
Abstract
Targeting specific molecular alterations in glioblastoma (GBM) might more effectively kill tumor cells and increase survival. Vandetanib inhibits epidermal growth factor receptor and vascular endothelial growth factor receptor 2. Sirolimus inhibits mammalian target of rapamycin (mTOR), a member the phosphoinositide 3-Kinase signaling pathway. We sought to determine the maximum tolerated dose (MTD) and dose-limiting toxicity (DLT) of vandetanib combined with sirolimus. Twenty-two patients (14 men; 8 women) with recurrent GBM enrolled. Median age and KPS were 52.5 years and 90 %, respectively. Patients were naive to anti-VEGF and anti-EGF therapy and mTOR inhibitors, and not on CYP3A4-inducing drugs. Vandetanib and sirolimus were orally administered on a continuous daily dosing schedule in escalating dose cohorts. Ten patients enrolled in the dose escalation phase. Twelve more enrolled at the MTD to explore progression-free survival at 6 months (PFS6) in a single arm, single stage phase II-type design. In total, 19 patients received at least one dose at the MTD, and 15 completed at least 1 cycle at MTD. MTD was 200 mg vandetanib plus 2 mg sirolimus. The DLT was elevated AST/SGOT. The most common toxicities were lymphopenia, fatigue, rash, and hypophosphatemia. For 19 patients who received at least one dose at the MTD, including seven from the phase I group, two had a partial response [10.5 %; 95 % CI (1, 33 %)] and PFS6 was 15.8 % [95 % CI (3.9, 34.9 %)]. Vandetanib and sirolimus can be safely co-administered on a continuous, daily dosing schedule.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM) is the most common primary malignant brain tumor and recurrence is nearly inevitable [1]; average survival is approximately 1 year despite surgical resection, radiation, and temozolomide [2]. Radiation and temozolomide non-specifically target rapidly proliferating cells by causing DNA damage. An alternative therapeutic strategy is to target the specific molecular alterations upon which GBM cells are dependent on survival. Multiple signaling pathways are commonly aberrant in any given tumor, and targeting individual members is not sufficient to kill GBM cells [3–5]. For example, despite the high prevalence of epidermal growth factor receptor (EGFR) amplification and mutation in GBM, EGFR inhibition with gefitinib or erlotinib did not improve patient outcomes [6–8], although this may have been due to inadequate drug levels reaching tumor cells [9]. This disappointment with single agent targeted therapy has in turn led to an understanding that concomitant use of multiple targeted agents may be necessary to increase patient survival.
We performed a phase I clinical trial with a dose expansion cohort to test the combination of two targeted therapies for recurrent GBM: vandetanib targets pathways that enable proliferation, invasion, and angiogenesis, while sirolimus impacts cell growth and metabolism. Vandetanib is a receptor tyrosine kinase (RTK) inhibitor that targets two frequently aberrant signaling pathways in GBM [3]: vascular endothelial growth factor (VEGF) and epidermal growth factor (EGF) signaling. Vascular endothelial growth factor receptor 2 (VEGFR2, KDR) is highly expressed on both tumor endothelial and glioma cells and EGFR is commonly amplified and mutated in GBM. In enzyme assays, vandetanib inhibits vascular endothelial growth factor receptor2 tyrosine kinase activity (IC50 = 40 nM), and shows additional inhibitory activity at sub-micromolar concentrations against rearranged during transfection (RET) receptor tyrosine kinase (inhibitory concentration [IC50] = 100 nM), Flt-4 (VEGF receptor3: IC50 = 110 nM) and EGF receptor (IC50 = 500 nM) tyrosine kinases [10, 11]. In preclinical testing, vandetanib prolonged survival of mice with orthotopic glioma xenografts when administered immediately after implantation. In subcutaneous xenografts, vandetanib caused partial tumor regression in most models examined [12]. A prior phase I trial with vandetanib as a single agent in patients with solid tumors established a maximum tolerated dose of 300 mg daily [13].
Pathways that converge on PI3K/Akt/mTOR (phosphatidylinositol 3-kinase/mammalian target of rapamycin) signaling play an important role in GBM. Mutations that activate PI3K occur in ~15 % of primary GBM, and PI3K is downstream of multiple aberrant receptor tyrosine kinases (e.g. EGFR, PDGFRA, MET), as well as RAS, and it is regulated by PTEN. Signaling in this pathway is altered in 87 % of primary GBM [3], making it a logical therapeutic target. Sirolimus targets the pathway by directly inhibiting mTOR, a key regulator of cell growth and metabolism. In preclinical testing, mTOR inhibitors have shown antitumor activity in malignant glioma [14, 15].
Vandetanib is approved by the United States Food and Drug Administration (FDA) for medullary thyroid cancer. It is metabolized by CYP3A4, and to a minor extent, FMO1 and FMO3. While co-administration of CYP3A inducers is to be avoided, no clinically significant interaction was observed between vandetanib and the potent CYP3A4 inhibitor, itraconazole. The most common Common Terminology Criteria for Adverse Events version 3.0 (CTCAE v3.0) adverse reactions observed in treatment with vandetanib include rash, nausea, headache, decreased appetite, abdominal pain and grade 3–4 events of diarrhea/colitis, hypertension and hypertensive crisis, QT prolongation, and fatigue [16]. Common laboratory abnormalities include decreased calcium, increased ALT/SGPT, and decreased glucose.
Sirolimus is an FDA-approved immunosuppressant used for the treatment and prophylaxis of renal transplant rejections. It is metabolized in the liver via the CYP3A and P-glycoprotein 1 systems. Patients treated with it have an increased susceptibility to infections and lymphoma. Additionally, common adverse reactions in sirolimus-treated patients include hypertension, thromboembolism, hypercholesterolemia, hyperlipidemia, hepatotoxicity, anemia, thrombocytopenia, arthralgia, nausea, and headaches [17].
The rationale for the present study combining vandetanib and sirolimus was to assess the safety and maximum tolerated dose (MTD) of vandetanib and sirolimus in patients with recurrent GBM. Additionally, in an expansion phase of the trial, we sought to assess the proportion of recurrent GBM patients treated at the MTD that were alive and progression-free at 6 months.
Materials and methods
The study was approved by the institutional review board at Dana-Farber/Harvard Cancer Center (Protocol 07-396). All patients signed informed consent as per institutional guidelines. The study was in accordance with the ethical principles outlined in the Declaration of Helsinki. Between 2/19/2009 and 11/30/2012, 22 patients were enrolled at Massachusetts General Hospital and Dana-Farber Cancer Institute.
Patient eligibility
Patients were eligible for the present study if they were adults (age 18 or greater) with recurrent GBM on no CYP3A4-inducing drugs, previously treated with standard therapy including resection if feasible, radiation and temozolomide, no more than three prior chemotherapies, naïve to prior anti-VEGF, anti-EGF therapy or mTOR inhibitors and had a Karnofsky performance status (KPS) ≥60 % and mini-mental status examination (MMSE) score >15. Patients must have been at least 3 months from the completion of radiation or radiosurgery and must have been maintained on a stable or decreasing corticosteroid regimen for at least 3 days prior to the start of treatment. Laboratory prerequisites included: normal ANC and platelets, renal, liver, and cardiac function. The patient could not be taking any concomitant medication that might cause QTc prolongation, induce Torsades de Pointes or induce CYP3A4 function. Uncontrolled hypertension and hyperlipidemia were also exclusions to enrollment.
Treatment and study design
We executed a standard phase I dose escalation in cohorts of 3 plus 3 patients, followed by an exploratory analysis in a dose expansion cohort. In the expansion cohort, we sought, a priori, to study 15 patients who completed at least 1 cycle at the MTD (see statistical analysis below). Cycle length and the DLT period were 28 days. Vandetanib was supplied by AstraZeneca (Wilmington, DE). The starting dose was 100 mg. Three to six evaluable patients (receiving at least 28 days of vandetanib) per cohort were treated at different dose levels of vandetanib (100 mg daily, 200 mg daily, and a plan to go to 300 mg daily) in combination with sirolimus (10 mg loading and 2 mg daily, and a plan to go to 15 mg loading and 5 mg daily). The study was designed such that cohort one was to have completed at least 4 weeks of treatment before the next cohort on the higher dose began therapy. The MTD was defined as the dose combination that caused DLT in no more than one of six patients having received at least 28 days of treatment. If one out of three patients had a dose limiting toxicity, the cohort was to be increased to six patients. If no others in that cohort had a DLT, then that dose was to be declared the MTD; if two or more patients out of six had a dose limiting toxicity, the dose of vandetanib was to be de-escalated by 100 mg daily and that was to be declared the MTD. Patients were assessed for DLTs each week for the first two cycles, and every other week with subsequent cycles or with each laboratory evaluation in cases of hematologic toxicities.
Parameters for declaring a DLT were defined as any of the following occurring during the combination therapy: Grade 3 or 4 thrombocytopenia or neutropenia for more than a week; grade 3 or 4 non-hematologic toxicities for more than a week felt to be related to the study medications; grade 3 or 4 nausea, emesis, or diarrhea for more than 1 week that is not controllable with medications; single QTc value of ≥550 ms or a sustained increase of ≥100 ms from baseline. All toxicities were graded according to National Cancer Institute’s CTCAE v3.0. A patient’s first episode of venous thromboembolism (deep venous thrombosis [DVT] or pulmonary embolus [PE]) was not considered a DLT, as patients with GBM have a significantly increased likelihood of developing a PE and/or DVT. Patients with thromboembolism were to be anticoagulated with warfarin, low molecular weight heparins or have an IVC filter placed and were to remain on study. Subsequent episodes of DVT or PE were to be considered a DLT. Response was assessed by MacDonald criteria [18].
Statistical considerations
The primary objective of the study was to assess safety and determine the MTD of the combination in the dose escalation (Phase I) study. In the phase I study, patients were to receive at least 28 days of vandetanib and sirolimus (unless a DLT was observed) and have all study procedures performed; if any patient did not meet these criteria, then a replacement would be entered onto the study. A secondary objective was to assess efficacy in a single arm expansion cohort of patients treated at the MTD. A sample size of 15 participants who were treated at the MTD for at least 28 days was calculated with 91 % power to reject a null hypothesis of less than or equal to 15 % alive and progression free at 6 months (APF6) rate using a one-sided alpha = 0.10 test if the true APF6 rate was at least 47 %. The study had an 80 % probability of observing, in at least one participant, toxicities that occur with a true rate of at least 10 %.
Results
Patient characteristics
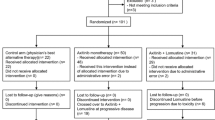
Twenty-two patients with recurrent GBM received vandetanib and sirolimus as described in Table 1. Median age was 52.5 years (range 33–71 years) and median KPS was 90 % (range 60–100 %). Eight patients (36 %) were female. The median number of prior therapies was 2 (range 1–5), including re-resections. Ten patients were part of the dose escalation cohort. Two of these were treated at the MTD for less than 1 cycle; one of these two was the patient who experienced a DLT in the dose escalation phase of the trial. Once MTD was determined, additional patients enrolled so that a total of 15 patients completed at least one cycle at the MTD. This took a total of 12 additional enrollees after the dose escalation phase. In all, a total of 19 patients initiated treatment at MTD, and of these, a total of 15 completed at least 1 cycle at MTD (Table 1).
Adverse events and toxicities
There were no toxicities for the first three patients, treated at 100 mg vandetanib and 2 mg sirolimus (after 10 mg loading dose of sirolimus). One patient of the first three treated at 200 mg vandetanib and 2 mg sirolimus had a study-defined DLT of grade 3 elevated AST/SGOT. None of the next three evaluable patients treated at 200 mg had toxicities within the first cycle, and by this trial’s design (see Treatment and study design above), the MTD was defined as 200 mg vandetanib with 2 mg sirolimus (after a 10 mg sirolimus loading dose). A total of nineteen patients initiated treatment at the MTD, fifteen of whom completed at least 1 cycle (Table 1). 10 out of 19 patients [95 % CI (30, 75 %)] treated at the MTD had potentially treatment-related grade 2, 3, and 4 adverse events that were encountered in any cycle as summarized in Table 2. The most commonly reported grade ≥2 adverse events probably or definitely related to the study drugs in this dose expansion cohort were fatigue (five patients), rash (four patients), and hypophosphatemia (three patients). There were no deaths related to the study drugs. Considering possible associations with vandetanib and/or sirolimus, additional toxicities were: leukopenia (one patient, grade 3), lymphopenia (three patients, grade 3; one patient, grade 4), photosensitivity with pruritis (one patient, grade 3), hypophosphatemia (one patient, grade 3), fatigue (one patient, grade 3), colitis (one patient, grade 3), elevated AST-SGOT (one patient, grade 3), prolonged QTc (one patient, grade 3), headache (one patient, grade 3), and thromboembolism (two patients, grade 4).
Response and survival
All analyses included patients who received at least a single dose of vandetanib and sirolimus. In the dose expansion cohort, 2 out of 19 patients achieved a partial response [10.5 %; 95 % CI (1, 33 %)], median progression-free survival was 1.9 months [95 % CI (0.1, 1.9 months)]; 6-month progression free survival was 15.8 % [95 % CI (3.9, 34.9 %)]. Median overall survival was 7.2 months [95 % CI (3.2, 8.8 months)] and 6-month overall survival was 63 % [95 % CI (38, 80 %)].
For patients studied at all dose levels, median overall survival was 7.7 months [95 % CI (4.7, 9.3 months)]; four patients were still alive at the time of analysis (Fig. 1). Median progression-free survival was 2.1 months [95 % CI (0.9, 3.1 months)] and 6-month progression-free survival was 18.2 %; one of these patients had received treatment at the lowest dose level (Fig. 2).

Overall survival. Kaplan–Meier survival curve for all patients having received at least one dose of vandetanib and sirolimus at the maximum tolerated dose

Progression-free survival. Kaplan–Meier progression-free survival curve for all patients having received at least one dose of vandetanib and sirolimus at the maximum tolerated dose
If we were to consider those patients with at least one cycle of treatment (as opposed to a single dose) at the MTD (15 patients), the median survival was 8.6 months [95 % CI (4.87, NA)] and the median progression free survival was 2.77 months [95 % CI (1.83, 3.27)].
Discussion
The phase I dose limiting toxicity of increased AST/SGOT has been observed in other studies of vandetanib [19]. The hematologic and dermatologic toxicities as well as fatigue are reported to occur with vandetanib or the sirolimus prodrug, temsirolimus [14, 19]. Although hypertension has been described in other studies of vandetanib [19–21], it was not noted in this study; significant QTc prolongation was observed in a single patient. Rash, as seen in other trials of vandetanib monotherapy in patients with solid tumors [20, 21], was similarly observed here. Diarrhea, which is also commonly encountered with vandetanib treatment [20, 21], was not a prominent toxicity in the patients enrolled in this trial. The two thromboembolic events in this study was not disproportionate in the context of the hypercoagulable state associated with glioblastoma [22], although they were deemed possibly related to the study drugs.
The progression-free survival proportion at 6 months observed in the dose expansion cohort of the study was greater than the 6.5 % reported for vandetanib monotherapy [23], 2.4–7.8 % for temsirolimus monotherapy [14, 24], and 3.1 % reported for erlotinib, an EGFR inhibitor, combined with sirolimus [25]. While this might suggest that the vandetanib and sirolimus combination has more activity than these agents, comparisons are difficult given the small numbers of patients studied. Responses in this study were comparable to the historical controls of patients with recurrent GBM treated with cytotoxic chemotherapy [26], but again, the caveat is that our sample size was low.
This study had several limitations. We did not measure pharmacokinetic (pK) or pharmacodynamic (pD) parameters and we did not allow intra-patient dose variation of the study drugs. Detailed pK and pD data is available with vandetanib monotherapy [16, 27] and sirolimus [17, 28, 29]. As monotherapy, vandetanib has been well tolerated and efficacious at doses up to 300 mg daily [30] however it has not been previously studied in combination with an mTOR inhibitor. Our study did not pre-select patients based on pathology that demonstrated activation of the VEGFR, EGFR, or PI3K/Akt/mTOR pathways. Tumor tissue specimens were not available for retrospective analysis of association with clinical outcomes. By treating unselected patients, the outcomes data from the dose expansion cohort of the study may have underestimated the response proportion.
In conclusion, the combination of vandetanib and sirolimus appears safe and feasible. The most commonly observed toxicities were lymphopenia, rash, hypophosphatemia, and fatigue. While the APF6 of 15.8 % for patients treated at MTD, and 18.2 % for all enrolled patients, is modest, our findings suggest that the combination of vandetanib and sirolimus is safe to use in further studies in patients with evidence of activation of these pathways. In particular, this regimen could be considered in combination with other agents targeting aberrant pathways in GBM.
References
Dolecek TA, Propp JM, Stroup NE, Kruchko C (2012) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro-Oncol 14(Suppl 5):vi–49. doi:10.1093/neuonc/nos218
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis M, Olson JJ, Mikkelsen T, Lehman N, Aldape K, Yung WKA, Bogler O, Vandenberg S, Berger M, Prados M, Muzny D, Morgan M, Scherer S, Sabo A, Nazareth L, Lewis L, Hall O, Zhu Y, Ren Y, Alvi O, Yao J, Hawes A, Jhangiani S, Fowler G, San Lucas A, Kovar C, Cree A, Dinh H, Santibanez J, Joshi V, Gonzalez-Garay ML, Miller CA, Milosavljevic A, Donehower L, Wheeler DA, Gibbs RA, Cibulskis K, Sougnez C, Fennell T, Mahan S, Wilkinson J, Ziaugra L, Onofrio R, Bloom T, Nicol R, Ardlie K, Baldwin J, Gabriel S, Lander ES, Ding L, Fulton RS, McLellan MD, Wallis J, Larson DE, Shi X, Abbott R, Fulton L, Chen K, Koboldt DC, Wendl MC, Meyer R, Tang Y, Lin L, Osborne JR, Dunford-Shore BH, Miner TL, Delehaunty K, Markovic C, Swift G, Courtney W, Pohl C, Abbott S, Hawkins A, Leong S, Haipek C, Schmidt H, Wiechert M, Vickery T, Scott S, Dooling DJ, Chinwalla A, Weinstock GM, Mardis ER, Wilson RK, Getz G, Winckler W, Verhaak RG, Lawrence MS, O’Kelly M, Robinson J, Alexe G, Beroukhim R, Carter S, Chiang D, Gould J, Gupta S, Korn J, Mermel C, Mesirov J, Monti S, Nguyen H, Parkin M, Reich M, Stransky N, Weir BA, Garraway L, Golub T, Meyerson M, Chin L, Protopopov A, Zhang J, Perna I, Aronson S, Sathiamoorthy N, Ren G, Yao J, Wiedemeyer WR, Kim H, Kong SW, Xiao Y, Kohane IS, Seidman J, Park PJ, Kucherlapati R, Laird PW, Cope L, Herman JG, Weisenberger DJ, Pan F, Van Den Berg D, Van Neste L, Yi JM, Schuebel KE, Baylin SB, Absher DM, Li JZ, Southwick A, Brady S, Aggarwal A, Chung T, Sherlock G, Brooks JD, Myers RM, Spellman PT, Purdom E, Jakkula LR, Lapuk AV, Marr H, Dorton S, Choi YG, Han J, Ray A, Wang V, Durinck S, Robinson M, Wang NJ, Vranizan K, Peng V, Van Name E, Fontenay GV, Ngai J, Conboy JG, Parvin B, Feiler HS, Speed TP, Gray JW, Brennan C, Socci ND, Olshen A, Taylor BS, Lash A, Schultz N, Reva B, Antipin Y, Stukalov A, Gross B, Cerami E, Wang WQ, Qin LX, Seshan VE, Villafania L, Cavatore M, Borsu L, Viale A, Gerald W, Sander C, Ladanyi M, Perou CM, Hayes DN, Topal MD, Hoadley KA, Qi Y, Balu S, Shi Y, Wu J, Penny R, Bittner M, Shelton T, Lenkiewicz E, Morris S, Beasley D, Sanders S, Kahn A, Sfeir R, Chen J, Nassau D, Feng L, Hickey E, Zhang J, Weinstein JN, Barker A, Gerhard DS, Vockley J, Compton C, Vaught J, Fielding P, Ferguson ML, Schaefer C, Madhavan S, Buetow KH, Collins F, Good P, Guyer M, Ozenberger B, Peterson J, Thomson E (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455(7216):1061–1068
Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, Chin L, DePinho RA (2007) Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318:287–290
Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD, Betensky RA, Louis DN, Iafrate AJ (2011) Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20:810–817. doi:10.1016/j.ccr.2011.11.005
Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, Wikstrand CJ, Van Duyn LB, Dancey JE, McLendon RE, Kao JC, Stenzel TT, Ahmed Rasheed BK, Tourt-Uhlig SE, Herndon JE 2nd, Vredenburgh JJ, Sampson JH, Friedman AH, Bigner DD, Friedman HS (2004) Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol 22:133–142. doi:10.1200/JCO.2004.08.110
Prados MD, Chang SM, Butowski N, DeBoer R, Parvataneni R, Carliner H, Kabuubi P, Ayers-Ringler J, Rabbitt J, Page M, Fedoroff A, Sneed PK, Berger MS, McDermott MW, Parsa AT, Vandenberg S, James CD, Lamborn KR, Stokoe D, Haas-Kogan DA (2009) Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J Clin Oncol 27:579–584. doi:10.1200/JCO.2008.18.9639
Prados MD, Lamborn KR, Chang S, Burton E, Butowski N, Malec M, Kapadia A, Rabbitt J, Page MS, Fedoroff A, Xie D, Kelley SK (2006) Phase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant glioma. Neuro-Oncology 8:67–78. doi:10.1215/S1522851705000451
Vivanco I, Robins HI, Rohle D, Campos C, Grommes C, Nghiemphu PL, Kubek S, Oldrini B, Chheda MG, Yannuzzi N, Tao H, Zhu S, Iwanami A, Kuga D, Dang J, Pedraza A, Brennan CW, Heguy A, Liau LM, Lieberman F, Yung WK, Gilbert MR, Reardon DA, Drappatz J, Wen PY, Lamborn KR, Chang SM, Prados MD, Fine HA, Horvath S, Wu N, Lassman AB, DeAngelis LM, Yong WH, Kuhn JG, Mischel PS, Mehta MP, Cloughesy TF, Mellinghoff IK (2012) Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov 2:458–471. doi:10.1158/2159-8290.CD-11-0284
Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Chester R, Jackson JA, Boffey SJ, Valentine PJ, Curwen JO, Musgrove HL, Graham GA, Hughes GD, Thomas AP, Stokes ES, Curry B, Richmond GH, Wadsworth PF, Bigley AL, Hennequin LF (2002) ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res 62:4645–4655
Carlomagno F, Vitagliano D, Guida T, Ciardiello F, Tortora G, Vecchio G, Ryan AJ, Fontanini G, Fusco A, Santoro M (2002) ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res 62:7284–7290
Rich JN, Sathornsumetee S, Keir ST, Kieran MW, Laforme A, Kaipainen A, McLendon RE, Graner MW, Rasheed BK, Wang L, Reardon DA, Ryan AJ, Wheeler C, Dimery I, Bigner DD, Friedman HS (2005) ZD6474, a novel tyrosine kinase inhibitor of vascular endothelial growth factor receptor and epidermal growth factor receptor, inhibits tumor growth of multiple nervous system tumors. Clin Cancer Res 11:8145–8157. doi:10.1158/1078-0432.ccr-05-0319
Tamura T, Minami H, Yamada Y, Yamamoto N, Shimoyama T, Murakami H, Horiike A, Fujisaka Y, Shinkai T, Tahara M, Kawada K, Ebi H, Sasaki Y, Jiang H, Saijo N (2006) A phase I dose-escalation study of ZD6474 in Japanese patients with solid, malignant tumors. J Thorac Oncol 1:1002–1009
Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, Peralba JM, Jenkins RB, Dakhil SR, Morton RF, Jaeckle KA, Scheithauer BW, Dancey J, Hidalgo M, Walsh DJ (2005) Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central cancer treatment group study. J Clin Oncol 23:5294–5304
Arcella A, Biagioni F, Antonietta Oliva M, Bucci D, Frati A, Esposito V, Cantore G, Giangaspero F, Fornai F (2013) Rapamycin inhibits the growth of glioblastoma. Brain Res 1495:37–51. doi:10.1016/j.brainres.2012.11.044
FDA (2011) Caprelsa (vandetanib) full prescribing information
FDA (2008) Rapamune (Sirolimus) full prescribing information
Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG (1990) Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol 8:1277–1280
Holden SN, Eckhardt SG, Basser R, de Boer R, Rischin D, Green M, Rosenthal MA, Wheeler C, Barge A, Hurwitz HI (2005) Clinical evaluation of ZD6474, an orally active inhibitor of VEGF and EGF receptor signaling, in patients with solid, malignant tumors. Ann Oncol 16:1391–1397. doi:10.1093/annonc/mdi247
Natale RB, Bodkin D, Govindan R, Sleckman BG, Rizvi NA, Capo A, Germonpre P, Eberhardt WE, Stockman PK, Kennedy SJ, Ranson M (2009) Vandetanib versus gefitinib in patients with advanced non-small-cell lung cancer: results from a two-part, double-blind, randomized phase II study. J Clin Oncol 27:2523–2529. doi:10.1200/JCO.2008.18.6015
Natale RB, Thongprasert S, Greco FA, Thomas M, Tsai CM, Sunpaweravong P, Ferry D, Mulatero C, Whorf R, Thompson J, Barlesi F, Langmuir P, Gogov S, Rowbottom JA, Goss GD (2011) Phase III trial of vandetanib compared with erlotinib in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol 29:1059–1066. doi:10.1200/JCO.2010.28.5981
Semrad TJ, O’Donnell R, Wun T, Chew H, Harvey D, Zhou H, White RH (2007) Epidemiology of venous thromboembolism in 9489 patients with malignant glioma. J Neurosurg 106:601–608. doi:10.3171/jns.2007.106.4.601
Kreisl TN, McNeill KA, Sul J, Iwamoto FM, Shih J, Fine HA (2012) A phase I/II trial of vandetanib for patients with recurrent malignant glioma. Neuro-Oncology 14:1519–1526. doi:10.1093/neuonc/nos265
Chang SM, Wen P, Cloughesy T, Greenberg H, Schiff D, Conrad C, Fink K, Robins HI, De Angelis L, Raizer J, Hess K, Aldape K, Lamborn KR, Kuhn J, Dancey J, Prados MD (2005) Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Investig New Drugs 23:357–361. doi:10.1007/s10637-005-1444-0
Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE 2nd, Marcello J, Norfleet JA, McLendon RE, Sampson JH, Friedman HS (2010) Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol 96:219–230. doi:10.1007/s11060-009-9950-0
Wong ET, Hess KR, Gleason MJ, Jaeckle KA, Kyritsis AP, Prados MD, Levin VA, Yung WK (1999) Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol 17:2572–2578
Martin P, Oliver S, Kennedy SJ, Partridge E, Hutchison M, Clarke D, Giles P (2012) Pharmacokinetics of vandetanib: three phase I studies in healthy subjects. Clin Ther 34:221–237. doi:10.1016/j.clinthera.2011.11.011
Mahalati K, Kahan BD (2001) Clinical pharmacokinetics of sirolimus. Clin Pharmacokinet 40:573–585
Gallant-Haidner HL, Trepanier DJ, Freitag DG, Yatscoff RW (2000) Pharmacokinetics and metabolism of sirolimus. Ther Drug Monit 22:31–35
Heymach JV, Paz-Ares L, De Braud F, Sebastian M, Stewart DJ, Eberhardt WE, Ranade AA, Cohen G, Trigo JM, Sandler AB, Bonomi PD, Herbst RS, Krebs AD, Vasselli J, Johnson BE (2008) Randomized phase II study of vandetanib alone or with paclitaxel and carboplatin as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol 26:5407–5415. doi:10.1200/JCO.2008.17.3138
Acknowledgments
We gratefully acknowledge Faye Safavi and Karly Griffin for aiding with data collection. This research was conducted with support from the Investigator-Sponsored Study Program of AstraZeneca. Authors received grant support from the NIH (K08 NS062907 and K12 CA090354 Career Development Awards to MGC; K24 CA125440 and K12 CA090354 to TTB), Brain Tumor Center of Memorial Sloan-Kettering Cancer Center, American Brain Tumor Association Fellowship, and American Association for Cancer Research/National Brain Tumor Foundation Fellowship to MGC; and Richard Floor Bequest (FHH). These data were presented in part at the annual meeting of the American Academy of Neurology (2012).
Conflict of interest
This research was conducted with support from the Investigator-Sponsored Study Program of AstraZeneca. PYW receives research support from AstraZeneca. RB owns shares in AstraZeneca. TTB receives research support from AstraZeneca. All other authors have no conflict of interest.
Ethical standards
All studies comply with the laws of the United States. The study was approved by the institutional review board at Dana-Farber/Harvard Cancer Center (Protocol 07-396).
Author information
Authors and Affiliations
Corresponding author
Additional information
Clinicaltrials.gov identifier: NCT00821080.
Rights and permissions
About this article

Cite this article
Chheda, M.G., Wen, P.Y., Hochberg, F.H. et al. Vandetanib plus sirolimus in adults with recurrent glioblastoma: results of a phase I and dose expansion cohort study. J Neurooncol 121, 627–634 (2015). https://doi.org/10.1007/s11060-014-1680-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-014-1680-2