
Abstract
Acute myeloid leukaemia (AML) is a complex haematological malignancy characterised by diverse genetic alterations leading to abnormal proliferation of myeloid precursor cells. One of the most significant genetic alterations in AML involves mutations in the FLT3 gene, which plays a critical role in haematopoiesis and haematopoietic homeostasis. This review explores the current understanding of FLT3 gene mutations and isoforms and the importance of the FLT3 protein in AML. FLT3 mutations, including internal tandem duplications (FLT3-ITD) and point mutations in the tyrosine kinase domain (FLT3-TKD), occur in 25–30% in AML and are associated with poor prognosis. FLT3-ITD mutations lead to constitutive activation of the FLT3 signalling pathway, promoting cell survival and proliferation. FLT3-TKD mutations affect the tyrosine kinase domain and affect AML prognosis in various ways. Furthermore, FLT3 isoforms, including shorter variants, contribute to the complexity of FLT3 biology. Additionally, nonpathological polymorphisms in FLT3 are being explored for their potential impact on AML prognosis and treatment response. This review also discusses the development of molecular treatments targeting FLT3, including first-generation and next-generation tyrosine kinase inhibitors, highlighting the challenges of resistance that often arise during therapy. The final chapter describes FLT3 protein domain rearrangements and their relevance to AML pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction and characterization of AML and its genetic landscape
One of the most severe haematological malignancies is acute myeloid leukaemia (AML), a heterogeneous clonal disorder characterised by the rapid proliferation of abnormal myeloid precursor cells in the bone marrow [1]. The development of this disease starts with accumulation of different classes of DNA mutations first in haematopoietic stem cells to form preleukaemic cells that eventually progress to AML [2]. AML can affect people of all ages but is more common in older adults. Fifty years ago, this disease was incurable but is now cured in 35 to 45% of adult patients. Patients aged 60 to 75 years have an enrichment of adverse karyotypic/molecular profiles and poor outcomes, with a median survival of ∼2 years [3, 4]. On the other hand, younger patients with AML who are otherwise healthy and have favourable genetic profiles may have a greater chance of achieving complete remission with intensive chemotherapy. With a good response to treatment, 50 to 80% or more of patients may have relatively high five-year survival rates [5,6,7].
The new 5th WHO classification (starting in 2022) divides AML patients according to genetic abnormalities and defines AML by the WHO differentiation. The mutations in AML can be divided into groups according to the functional class of the mutated genes: (1) signalling and kinase pathway, (2) epigenetic modifiers, (3) nucleophosmin, (4) transcription factors, (5) tumour suppressors, (6) spliceosome complex and (7) cohesin complex [8, 9]. The identification and classification of mutations according to the present mutation status have essential impacts on patient prognosis and treatment choice. In the last decade, there has been an intensified focus on the fms-like tyrosine kinase 3 (FLT3) gene because its mutation seems to be crucial for AML, and approximately 30% of AML patients harbour some form of FLT3 mutation [10,11,12].
The main therapeutic approach used for approximately forty years is a combination of anthracycline and cytarabine. The treatment utilised a protocol known as “3 + 7,” in which patients received a combination of anthracyclines for three days and cytarabine for seven days. Most recently, there is a new promise for patients in specific treatment in the form of inhibitors or cell therapy [13]. FLT3 mutations in AML have led to the development of numerous tyrosine kinase inhibitors (TKIs). The most effective FLT3 inhibitors include midostaurin, gilteritinib, crenolanib, sorafenib and quizartinib. However, patients often develop resistance, resulting in short and unsustainable responses [14].
Generally, mutations in AML can affect genes involved in signalling pathways, transcriptional regulation, epigenetic modifications, and cell cycle control. The identification of recurrent genetic alterations has allowed for the classification of AML into distinct subgroups, each with unique clinical and prognostic implications. Moreover, cytogenetic abnormalities, such as chromosomal translocations and rearrangements, are common in AML. These include the translocations t(8;21), inv(16)/t(16;16), and t(15;17), which result in fusion genes involving RUNX1-RUNX1T1, CBFB-MYH11, and PML-RARA, respectively [15]. These fusion genes disrupt normal gene expression and contribute to leukaemogenesis. Other recurrent genetic alterations in AML include mutations in genes such as NPM1, DNMT3A, IDH1/2, TET2, RUNX1, and TP53. Furthermore, recent research has shown the increasing importance of mutations in genes involved in growth signalling pathways (NRAS, KRAS, and PTPN11) [16]. These mutations affect various cellular processes, including DNA methylation, chromatin remodelling, and transcriptional regulation. These mutations contribute to the dysregulation of haematopoietic stem and progenitor cells, leading to the development of AML. As mentioned above, one of the most common genetic alterations in AML involves the FLT3 gene.
The role of FLT3 in AML
FLT3 plays a critical role in haematopoiesis, and the crucial importance of FLT3 is the regulation of haematopoietic stem and progenitor cells and maintenance of haematopoietic homeostasis. FLT3 is expressed in haematopoietic stem cells and in granulocyte/macrophage progenitor cells [17]. Experimental data suggest that FLT3 may also act on early lymphoid precursors, causing their development and thereby promoting the development of various immune cells, such as T cells, B cells, and natural killer (NK) cells; however, the exact mechanism is not known [18, 19]. FLT3 expression on leukaemic blasts can indicate the presence of FLT3 in most patients with AML [20]. Despite the major importance of FLT3 alterations in association with AML, they can also be detected in other diseases, as approximately 5% of acute lymphoblastic leukaemia (ALL) patients and few chronic lymphoblastic leukaemia (CLL) patients also exhibit FLT3 mutations [21, 22].
FLT3 mutations, particularly internal tandem duplications (FLT3-ITD) and point mutations in the tyrosine kinase domain (FLT3-TKD), are game changers in AML, as they are both associated with a poor prognosis. FLT3 mutations result in constitutive activation of the FLT3 signalling pathway, promoting cell survival and proliferation.
FLT3 gene structure and function
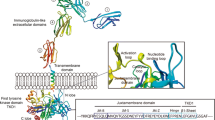
FLT3, also known as CD135, was identified 30 years ago, and its location is on chromosome 13q12.2. It belongs to the class III receptor tyrosine kinase (RTK) family and is a member of the RTK family together with the KIT, FMS, and PDGF receptors. All these proteins share structural characteristics, such as 5 immunoglobulin-like domains in the extracellular region, a juxtamembrane (JM) domain, 2 kinase domains (TK1 and TK2) separated by a kinase insert (KI) domain, and a C-terminal domain in the intracellular region – Fig. 1A [12, 23, 24].
In 1993, the full-length human FLT3 gene was successfully cloned from two different libraries: one from a pre-B-cell library [25] and the other from a CD34 + haematopoietic stem cell-enriched library [26]. The human FLT3 gene consists of 24 exons with a total length of 3,848 base pairs. Notably, certain sections of exon 1 and exon 24 remain untranslated [27, 28]. The entire genomic DNA sequence encompassing the FLT3 gene spans more than 97 kilobases [25]. The translation initiation codon is found in the latter part of the first exon. The signal peptide, which is responsible for guiding the protein to its proper location, is encoded by the first two exons. The extracellular immunoglobulin-like domains (ECD), which are crucial for receptor function, are encoded by exons 3 to 12. Exon 13 encodes the transmembrane domain, which anchors the protein to the cell membrane, and exons 14 through 24 are responsible for encoding the intracellular regions, specifically exons 14 and 15 for the JM domain, exons 15–17 for the TK1 domain, and 18–23 exons for the TK2 domain with a short kinase insert between TK1 and TK2 [28, 29]. The FLT3 receptor binds to its ligand, the FLT3 ligand (FL), which is produced by cells in the haematopoietic microenvironment. The interaction between FLT3 and FL triggers the activation of signalling pathways that regulate the growth and differentiation of haematopoietic cells. The interaction between the FLT3 receptor and FL leads to receptor dimerization and autophosphorylation of tyrosine residues in the intracellular domain [30,31,32]. The activated FLT3 receptor subsequently triggers a cascade of signalling pathways, including RAF/MEK/ERK, PI3K/AKT, and STAT pathways. Activated FLT3 (Fig. 1B) stimulates the proliferation of haematopoietic cells and blocks apoptotic processes with a possible triggering of hyperactivation leading to malignant proliferation [33, 34]. When the ligand is not present, the kinase activity of FLT3 is subject to negative modulation by tyrosine phosphatases, which catalyse the removal of phosphate groups from tyrosine residues within the unbound JM domain. This dephosphorylation event facilitates the JM domain to assume its autoinhibitory conformation, thereby suppressing the kinase activity of FLT3 [35, 36].

FLT3 receptor A: In a nonactivated state lack tyrosine kinase activity and cannot initiate signalling pathways within the cell. B: Upon binding of a ligand such as the FLT3 ligand (FL), the two FLT3 receptors dimerize. This results in autophosphorylation of the tyrosine kinase domains within the receptor. The green spots represent the phosphorylated tyrosines. Src-like protein (SLAP) is shown binding to phosphorylated tyrosine residues on the intracellular domain of FLT3. This interaction serves as a regulatory mechanism to inhibit further signalling events downstream of FLT3 activation. The figure was created with BioRender.com
FLT3 mutations in AML
The emergence of FLT3 mutations in AML typically occurs at later stages of disease development, rather than being among the initial mutations. Additionally, an extra 10% of patients acquire these mutations during the progression of the disease of AML. They arise during clonal evolution, contributing to disease heterogeneity and progression. On top of that, FLT3 alternations are prevalent upon AML diagnosis, exerting a significant influence on disease characterization, progression, and clinical management [37,38,39]. The two main types of FLT3 mutations found in AML are internal tandem duplications (FLT3-ITD), which affect the JM domain and its close surroundings, and point mutations in TKD or JM domain. FTL3 mutations are not detectable during standard sample analysis, but the FLT3 receptor is overexpressed on the cell surface of cell blasts and can contribute to the survival and proliferation of leukaemic clones [40, 41].
FLT3 internal tandem duplication (FLT3-ITD) mutation
From a molecular point of view, the FLT3-ITD mutation involves the insertion of a tandem repeat in a segment of DNA within the JM domain of the FLT3 gene [42]. This tandem duplication leads to the production of a longer FLT3 protein with an extended JM domain. The duplicated segment contains the activation loop of the tyrosine kinase domain, resulting in constitutive activation of FLT3 kinase activity [43, 44]. It is detected in approximately 25–30% of AML patients [45]. The presence and allelic burden (the ratio of mutant to wild-type (WT) FLT3-ITD alleles) can vary among patients and could have some impact on the prognosis and therapeutic implications. However, according to the 2022 ELN AML recommendations, the FLT3-ITD allelic ratio is no longer considered in the risk classification [11]. Furthermore, the FLT3-ITD mutation is associated with an adverse prognosis in AML. Patients with FLT3-ITD mutations often have a higher relapse rate than do those without this mutation [46]. Currently, we can find multiple studies where it is mentioned that the size of the ITD can have clinical implications, and generally, AML patients with larger ITD mutations tend to have a poorer prognosis compared to those with shorter ITD mutations. On the other hand, there are multiple studies that prove that the size of the ITD mutation has no significant effect on treatment response or survival. The consensus remains that FLT3-ITD presence is a sign of poor prognosis for AML patients, and the relationship between ITD size and clinical outcomes is complex and can be influenced by other factors such as the specific location of the mutation, the presence of other genetic abnormalities, and individual patient characteristics [47,48,49]. Recently, studies have also begun to appear about FLT3-VSI (very short in-frame insertions) and reveal interesting trends. Surprisingly, a nonsignificant trend for decreased survival was observed in patients with simple FLT3 VSIs compared with composite cases with combined VSI and ITD abnormalities [50]. A significant diagnostic marker for this mutation is also a higher leucocyte count (due to an increased production of blasts that is stimulated by FLT3) at the time of diagnosis [51, 52]. AML patients with FLT3-ITD mutations generally have shorter overall survival than patients with FLT3 wild-type mutations [44]. FLT3-ITD status is routinely evaluated in the diagnostic workup of AML patients and helps guide treatment decisions to optimise patient outcomes [53]. Approximately 30% of ITDs are localised within the TKD region. Patients carrying ITD mutations in the TKD (TKD-ITD) demonstrate a poorer prognosis for survival in contrast to those with JMD-ITD, the underlying cause of which remains elusive [54].
FLT3-ITD is often associated with other molecular and cytogenetic abnormalities in AML, such as NPM1 mutations, DNMT3A mutations, and adverse cytogenetics [55, 56]. The co-occurrence of FLT3-ITD and other molecular or cytogenetic abnormalities can affect the prognosis of AML patients [57]. The most common cooccurring mutation in patients with FLT3-ITD mutations was NPM1 mutation, and these patients had a more favourable prognosis than did those with FLT3-ITD mutations alone. This combination is associated with a higher rate of complete remission and improved overall survival [58]. Moreover, the presence of DNMT3A mutations combined with FLT3-ITD mutations can be associated with a less favourable prognosis. Patients with both mutations may have a higher risk of relapse and a poorer response to standard treatments [59]. The combination of FLT3-ITD and TP53 in AML patients indicates that the prognosis tends to be particularly unfavourable. This combination of mutations may lead to a more aggressive disease course, reduced response to standard therapies, and shorter overall survival [60].
FLT3-point mutations
The FLT3-TKD mutation is a determined genetic alteration that affects mainly the tyrosine kinase domain. In the JM domain of the FLT3 gene, point mutations and deletions may also be observed. However, due to the low frequency of the alterations in the JM domain, there is only limited information on molecular and clinical associations [61]. The D835Y/V substitution constitutes approximately 50% of FLT3-TKD mutations in which the aspartic acid at position 835 is typically replaced by valine or tyrosine. Other FLT3 mutations that occur less commonly in AML include N676K, Y842C, K663Q, V592A, N841I, N841K, S451F, Y572C, R834Q, V579A, and F594L [28, 62, 63]. These substitutions can impact the structure or activity of the FLT3 kinase. The prevalence of the FLT3-TKD mutation is lower than that of the FLT3-ITD mutation, which is found in approximately 5–10% of AML patients. The impact of the FLT3-TKD mutation on AML prognosis is not as clear as that of the FLT3-ITD mutation. Some studies suggest an adverse prognosis, while other reports indicate variable outcomes depending on additional genetic or molecular factors [64,65,66]. It is important to note that the specific FLT3-TKD mutation can vary among individual patients and may differ in terms of amino acid substitution. The specific effects of individual FLT3-TKD mutations on the biology and prognosis of AML are still being investigated. The identification of the FLT3-TKD mutation was performed as part of the diagnostic workup for AML [43, 44].
FLT3 gene isoforms, polymorphisms, and splicing aberrations
The isoforms represent variant forms of a gene or its protein, offering a nuanced perspective on genetic diversity and functional adaptability. The isoforms arise from alternative splicing, alternative transcription initiation or termination, or other mechanisms. These isoforms often have similar sequences but may differ in certain regions. In addition to the primary, full-length FLT3 receptor, several shorter isoforms that lack certain functional domains have been described. The exact physiological roles of these shorter isoforms are not completely understood [67, 68]. However, alternative splicing and the creation of various isoforms are common mechanisms that allow for the functional diversity of proteins, and it is likely that different FLT3 isoforms may have slightly different roles in cell signalling and haematopoiesis [69]. Aberrant splicing of the FLT3 gene results in the formation of the common mutant isoforms FLT3-ITD and FLT3-TKD, the importance of which is mentioned above. Moreover, FLT3 splicing results in other isoforms are less strongly associated with AML. One well-studied isoform is commonly named isoform 2 (XM_011535018.3), which lacks exon 19 and leads to the formation of a truncated and nonfunctional tyrosine kinase domain [70].

Overview of isoforms and polymorphism-like isoforms of FLT3 gene. A: This scheme is alignment of NCBI annotated mRNA sequences of FLT3 gene with marked differences. Lacking exons are grey, shorter sequence of exon is in light blue and *purple is the same exon sequence on different positions compared to other isoforms. Exons 14 and 15 bounded by a red rectangle is the locus where ITD mutation is occurring. B: On this scheme are isoforms that are annotated polymorphism-like isoforms in the NCBI database and also in the LOVD database. Here are shown positions of 11 following listed isoforms and how this polymorphism affects amino acid type production: (1) FLT3(NM_004119.2):c.580G > A (p.(Val194Met); (2) FLT3(NM_004119.2):c.1301 A > G (p.N434S); (3) FLT3(NM_004119.2):c.970G > A (p.(Asp324Asn)); (4) FLT3(NM_004119.2):c.1032 C > T (p.I344=); 5.FLT3(NM_004119.3):c.288 C > T (p.D96=); 6.FLT3(NM_004119.2):c.1815T > C (p.F605=); 7. FLT3(NM_004119.2):c.615-15 C > A (p.(=); 8. FLT3(NM_004119.2):c.743-9 A > G; 9. FLT3(NM_004119.2):c.592 C > T (p.L198F); 10. FLT3(NM_004119.2):c.73 A > G (p.I25V); 11. FLT3(NM_004119.2):c.499 A > G (p.T167A). The figure was created with BioRender.com
Previously, we introduced two isoforms of the FLT3 gene linked to AML disease, which are specifically associated with DNA mutations. Additionally, there are likely nonpathological variants, commonly referred to as polymorphisms, which are prevalent in the general population. Global databases about human genome variability currently annotate twenty polymorphism-like isoforms, where eleven of them are annotated in NCBI database (Fig. 2) [71]. Although the importance of their function remains uncertain, ongoing studies are elucidating a more precise comprehension of the significance of these polymorphisms. Clinical trials have indicated the potential discovery of polymorphisms associated with improved prognosis in adult AML patients. However, these findings mostly fall below a statistically significant threshold [72]. In a yet unpublished study by Alsheikh et al., in silico analysis of various algorithms revealed 20 novel nonsynonymous single nucleotide polymorphisms (nsSNPs) in the FLT3 gene that are considered damaging and contribute to AML. Moreover, 12 SNPs in the 3’UTR were predicted to have 69 different functional classes. Among them, 31 alleles affected a conserved miRNA site, while 37 derived alleles created a new miRNA site [73]. As was shown for other genes, it could be expected that particular polymorphisms of FLT3 may alter or modify disease and treatment outcomes. It was observed that the DCK 201-T and CDA-79-A variants are connected to conventional treatment toxicity in AML. Patients with the variant allele of DCK 201-T or the wild-type allele of CDA-79-A exhibited a significantly elevated risk of liver impairment (p = 0.014 and < 0.001, respectively) and renal impairment (p = 0.004 and 0.002, respectively). Moreover, patients with the variant allele of DCK 360-G or the wild-type allele of CDA-79-A demonstrated a notably greater association with neurological toxicity (p = 0.002 and 0.025, respectively) [74,75,76].
Furthermore, alternative splicing aberrations on FLT3 refer to irregularities in gene expression that can result in different isoforms of the FLT3 receptor. Some of these isoforms may lack the ECD due to these aberrations. The ECD of FLT3 is essential for ligand binding and receptor dimerization, both of which are necessary for receptor activation. This leads to a loss of function of the receptor, resulting in impaired signalling. Alternatively, the absence of the ECD could result in constitutive activation of the receptor, leading to uncontrolled cell proliferation [77, 78].
FLT3 protein and domain rearrangements
The FLT3 gene in humans encodes a protein comprising 993 amino acids. More than half of the protein is taken up by the extracellular domain, within which the transmembrane region is positioned between amino acids 542 and 564 [28].
After translation, the FLT3 protein undergoes a number of posttranslational changes, including glycosylation, phosphorylation, and ubiquitination. The glycosylation process consists of two steps. The FLT3 protein is partially glycosylated in the endoplasmic reticulum, where it enters during translation, where it forms the immature receptor. The other and final glycosylation occurs in the Golgi complex. The mature receptor is formed and translocated to the cell surface. It has at least nine N-linked glycosylation sites [79]. The first one can be found after the signal peptide at N43. Ig-like domain 1 (D1) contains two domains, N100 and N151. The second Ig-like domain (D2) contains no glycosylation site. The other Ig-like domains 3–5 each contain two glycosylation sites: D3 N306 and N323, D4 N351 and N354, and D5 N473 and N502 [79].
The FLT3 protein is phosphorylated at serine, threonine, and tyrosine residues. More than 10 tyrosine residues are phosphorylated upon ligand-induced activation of the receptor—the positions of known phosphorylation sites are Y572, Y589, Y591, and Y599 in the JM domain and Y726, Y768, Y793, Y842, Y955, and Y969 in the kinase domain. The intracellular domain contains these phosphorylation sites, which serve as docking sites for interactions with signalling proteins to advance the receptor signal throughout the cell [77]. However, the phosphorylation sites of serine and threonine in FLT3 have not been sufficiently investigated. Phosphorylation and its regulatory role have already been described for other related RTKs, and these proteins could also play important regulatory roles [80].
Protein domain rearrangements, specifically ITDs, are a significant subtype of FLT3 mutation and are characterised by the insertion of a variable number of amino acids within the juxtamembrane domain (JM domain) of the FLT3 receptor [81]. The exact length and sequence of the inserted segment can vary between patients, making the ITD mutations heterogeneous [82] (Fig. 3).

Conformation of intracellular part of FLT3 – the native FLT3 kinase and juxtamembrane domains (the model was built using the Protein Builder tool available in BioRender.com from the PDB entries: 1RJB [35])
Possibilities of molecular treatment for FLT3 and its development
Over the last few years, there has been significant progress in studying the fundamental disease-causing mechanisms of acute myeloid leukaemia (AML). This research has greatly enhanced the understanding of the condition of AML patients. The key determinants of how AML responds to chemotherapy and its long-term prognosis are cytogenetic and molecular abnormalities. However, these factors not only aid in predicting outcomes but also hold promise as potential targets for therapeutic interventions [83, 84]. Due to its prevalent mutations in acute myeloid leukaemia (AML), FLT3 has emerged as a promising therapeutic target, resulting in the development of numerous inhibitors [85]. FLT3 inhibitors are categorised as first-generation or next-generation tyrosine kinase inhibitors (TKIs), primarily because of their potency and specificity for FLT3 and its downstream targets [86]. The initial group, which included sunitinib, sorafenib, and midostaurin, exhibited relatively broad activity beyond FLT3, affecting other potential targets, such as KIT, PDGFR, VEGFR, RAS/RAF, and JAK2 kinases. While this versatility may contribute to heightened toxicity and clinical effectiveness in patients with non-FLT3-mutated acute myeloid leukaemia (AML), it leads to reduced efficacy in patients with FLT3-mutated AML with substantial allelic burden. In contrast, subsequent generations of inhibitors, such as quizartinib, crenolanib, and gilteritinib, exhibit enhanced FLT3 specificity and potency. These agents boast lower half-maximal inhibitory concentrations (IC50) and fewer side effects stemming from off-target interactions [87,88,89]. Furthermore, these FLT3 inhibitors fall into the Type I and Type II classifications, reflecting the distinct mechanisms through which they interact with the FLT3 receptor. Type I inhibitors bind the FLT3 receptor in both active and inactive conformation, either near the activation loop or the ATP-binding pocket, and are active against ITD and TKD mutations. Type II inhibitors bind the FLT3 receptor in the inactive conformation in a region adjacent to the ATP-binding domain [14].
Numerous FLT3 inhibitors, including midostaurin, sorafenib, quizartinib, and gilteritinib, have gained approval for cancer treatment. However, patients often develop resistance shortly after beginning FLT3 inhibitor therapy, leading to brief and unsustainable responses. This resistance arises from compensatory activation of FLT3 downstream pathways, safeguarding the bone marrow microenvironment, specific gene mutations, and activation of alternative proteins, collectively undermining the clinical impact of treatment [90, 91].
Among the most effective inhibitors are midostaurin, gilteritinib, crenolanib and quizartinib [14]; unfortunately, several mechanisms of resistance have already been identified [92]. The most commonly observed mechanism of resistance is secondary FLT3 mutation. While the FLT3 inhibitor initially targets and suppresses the primary FLT3 mutation (FLT3-ITD), AML cells can acquire additional mutations in the FLT3 gene that render the inhibitor less effective. These secondary mutations can alter the structure of the FLT3 kinase domain and reduce inhibitor binding [93]. Type II inhibitors have no affinity for FLT3-TKD. Mutations in TKD can confer resistance by decreasing the binding affinity of type II inhibitors to the ATP binding site. For instance, the activation loop of the TKD has the most prevalent TKD mutation, residue D835, which may result in a decreased binding affinity for type II inhibitors. A mutation that affects the TKD residue F691 can potentially result in resistance to type I and type II inhibitors [94]. Furthermore, AML cells can activate alternative signalling pathways that bypass the need for FLT3 signalling. This process can involve the upregulation of other receptor tyrosine kinases or downstream signalling molecules, allowing cells to continue proliferating and surviving despite FLT3 inhibition [95]. The clonal evolution of the AML escape subclone or a pharmacokinetic factor can also play a significant role [96]. Their deeper description is beyond the scope of this review, but it does not detract from their importance.
Conclusion
In conclusion, gene mutations and isoforms of FLT3 in acute myeloid leukaemia (AML) play intricate and pivotal roles in AML pathogenesis [97]. FLT3 mutations, particularly internal tandem duplications (FLT3-ITD) and tyrosine kinase domain mutations (FLT3-TKD), are common in AML and are associated with various prognoses. FLT3-ITD mutations lead to constitutive activation of the FLT3 signalling pathway, promoting cell survival and proliferation, while the impact of FLT3-TKD mutations on AML prognosis varies [98]. While there is growing research into new FLT3 mutations, the significance of FLT3 expression levels both for wild-type patients, which are highly expressed in most acute leukaemias, and for patients with mutated FLT3 has received limited attention thus far [99, 100]. The existence of FLT3 isoforms adds to the complexity of FLT3 biology, and ongoing research is elucidating the specific roles of these isoforms in cell signalling and haematopoiesis [79]. Nonpathological polymorphisms in FLT3 are also being investigated for their potential impact on AML prognosis and treatment response [101]. The development of molecular treatments targeting FLT3, including both first-generation and next-generation tyrosine kinase inhibitors, holds promise for AML patients [102]. However, the challenge of resistance to these therapies remains a significant hurdle. The study of FLT3 protein domain rearrangements further deepens our understanding of AML pathogenesis [103]. Overall, comprehending the nuances of FLT3 mutations, isoforms, and protein function is essential for the development of targeted therapies and the improvement of outcomes for AML patients. As research in this field continues to evolve, novel insights and therapies may continue to emerge, offering new hope for those affected by this challenging disease.
Data availability
No datasets were generated or analysed during the current study.
References
Saultz JN, Garzon R (2016) Acute myeloid leukemia: a concise review. J Clin Med 5(3):33. https://doi.org/10.3390/jcm5030033
Assi SA, Bonifer C, Cockerill PN (2019) Rewiring of the Transcription Factor Network in Acute myeloid leukemia. Cancer Inf 18:1176935119859863. https://doi.org/10.1177/1176935119859863
Döhner H, Weisdorf DJ, Bloomfield CD (2015) Acute myeloid leukemia. N Engl J Med 373(12):1136–1152. https://doi.org/10.1056/NEJMra1406184
Tey SK, Lane SW (2022) Better the cure you know: why patients with AML ≥ 60 years of age should be offered early allogeneic stem cell transplantation. Blood Adv 6(5):1619–1622. https://doi.org/10.1182/bloodadvances.2021004829
Jaime-Pérez JC, Padilla-Medina JR, Fernández LT et al (2018) Outcomes of adolescents and young adults with Acute myeloid leukemia treated in a single latin American Center. Clin Lymphoma Myeloma Leuk 18(4):286–292. https://doi.org/10.1016/j.clml.2018.02.002
Hossain MJ, Xie L, Caywood EH (2015) Prognostic factors of childhood and adolescent acute myeloid leukemia (AML) survival: evidence from four decades of US population data. Cancer Epidemiol 39(5):720–726. https://doi.org/10.1016/j.canep.2015.06.009
Lalayanni C, Demosthenous C, Iskas M et al (2022) Adolescents and young adults (AYA) with acute myeloid leukemia (AML): real-world long-term results and age-specific outcomes. Leuk Lymphoma 63(13):3128–3137. https://doi.org/10.1080/10428194.2022.2113527
Huber S, Baer C, Hutter S et al (2023) AML classification in the year 2023: how to avoid a babylonian confusion of languages. Leukemia 37(7):1413–1420. https://doi.org/10.1038/s41375-023-01909-w
DiNardo CD, Cortes JE (2016) Mutations in AML: prognostic and therapeutic implications. Hematol Am Soc Hematol Educ Program 2016(1):348–355. https://doi.org/10.1182/asheducation-2016.1.348
Johansson B, Harrison CJ (2010) Acute myeloid leukemia. Cancer Cytogenetics, 1st edn. Wiley, New York, pp 45–139. https://doi.org/10.1002/9781118010136.ch5.
Döhner H, Wei AH, Appelbaum R et al (2022) Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 22(12):1345–1377. https://doi.org/10.1182/blood.2022016867
Patnaik MM (2018) The importance of FLT3 mutational analysis in acute myeloid leukemia. Leuk Lymphoma 59(10):2273–2286. https://doi.org/10.1080/10428194.2017.1399312
Lim SH, Dubielecka PM, Raghunathan VM (2017) Molecular targeting in acute myeloid leukemia. J Transl Med 15(1):183. https://doi.org/10.1186/s12967-017-1281-x
Zhao JC, Agarwal S, Ahmad H et al (2022) A review of FLT3 inhibitors in acute myeloid leukemia. Blood Rev 52:100905. https://doi.org/10.1016/j.blre.2021.100905
Meraj F, Jamal S, Javed O et al (2023) Cytogenetic profiling in paediatric Acute Leukaemia; a Report on 746 newly diagnosed paediatric cases analyzing the spectrum of recurring chromosomal rearrangements in B cell lymphoblastic and acute myeloid leukaemia. J Ayub Med Coll Abbottabad 35(2):196–202. https://doi.org/10.55519/JAMC-02-11634
Yu J, Li Y, Zhang D, Wan D, Jiang Z (2020) Clinical implications of recurrent gene mutations in acute myeloid leukemia. Exp Hematol Oncol 9:4. https://doi.org/10.1186/s40164-020-00161-7
Kikushige Y, Yoshimoto G, Miyamoto T et al (2008) Human Flt3 is expressed at the hematopoietic stem cell and the granulocyte/macrophage progenitor stages to maintain cell survival. J Immunol 180(11):7358–7367. https://doi.org/10.4049/jimmunol.180.11.7358
Blom B, Spits H (2006) Development of human lymphoid cells. Annu Rev Immunol 24:287–320. https://doi.org/10.1146/annurev.immunol.24.021605.090612
Hernández-Barrientos D, Pelayo R, Mayani H (2023) The hematopoietic microenvironment: a network of niches for the development of all blood cell lineages. J Leukoc Biol 114(5):404–420. https://doi.org/10.1093/jleuko/qiad075
Fenski R, Flesch K, Serve S et al (2000) Constitutive activation of FLT3 in acute myeloid leukaemia and its consequences for growth of 32D cells. Br J Haematol 108(2):322–330. https://doi.org/10.1046/j.1365-2141.2000.01831.x
Okabe A, Guirales F, Zhao D, Tirado CA (2021) FLT3 gene involvement in B-cell Acute Lymphoblastic Leukemia (B-ALL). J Assoc Genet Technol 47(1):6–14
Rosnet O, Bühring HJ, deLapeyrière O et al (1996) Expression and signal transduction of the FLT3 tyrosine kinase receptor. Acta Haematol 95(3–4):218–223. https://doi.org/10.1159/000203881
Sakaguchi M, Yamaguchi H, Kuboyama M et al (2019) Significance of FLT3-tyrosine kinase domain mutation as a prognostic factor for acute myeloid leukemia. Int J Hematol 110(5):566–574. https://doi.org/10.1007/s12185-019-02720-z
Yamamoto Y, Kiyoi H, Nakano Y et al (2001) Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 97(8):2434–2439. https://doi.org/10.1182/blood.v97.8.2434
Rosnet O, Schiff C, Pébusque MJ et al (1993) Human FLT3/FLK2 gene: cDNA cloning and expression in hematopoietic cells. Blood 82(4):1110–1119
Smith CC, Wang Q, Chin CS et al (2012) Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 485(7397):260–263. https://doi.org/10.1038/nature11016
Lejman M, Dziatkiewicz I, Jurek M (2022) Straight to the point-the novel strategies to Cure Pediatric AML. Int J Mol Sci 23(4):1968. https://doi.org/10.3390/ijms23041968
Kazi JU, Rönnstrand L (2019) FMS-like tyrosine kinase 3/FLT3: from Basic Science to Clinical implications. Physiol Rev 99(3):1433–1466. https://doi.org/10.1152/physrev.00029.2018
Cumbo C, Tarantini F, Anelli L et al (2022) FLT3 mutational analysis in acute myeloid leukemia: advantages and pitfalls with different approaches. Blood Rev 54:100928. https://doi.org/10.1016/j.blre.2022.100928
Fukuda S, Broxmeyer HE, Pelus LM (2005) Flt3 ligand and the Flt3 receptor regulate hematopoietic cell migration by modulating the SDF-1alpha(CXCL12)/CXCR4 axis. Blood 105(8):3117–3126. https://doi.org/10.1182/blood-2004-04-1440
Gebru MT, Wang HG (2020) Therapeutic targeting of FLT3 and associated drug resistance in acute myeloid leukemia. J Hematol Oncol 13(1):155. https://doi.org/10.1186/s13045-020-00992-1
Heiss E, Masson K, Sundberg C et al (2006) Identification of Y589 and Y599 in the juxtamembrane domain of Flt3 as ligand-induced autophosphorylation sites involved in binding of src family kinases and the protein tyrosine phosphatase SHP2. Blood 108(5):1542–1550. https://doi.org/10.1182/blood-2005-07-008896
Steelman LS, Franklin RA, Abrams SL et al (2011) Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 25(7):1080–1094. https://doi.org/10.1038/leu.2011.66
Gilliland DG, Griffin JD (2002) The roles of FLT3 in hematopoiesis and leukemia. Blood 100(5):1532–1542. https://doi.org/10.1182/blood-2002-02-0492
Griffith J, Black J, Faerman C et al (2004) The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell 13(2):169–178. https://doi.org/10.1016/s1097-2765(03)00505-7
Du Z, Lovly CM (2018) Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer 17(1):58. https://doi.org/10.1186/s12943-018-0782-4
Levis M, Small D (2003) FLT3: ITDoes matter in leukemia. Leukemia 17(9):1738–1752. https://doi.org/10.1038/sj.leu.2403099
Small D (2006) FLT3 mutations: biology and treatment. Hematol Am Soc Hematol Educ Program 178 – 84. https://doi.org/10.1182/asheducation-2006.1.178
Kiyoi H, Kawashima N, Ishikawa Y (2020) FLT3 mutations in acute myeloid leukemia: therapeutic paradigm beyond inhibitor development. Cancer Sci 111(2):312–322. https://doi.org/10.1111/cas.14274
Ambinder AJ, Levis M (2021) Potential targeting of FLT3 acute myeloid leukemia. Haematologica 106(3):671–681. https://doi.org/10.3324/haematol.2019.240754
Jahn N, Terzer T, Sträng E et al (2020) Genomic heterogeneity in core-binding factor acute myeloid leukemia and its clinical implication. Blood Adv 4(24):6342–6352. https://doi.org/10.1182/bloodadvances.2020002673
Lo Iudice G, De Bellis E, Savi A et al (2022) Molecular dissection of a hyper-aggressive CBFB-MYH11/FLT3-ITD-positive acute myeloid leukemia. J Transl Med 20(1):311. https://doi.org/10.1186/s12967-022-03486-5
Todde G, Friedman R (2019) Conformational modifications induced by internal tandem duplications on the FLT3 kinase and juxtamembrane domains. Phys Chem Chem Phys 21(34):18467–18476. https://doi.org/10.1039/c9cp02938a
Kellner F, Keil A, Schindler K et al (2020) Wild-type FLT3 and FLT3 ITD exhibit similar ligand-induced internalization characteristics. J Cell Mol Med 24(8):4668–4676. https://doi.org/10.1111/jcmm.15132
Niswander LM, Graff ZT, Chien CD et al (2023) Potent preclinical activity of FLT3-directed chimeric antigen receptor T-cell immunotherapy against FLT3- mutant acute myeloid leukemia and KMT2A-rearranged acute lymphoblastic leukemia. Haematologica 108(2):457–471. https://doi.org/10.3324/haematol.2022.281456
Tao S, Wang C, Chen Y et al (2019) Prognosis and outcome of patients with acute myeloid leukemia based on FLT3-ITD mutation with or without additional abnormal cytogenetics. Oncol Lett 18(6):6766–6774. https://doi.org/10.3892/ol.2019.11051
Kim Y, Lee GD, Park J et al (2015) Quantitative fragment analysis of FLT3-ITD efficiently identifying poor prognostic group with high mutant allele burden or long ITD length. Blood Cancer J 5(8):e336. https://doi.org/10.1038/bcj.2015.61
Castaño-Bonilla T, Alonso-Dominguez JM, Barragán E et al (2021) Prognostic significance of FLT3-ITD length in AML patients treated with intensive regimens. Sci Rep 11(1):20745. https://doi.org/10.1038/s41598-021-00050-x
Lagunas-Rangel FA, Chávez-Valencia V (2017) FLT3-ITD and its current role in acute myeloid leukaemia. Med Oncol 34(6):114. https://doi.org/10.1007/s12032-017-0970-x
Tamburini J, Mouche S, Larrue C et al (2023) Very short insertions in the FLT3 gene are of therapeutic significance in acute myeloid leukemia. Blood Adv 7(24):7576–7580. https://doi.org/10.1182/bloodadvances.2023011916
Zalpoor H, Rezaei M, Yahyazadeh S, Ganjalikhani-Hakemi M (2022) Flt3-ITD mutated acute myeloid leukemia patients and COVID-19: potential roles of autophagy and HIF-1α in leukemia progression and mortality. Hum Cell 35(4):1304–1305. https://doi.org/10.1007/s13577-022-00718-0
Tien FM, Tsai CH, Huang SC et al (2022) Distinct clinico-biological features in AML patients with low allelic ratio FLT3-ITD: role of allogeneic stem cell transplantation in first remission. Bone Marrow Transpl 57(1):95–105. https://doi.org/10.1038/s41409-021-01454-z
Aitken MJL, Ravandi F, Patel KP, Short NJ (2021) Prognostic and therapeutic implications of measurable residual disease in acute myeloid leukemia. J Hematol Oncol 14(1):137. https://doi.org/10.1186/s13045-021-01148-5
Marhäll A, Heidel F, Fischer T, Rönnstrand L (2018) Internal tandem duplication mutations in the tyrosine kinase domain of FLT3 display a higher oncogenic potential than the activation loop D835Y mutation. Ann Hematol 97(5):773–780. https://doi.org/10.1007/s00277-018-3245-5
Mer AS, Heath EM, Madani Tonekaboni SA et al (2021) Biological and therapeutic implications of a unique subtype of NPM1 mutated AML. Nat Commun 12(1):1054. https://doi.org/10.1038/s41467-021-21233-0
Lachowiez CA, Reville PK, Kantarjian H et al (2022) Contemporary outcomes in IDH-mutated acute myeloid leukemia: the impact of co-occurring NPM1 mutations and venetoclax-based treatment. Am J Hematol 97(11):1443–1452. https://doi.org/10.1002/ajh.26694
Reikvam H (2023) Revisiting the prognostic role of FLT3 mutations in acute myelogenous leukemia. Expert Rev Hematol 16(5):317–323. https://doi.org/10.1080/17474086.2023.2202849
Döhner K, Thiede C, Jahn N et al (2020) Impact of NPM1/FLT3-ITD genotypes defined by the 2017 European LeukemiaNet in patients with acute myeloid leukemia. Blood 135(5):371–380. https://doi.org/10.1182/blood.2019002697
Varelas C, Papalexandri A, Iskas M et al (2023) PB1894: NPM1 mutated Acute myeloid leukemia: the co-mutation patterns may be Associated with Prognosis. Hemasphere 7(Suppl):e756808d. https://doi.org/10.1097/01.HS9.0000974400.75680.8d
Hammer ASB, Juul-Dam KL, Sandahl JD et al (2023) Hypodiploidy has unfavorable impact on survival in pediatric acute myeloid leukemia: an I-BFM Study Group collaboration. Blood Adv 7(6):1045–1055. https://doi.org/10.1182/bloodadvances.2022008251
Stasik S, Kramer M, Zukunft S et al (2022) Point mutations in the FLT3-ITD region are rare but recurrent alterations in adult AML and Associated with concomitant KMT2A-PTD. Front Oncol 12:862991. https://doi.org/10.3389/fonc.2022.862991
Mahmoudi A, Moradabadi A, Noroozi-Aghideh A (2021) Comparison of high-resolution melting analysis with direct sequencing for detection of FLT3-TKD, FLT3-ITD and WT1 mutations in acute myeloid leukemia. Cancer Treat Res Commun 28:100432. https://doi.org/10.1016/j.ctarc.2021.100432
Whitman SP, Ruppert AS, Radmacher MD et al (2008) FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood 111(3):1552–1559. https://doi.org/10.1182/blood-2007-08-107946
Shimony S, Stahl M, Stone RM (2023) Acute myeloid leukemia: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol 98(3):502–526. https://doi.org/10.1002/ajh.26822
Short NJ, Kantarjian H, Ravandi F, Daver N (2019) Emerging treatment paradigms with FLT3 inhibitors in acute myeloid leukemia. Ther Adv Hematol 10:2040620719827310. https://doi.org/10.1177/2040620719827310
Carter JL, Hege K, Yang J et al (2020) Targeting multiple signaling pathways: the new approach to acute myeloid leukemia therapy. Signal Transduct Target Ther 5(1):288. https://doi.org/10.1038/s41392-020-00361-x
Stirewalt DL, Radich JP (2003) The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer 3(9):650–665. https://doi.org/10.1038/nrc1169
Fatemeh S (2023) Early fate decissions in hematopoietic stem and progenitor cells. Through the lens of genomic and functional assays. Dissertation, Lund University
Lu PCW, Shahbaz S, Winn LM (2020) Benzene and its effects on cell signaling pathways related to hematopoiesis and leukemia. J Appl Toxicol 40(8):1018–1032. https://doi.org/10.1002/jat.3961
Dlamini Z, Shoba B, Hull R (2020) Splicing machinery genomics events in acute myeloid leukaemia (AML): in search for therapeutic targets, diagnostic and prognostic biomarkers. Am J Cancer Res 10(9):2690–2704
https://databases.lovd.nl/shared/variants/FLT3/unique - last updated June 15th 2021
Marrero RJ, Cao X, Wu H et al (2023) SAMHD1 single nucleotide polymorphisms impact outcome in children with newly diagnosed acute myeloid leukemia. Blood Adv 7(11):2538–2550. https://doi.org/10.1182/bloodadvances.2022009088
Alsheikh T, Ameer T, NjmEldin A et al (2023) June Twenty novel nsSNPs may affect FLT3 gene leading to Acute Myeloid Leukemia (AML) using in silico analysis. Biorxiv - The Preprint server for biology. https://www.biorxiv.org/content/https://doi.org/10.1101/2023.06.24.546344v1.full Accessed 26
Rasekh EO, Amin EA, Yassa ME et al (2022) The Prognostic Significance of Genetic Polymorphisms of Deoxycytidine Kinase and Cytidine Deaminase on the outcome of adult Acute myeloid leukemia patients with Cytarabine Based Chemotherapy. Int J Hematol 33(1):081–092
Kolonen A, Sinisalo M, Huhtala H et al (2022) Efficacy of conventional-dose cytarabine, idarubicin and thioguanine versus intermediate-dose cytarabine and idarubicin in the induction treatment of acute myeloid leukemia: long-term results of the prospective randomized nationwide AML-2003 study by the Finnish Leukemia Group. Eur J Haematol 109(3):257–270. https://doi.org/10.1111/ejh.13805
Alarcón-Payer C, Sánchez Suárez MDM, Martín Roldán A et al (2022) Impact of genetic polymorphisms and biomarkers on the effectiveness and toxicity of treatment of chronic myeloid leukemia and Acute Myeloid Leukemia. J Pers Med 12(10):1607. https://doi.org/10.3390/jpm12101607
Razumovskaya E, Masson K, Khan R, Bengtsson S, Rönnstrand L (2009) Oncogenic Flt3 receptors display different specificity and kinetics of autophosphorylation. Exp Hematol 37(8):979–989. https://doi.org/10.1016/j.exphem.2009.05.008
Georgoulia PS, Bjelic S, Friedman R (2020) Deciphering the molecular mechanism of FLT3 resistance mutations. FEBS J 287(15):3200–3220. https://doi.org/10.1111/febs.15209
Verstraete K, Vandriessche G, Januar M et al (2011) Structural insights into the extracellular assembly of the hematopoietic Flt3 signaling complex. Blood 118(1):60–68. https://doi.org/10.1182/blood-2011-01-329532
Morrison P, Takishima K, Rosner MR (1993) Role of threonine residues in regulation of the epidermal growth factor receptor by protein kinase C and mitogen-activated protein kinase. J Biol Chem 268(21):15536–15543
Ali AM, Salih GF (2023) Molecular and clinical significance of FLT3, NPM1, DNMT3A and TP53 mutations in acute myeloid leukemia patients. Mol Biol Rep 50(10):8035–8048. https://doi.org/10.1007/s11033-023-08680-2
Ding Y, Smith GH, Deeb K, Schneider T, Campbell A, Zhang L (2022) Revealing molecular architecture of FLT3 internal tandem duplication: development and clinical validation of a web-based application to generate accurate nomenclature. Int J Lab Hematol 44(5):918–927. https://doi.org/10.1111/ijlh.13930
Guijarro F, López-Guerra M, Morata J et al (2023) Germ line variants in patients with acute myeloid leukemia without a suspicion of hereditary hematologic malignancy syndrome. Blood Adv 7(19):5799–5811. https://doi.org/10.1182/bloodadvances.2023009742
Eckardt JN, Bornhäuser M, Wendt K, Middeke JM (2020) Application of machine learning in the management of acute myeloid leukemia: current practice and future prospects. Blood Adv 4(23):6077–6085. https://doi.org/10.1182/bloodadvances.2020002997
Papaemmanuil E, Gerstung M, Bullinger L et al (2016) Genomic classification and prognosis in Acute myeloid leukemia. N Engl J Med 374(23):2209–2221. https://doi.org/10.1056/NEJMoa1516192
Antar AI, Otrock ZK, Jabbour E, Mohty M, Bazarbachi A (2020) FLT3 inhibitors in acute myeloid leukemia: ten frequently asked questions. Leukemia 34(3):682–696. https://doi.org/10.1038/s41375-019-0694-3
Weisberg E, Roesel J, Furet P et al (2010) Antileukemic effects of Novel First- and second-generation FLT3 inhibitors: structure-Affinity comparison. Genes Cancer 1(10):1021–1032. https://doi.org/10.1177/1947601910396505
Ran F, Xie X, Wu Q et al (2023) Development of novel hydrazidoarylaminopyrimidine-based BTK/FLT3 dual inhibitors with potent in vivo anti-hematological malignancies effects. Eur J Med Chem 245(Pt 1):114913. https://doi.org/10.1016/j.ejmech.2022.114913
Grimwade D, Ivey A, Huntly BJ (2016) Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 127(1):29–41. https://doi.org/10.1182/blood-2015-07-604496
Ke YY, Singh VK, Coumar MS et al (2015) Homology modeling of DFG-in FMS-like tyrosine kinase 3 (FLT3) and structure-based virtual screening for inhibitor identification. Sci Rep 5:11702. https://doi.org/10.1038/srep11702
Jahn N, Jahn E, Saadati M et al (2022) Genomic landscape of patients with FLT3-mutated acute myeloid leukemia (AML) treated within the CALGB 10603/RATIFY trial. Leukemia 36(9):2218–2227. https://doi.org/10.1038/s41375-022-01650-w
Fiskus W, Sharma S, Saha S et al (2015) Pre-clinical efficacy of combined therapy with novel β-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia 29(6):1267–1278. https://doi.org/10.1038/leu.2014.340
Daver N, Cortes J, Ravandi F et al (2015) Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood 125(21):3236–3245. https://doi.org/10.1182/blood-2014-10-605808
Larrosa-Garcia M, Baer MR (2017) FLT3 inhibitors in Acute myeloid leukemia: current status and future directions. Mol Cancer Ther 16(6):991–1001. https://doi.org/10.1158/1535-7163.MCT-16-0876
Rodrigues ACBDC, Costa RGA, Silva SLR et al (2021) Cell signaling pathways as molecular targets to eliminate AML stem cells. Crit Rev Oncol Hematol 160:103277. https://doi.org/10.1016/j.critrevonc.2021.103277
Lagunas-Rangel FA (2023) DNA damage accumulation and repair defects in FLT3-ITD acute myeloid leukemia: implications for clonal evolution and disease progression. Hematol Oncol 41(1):26–38. https://doi.org/10.1002/hon.3076
Kishtagari A, Levine RL (2021) The role of somatic mutations in Acute myeloid leukemia pathogenesis. Cold Spring Harb Perspect Med 11(4):a034975. https://doi.org/10.1101/cshperspect.a034975
Beitinjaneh A, Jang S, Roukoz H, Majhail NS (2010) Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations in acute promyelocytic leukemia: a systematic review. Leuk Res 34(7):831–836. https://doi.org/10.1016/j.leukres.2010.01.001
Zheng R, Levis M, Piloto O et al (2004) FLT3 ligand causes autocrine signaling in acute myeloid leukemia cells. Blood 103(1):267–274. https://doi.org/10.1182/blood-2003-06-1969
Kuchenbauer F, Kern W, Schoch C et al (2005) Detailed analysis of FLT3 expression levels in acute myeloid leukemia. Haematologica 90(12):1617–1625
Rovatti PE, Gambacorta V, Lorentino F, Ciceri F, Vago L (2020) Mechanisms of Leukemia Immune Evasion and their role in Relapse after Haploidentical hematopoietic cell transplantation. Front Immunol 11:147. https://doi.org/10.3389/fimmu.2020.00147
Arai Y, Chi S, Minami Y, Yanada M (2022) FLT3-targeted treatment for acute myeloid leukemia. Int J Hematol 116(3):351–363. https://doi.org/10.1007/s12185-022-03374-0
Ferng TT, Terada D, Ando M et al (2022) The irreversible FLT3 inhibitor FF-10101 is active against a diversity of FLT3 inhibitor resistance mechanisms. Mol Cancer Ther 21(5):844–854. https://doi.org/10.1158/1535-7163.MCT-21-0317
Acknowledgements
This article was checked by language proofreading, we can provide certificate if requested.
Funding
The grant SVV no. 260 651, GAUK no. 350322, and the Cooperatio program supported the work.
Author information
Authors and Affiliations
Contributions
All the authors contributed to the intellectual content of this review and its writing. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Macečková, D., Vaňková, L., Holubová, M. et al. Current knowledge about FLT3 gene mutations, exploring the isoforms, and protein importance in AML. Mol Biol Rep 51, 521 (2024). https://doi.org/10.1007/s11033-024-09452-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09452-2