:537–592
Lagas JS, van Waterschoot RA, van Tilburg VA, Hillebrand MJ, Lankheet N, Rosing H, Beijnen JH, Schinkel AH (2009) Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin Cancer Res 15:2344–2351
Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T (1993) In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res 53:4595–4602
Letrent SP, Pollack GM, Brouwer KR, Brouwer KLR (1999) Effects of a potent and specific P-Glycoprotein inhibitor on the blood-brain barrier distribution and antinociceptive effect of morphine in the rat. Drug Metab Dispos 27:827–834
Milano V, Piao Y, LaFortune T, de Groot J (2009) Dasatinib-induced autophagy is enhanced in combination with temozolomide in glioma. Mol Cancer Ther 8:394–406
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lu-Emerson, C., Norden, A.D., Drappatz, J. et al. Retrospective study of dasatinib for recurrent glioblastoma after bevacizumab failure. J Neurooncol 104, 287–291 (2011). https://doi.org/10.1007/s11060-010-0489-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-010-0489-x