
Abstract
Preeclampsia (PE) is associated with high maternal and perinatal morbidity and mortality. The development of effective treatment strategies remains a major challenge due to the limited understanding of the pathogenesis. In this review, we summarize the current understanding of PE research, focusing on the molecular basis of mitochondrial function in normal and PE placentas, and discuss perspectives on future research directions. Mitochondria integrate numerous physiological processes such as energy production, cellular redox homeostasis, mitochondrial dynamics, and mitophagy, a selective autophagic clearance of damaged or dysfunctional mitochondria. Normal placental mitochondria have evolved innovative survival strategies to cope with uncertain environments (e.g., hypoxia and nutrient starvation). Cytotrophoblasts, extravillous trophoblast cells, and syncytiotrophoblasts all have distinct mitochondrial morphology and function. Recent advances in molecular studies on the spatial and temporal changes in normal mitochondrial function are providing valuable insight into PE pathogenesis. In PE placentas, hypoxia-mediated mitochondrial fission may induce activation of mitophagy machinery, leading to increased mitochondrial fragmentation and placental tissue damage over time. Repair mechanisms in mitochondrial function restore placental function, but disruption of compensatory mechanisms can induce apoptotic death of trophoblast cells. Additionally, molecular markers associated with repair or compensatory mechanisms that may influence the development and progression of PE are beginning to be identified. However, contradictory results have been obtained regarding some of the molecules that control mitochondrial biogenesis, dynamics, and mitophagy in PE placentas. In conclusion, understanding how the mitochondrial morphology and function influence cell fate decisions of trophoblast cells is an important issue in normal as well as pathological placentation biology. Research focusing on mitochondrial function will become increasingly important for elucidating the pathogenesis and effective treatment strategies of PE.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Preeclampsia (PE) is a multisystemic syndrome that affects approximately 5% of all pregnancies and is characterized by de novo hypertension after the 20th week of gestation [1]. Preeclampsia remains the leading cause of maternal and fetal mortality and morbidity worldwide. The precise mechanisms underlying PE are still poorly understood, but research proposes a two-stage theory [2]. The first stage comprises inadequate remodeling of the spiral arteries associated with shallow invasion of the trophoblast and then poor placentation. This is followed by maternal general endothelial dysfunction and imbalance between angiogenic and antiangiogenic factors that lead to systemic end-organ dysfunction (i.e., the second stage). Defective placentation results in the release of soluble antiangiogenic factors, soluble fms-like tyrosine kinase 1 (sFIt1), and promotes maternal endothelial dysfunction [3]. Not all PE patients have elevated sFLT1 but in those with elevated sFLT1 maternal blood pressure is elevated by potentiating a long-term vasoconstriction. There are two clinically-distinct PE phenotypes characterized by the onset of symptoms before (early-onset preeclampsia, eoPE) or after (late-onset preeclampsia, loPE) the 34th week of gestation [1]. Despite considerable heterogeneity, PE is classified according to the timing of disease onset and clinical phenotype. Furthermore, the mechanisms underlying the pathogenesis of PE include placental disorders such as a hypoxic intrauterine environment, increased inflammation, oxidative stress, ischemia, excessive apoptosis, immune imbalance, nutritional imbalance, and cell damage and death [4]. All these processes cause syncytiotrophoblast stress [5]. This stress is closely related to mitochondrial dysfunction.
Mitochondria are believed to be descended from ancient prokaryotes, i.e., an endo-symbiosed bacterium, that were engulfed by pre-eukaryotic cells several billion years ago [6]. Mitochondria generate energy as adenosine 5’-triphosphate (ATP) through oxidative phosphorylation (OXPHOS), promote their bioenergetics, and supply precursors for macromolecular synthesis. Thay also regulate the cytoplasmic oxidation-reduction (redox) homeostasis, porphyrin and heme biosynthesis, calcium homeostasis, and mitochondrial quality and quantity control through autophagic, mitophagic (i.e., selective autophagy of mitochondria), and apoptotic machinery [7, 8]. However, mitochondria are potential sources of endogenous reactive oxygen species (ROS) and are highly susceptible to oxidative stress, causing the irreversible cell damage and even the progressive death [9]. Placenta is an oxidative tissue, so mitochondria play an essential role in optimal trophoblast performance through the processes of mitochondrial biogenesis, fusion, fissions, and mitophagy. These dynamic processes determine mitochondrial morphology, overall quality, and relative abundance in cells and tissues. Indeed, evidence has been reported that mitochondrial impairment contributes to the pathogenesis of PE [10]. In other words, hypoxia, oxidative stress, or inflammation can trigger mitochondrial dysfunction but that mitochondrial dysfunction itself might contribute to the disease development. Therefore, mitochondrial function has also attracted attention in the fields of neurodegenerative and metabolic disorders and cancer research [11].
In 1994, Furui et al. for the first time demonstrated that reduced expression of the mitochondrial genes, cytochrome c oxidase and cytochrome oxidase subunit I messenger RNA, is involved in placental dysfunction in PE pregnancy [12]. Oh et al. suggested that increased autophagy may be involved in the pathophysiology of PE placenta [13]. Indeed, significant changes in the expression of proteins that control redox homeostasis, mitochondrial biogenesis, mitochondrial fusion and fission dynamics, autophagy, and mitophagy, a selective autophagic clearance of damaged or dysfunctional mitochondria, are observed in PE patients [10, 14, 15]. Furthermore, defective extravillous trophoblast (EVT) invasion established during early pregnancy is also considered to be associated with impaired mitochondrial biogenesis [16]. Jahan et al. highlighted altered mitochondrial function may be a common feature across distinct PE subtypes [17]. However, research on mitochondrial function and dynamics, energy metabolism, mitochondrial DNA (mtDNA) changes, mitophagy, and apoptosis in PE has yielded mixed or inconsistent results [18, 19].
In this review, we summarize the current understanding of mitochondrial dynamics, biogenesis, and mitophagy, focusing on the cytotrophoblasts and syncytiotrophoblasts of normal placenta and early-onset and late-onset PE placentas and discuss future research directions. We also focus on the differences in mitochondrial function and its compensatory mechanisms between normal and PE placentas to elucidate the root cause of the discrepancy.
Materials and methods
Search strategy and selection criteria
We conducted a narrative review of the literature that focuses on mitochondrial function in PE. Electronic databases including PubMed and Google Scholar were searched for literature published up to the July 31, 2023, combining the following keywords: “Biogenesis,” “Dynamics,” “Mitochondria,” “Mitophagy,” and “Preeclampsia.” Papers reporting patients’ data and in vitro and animal studies conducted to investigate the potential effect and underlying molecular mechanism of mitochondria-related genes were also included.
Results
Mitochondrial function in normal placenta
Mitochondrial function and their dynamics
The human placenta provides a timely supply of energy substrates, nutrients, and optimal oxygen to the fetus and synthesizes growth-promoting proteins and hormones [20]. The placenta contains trophoblast cells derived from the trophectoderm [20]. Cytotrophoblasts differentiate into syncytiotrophoblasts and EVT. Thus, human trophoblast cells have evolved as three major cell subpopulations with different functions: cytotrophoblasts, syncytiotrophoblasts, and EVT. During early pregnancy, EVT invade the uterine decidual spiral arterioles and contributes to the process of remodeling of the vessels to ensure optimal uteroplacental blood flow [4]. The mononuclear cytotrophoblasts fuse to form the outer multinucleated syncytiotrophoblasts. Syncytiotrophoblasts are responsible for regulating feto–maternal exchanges of gases, amino acids, nutrients, and waste products [20]. Proper placental function requires an adequate supply of energy (i.e., ATP as the molecular energy coin). ATP is produced mainly through mitochondrial OXPHOS when oxygen is available [9]. The OXPHOS system in mitochondria is a complex machinery comprising the electron transport chain (ETC) and ATP synthase [9]. The human placenta is a highly dynamic organ with high mitochondrial activity that regulates energy metabolism [21, 22]. . The cellular differentiation from cytotrophoblasts to syncytiotrophoblasts induces a range of molecular, biochemical, and morphological changes in constituent organelles such as mitochondria [23]. Mitochondria can move along the cytoskeleton and control their architecture, morphology, and distribution in order to adapt to changing external stimuli and metabolic demands [9]. The processes of mitochondrial fusion (e.g., mitofusin 1 (MFN1), mitofusin 2 (MFN2), and optic atrophy 1 (OPA1) genes) and fission (e.g., Dynamin related protein 1 (DRP1) gene) are mediated by members of the Dynamin-related GTPase family [24]. Mitochondria can exhibit a filamentous network structure (i.e., mitochondrial fusion) or fragmented morphology (i.e., mitochondrial fission) in response to different stimulus. Mitochondrial dynamics is critical for their optimal energy generation and regulates mitochondrial quality and quantity control. Mitochondrial fusion compensates for the defects by diluting the respiratory contents that were damaged to equilibrate respiratory chain complexes and metabolites, thereby repairing dysfunctional mitochondria, leading to subsequent mitochondrial activation and prevention of their elimination [9, 10, 25]. On the other hand, fission facilitates the division of damaged mitochondria, induces mitophagy, and results in removal of non-functional organelles [9, 10, 25, 26]. This is important for mitochondrial quality control. However, further mitochondrial fission may trigger apoptosis and lead to irreversible cell death. The Dynamin-related GTPase genes act as key molecular switches in mitochondrial dynamics-mediated cell-fate decisions. In order to cope with the ever-changing environments (e.g., nutrient and oxygen supply), mitochondria have evolved specific strategies, including bioenergetics and dynamics [27]. Upon nutrient starvation conditions, mitochondria enhance bioenergetics efficiencies and promote cell survival through extensive fusion [28]. On the other hand, hypoxia induces mitochondrial fragmentation and facilitates cell migration and movement [29]. Indeed, upregulation of DRP1 expression is well known to promote cell invasion of various cancer cells [30]. Environmental changes may significantly influence efficient energy production and cell survival by regulating fission - fusion dynamics and mitophagy.
Mitochondrial function in the cytotrophoblasts, syncytiotrophoblasts, and EVT
First, we summarize the mitochondrial function of normal placental trophoblast cells. The unique morphological, metabolic, bioenergetic, and molecular genetic properties of mitochondria differ between cytotrophoblasts and syncytiotrophoblasts [20]. Mitochondria from the cytotrophoblasts are larger in volume and more elongated (Table 1), whereas in syncytiotrophoblasts, mitochondria undergo fission to produce smaller organelles [20]. Cytotrophoblasts in contact with fetal blood vessels must provide nutrients to the fetus. The mitochondria of cytotrophoblasts are morphologically longer and larger than those of syncytiotrophoblasts, which significantly activates glycolysis and OXPHOS and increases ATP generation [9, 31, 32]. Conversely, syncytiotrophoblasts, a multinucleated layer that covers chorionic villi, no longer need to divide and proliferate, shifting the balance from mitochondrial fusion to fission by reducing the expression of the fusion proteins MFN1 and MFN2 [9, 32]. The mitochondrial morphology of syncytiotrophoblasts undergoes changes from complex filamentous networks to small globular compartments, leading to a gradual decrease in the expression of enzymes related to ATP synthesis, glucose metabolism, and lipid metabolism [20]. Therefore, alterations in mitochondrial morphology are often accompanied by changes in ATP production: mitochondria in cytotrophoblasts have a much higher energy production capacity than those in syncytiotrophoblasts [31]. Cytotrophoblast mitochondria exhibit normal cristae morphology and increased mitochondrial content, whereas in syncytiotrophoblasts, decreased expression of the mitochondrial respiratory chain dimeric complex V results in decreased mitochondrial potential, reduced mitochondrial content, and impaired cristae morphology [33]. The differences between the two populations are reflected in mitochondrial dynamics, shape, metabolism, and phenotype. The evidence on why and how differences in mitochondrial dynamics occur between cytotrophoblasts and syncytiotrophoblasts is limited. However, significant molecular changes were observed in two main sub-populations. Fisher et al. have identified key proteins that are significantly less expressed in syncytiotrophoblasts than in cytotrophoblasts [20]. Representative molecules are involved in the metabolism of carbohydrate and amino acid metabolism (branched chain keto acid dehydrogenase E1 subunit alpha (BCKDHA), branched chain amino acid transaminase 2 (BCAT2), and solute carrier family 25 member 11 (SLC25A11)), fatty acid metabolism (fatty acid metabolizing enzyme acyl-CoA dehydrogenase (ACAD), and acyl-CoA dehydrogenase very long chain (ACADVL)), energy metabolism (pyruvate dehydrogenase (PDH), and pyruvate dehydrogenase phosphatase regulatory subunit (PDPR)), mitochondrial metabolism (pyruvate carboxylase (PC), phospoenolpyruvate carboxykinase-2 (PCK2), succinate dehydrogenase complex flavoprotein subunit A (SDHA), solute carrier family 25 member 6 (SLC25A6), glucose regulated protein-78 (GRP78) or heat shock protein family A member 5 (HSPA5 ), and protein disulfide isomerase (PDI)), and mitochondrial bioenergetics (NADH:ubiquinone oxidoreductase subunit A12 (NDUFA12), ATP synthase F1 subunit beta (ATP5B), and ATP synthase F1 subunit alpha (ATP5A1)) and dynamics (mitofusin 1 (MFN1) and mitofusin 2 (MFN2)) [20]. Many of these proteins (i.e., PDH, PDPR, PC, PCK2, SDHA, SLC25A6, GRP78, HSPA5, PDI, NDUFA12, ATP5B, ATP5A1, MFN1, and MFN2) are closely associated with decreased mitochondrial OXPHOS and mitochondrial fission in syncytiotrophoblasts [20]. Moreover, the metabolism (i.e., BCKDHA, BCAT2, SLC25A11, ACAD, and ACADVL) of carbohydrates, fatty acids, and amino acids is upregulated in cytotrophoblasts [20]. This is supported by the study of Kolahi et al. who demonstrated higher rates of ATP generation, mitochondrial respiration, and glycolysis in cytotrophoblasts when compared to syncytiotrophoblasts [34]. Large amounts of ATP, carbohydrates, fatty acids, and amino acids can be stored in cytotrophoblasts to provide fuel necessary for the rapidly developing fetus [20].
Second, during early pregnancy, EVT invade the uterine myometrium and migrate along the lumina of spiral arterioles under hypoxic conditions [19]. Extravillous trophoblasts specifically require energy to develop the placenta [19, 35, 36]. To achieve this goal, EVT first promote enhanced oxidative respiration and increased ATP production through the OPA1-mediated mitochondrial fusion [35, 36]. In other words, energy metabolism is directed toward OXPHOS activation. EVT are then exposed to changes in environmental factors (e.g., hypoxia and nutrient starvation). Hypoxia promotes mitochondrial fission and shifts the metabolic flow from OXPHOS in mitochondria to glycolysis, and facilitates further ATP and biomass generation in EVT through hypoxia-inducible factor-1 (HIF-1)-mediated DRP1 activation [29, 37]. Extravillous trophoblasts can reprogram their metabolism to support their invasion by controlling mitochondrial function. Mitochondrial fission is known to promote cell migration and invasion [38], so hypoxia provides a favorable microenvironment for EVT migration [39]. A proper change in mitochondrial dynamics over time is a physiological event for ensuring optimal placentation in EVT. Because it is ethically difficult to obtain EVT for research in the early first trimester, Gillmore et al. used placental mesenchymal stromal cells (pMSCs) instead of EVT to study mitochondrial dynamics [29]. They demonstrated that hypoxia-mediated mitochondrial fission is an adaptive mechanism for suppressing OXPHOS, conserving oxygen, and supplying energy through enhanced glycolysis and reduced ROS generation [29]. These data of pMSCs are important to complement the biological properties of EVT [29]. In addition, persistent hypoxia and nutrient starvation cause rapid aging of trophoblast cells, and the activation of mitophagy is required to eliminate dysfunctional mitochondria. However, impaired mitophagy may cause apoptosis [21, 40] (see the subsection 3.2.3). Mitochondria can respond to intrinsic (e.g., the unique gene expression patterns that are determined during cellular differentiation into cytotrophoblasts, syncytiotrophoblasts, and EVT [20]) and extrinsic or environmental (e.g., hypoxia and nutrient starvation) factors by timely altering their fission-fusion cycles [25]. Such changes in mitochondrial dynamics may be a mechanism acquired through the evolution of the placentation in viviparous mammals. Studies on mitochondrial function after trophoblast differentiation in the normal placenta provide valuable insights into the pathogenesis of pathological pregnancy.
Mitochondrial function in preeclamptic placenta
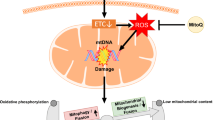
Recent reviews have suggested that mitochondrial dysfunction plays a key role in the pathogenesis of PE [41,42,43]. This review adds new findings about the disruption of the balance between mitochondrial function and its compensatory action in PE patients and animal models. See Fig. 1 for a conceptual summary of mitochondrial function in PE placenta.

Mitochondrial function and its conceptual diagram in preeclamptic placenta. This table summarizes mitochondrial functions into different categories: energy metabolism, redox homeostasis, mitochondrial dynamics, mtDNA content, and mitophagy
Impaired energy metabolism
Proteome bioinformatics analysis showed that mitochondria-related molecules were significantly altered in eoPE placentas compared to normal placentas (Fig. 1, second column, Mitochondrial activity). Changes in the expression of proteins related to mitochondrial machinery have been identified in PE placentas, including: the downregulation of two proteins related to mitochondrial complex I such as the iron-sulfur protein (IP) components (Ubiquinone Oxidoreductase Core Subunit S3 (NDUFS3)) and core subunit V1 (NADH:Ubiquinone Oxidoreductase Core Subunit V1 (NDUFV1)), and the upregulation of proteins related to the core (NADH:Ubiquinone Oxidoreductase Core Subunit S7 (NDUFS7)), subcomplex (NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 7 (NDUFB7)) and accessory (NADH:Ubiquinone Oxidoreductase Subunit B8 (NDUFB8)) subunits of the mitochondrial membrane respiratory chain NADH dehydrogenase of complex I [44, 45]. Dysfunction of the respiratory ETC in PE was evidenced by decreased function of mitochondrial complex proteins [44, 45]. Decreased ETC activity in dysfunctional mitochondria also reduces ATP production. Moreover, the mRNA expression [46] and activity [9] of the electron transfer complex IV, cytochrome c oxidase, have been reported to be significantly decreased in PE placentas compared to control placentas [47]. The mRNA expression level of cytochrome c oxidase decreased significantly depending on the severity of the disease [46]. Additionally, several reports have investigated the function of the TCA cycle in PE placentas by measuring the activity of citrate synthase linked to the ETC [9, 45, 48, 49]. Citrate synthase has a key role in the TCA cycle of all organisms that utilizes acetyl-CoA and oxaloacetate to form citrate [50]. Citrate synthase activity in PE placentas was unchanged [48, 49] or even increased [45] compared with control placentas [9]. Increased citrate synthase activity may compensate for abnormal ETC function in PE placentas. This is supported by the study of Holland et al. who demonstrated that mitochondrial respiration levels are significantly elevated in loPE placentas compared with control placentas [48]. These data may reflect a compensatory mechanism to balance energy metabolism.
Second, it has been reported that the energy deficit caused by reduced ETC activity, especially in cytotrophoblasts, are compensated by the metabolic shift from OXPHOS towards glycolysis [16, 51] (Fig. 1, second column, Metabolism and Molecular markers). Pyruvate dehydrogenase converts pyruvate to acetyl-coenzyme A, which activates metabolism through the TCA cycle for ATP generation in mitochondria [51]. In cytotrophoblasts, elevation of pyruvate dehydrogenase kinase (PDK), which inactivates PDH, shifts intracellular metabolism toward glycolysis [51]. Pyruvate dehydrogenase kinase has been shown to be activated by hypoxia-induced HIF-1 [51]. Therefore, profound changes in cytotrophoblast metabolism in PE are characterized by a switch from OXPHOS toward glycolysis [52]. Additionally, glycolysis is induced in trophoblast cells through AMP-activated protein kinase (AMPK)-dependent energy regulation [16, 53]. AMP-activated protein kinase is activated by low ATP levels and contributes to the regulation of mitochondrial homeostasis [54]. During pregnancy, AMPK coordinates the proper placental growth, differentiation, and nutrient transport to maintain maternal and fetal energy homeostasis [55]. Furthermore, AMPK can also promote mitophagy specifically through DRP1-dependent mitochondrial fission [56, 57]. However, excessive and persistent activation of AMPK progressively worsens placental function over time through further mitochondrial fission [16, 55]. In addition, prolonged glycolysis causes depletion of cell ATP pool and extensive cell damage and apoptosis, which may be associated with the progression of PE. Therefore, the ability to fine-tune the metabolic shift from glycolysis to OXPHOS may be reduced in PE.
Finally, dysregulation of mitochondrial lipid oxidation is commonly observed in PE placentas [10]. Human cells rely on fatty acid β-oxidation as the energy source. For example, enzymes involved in fatty acid β-oxidation pathway in mitochondria, such as acyl-coenzyme A dehydrogenase very long chain (ACADVL) and 3-hydroxyacyl coenzyme A dehydrogenase (HADH), are upregulated in human PE placentas, causing lipid accumulation and excessive ROS generation [58]. Conversely, with increasing severity, long-chain HADH levels and fatty acid oxidation capacity were significantly reduced in PE placentas, suggesting a reduction in the mitochondrial import of fatty acids and further metabolism [59]. Decreased fatty acids β-oxidation causes decreased ATP production and contributes to placental insufficiency in PE patients. Although the expression of enzymes related to fatty acid β-oxidation may differ depending on the severity of PE, dysregulation of mitochondrial lipid peroxidation can cause mitochondrial dysfunction [10].
Impaired redox homeostasis
Figure 1, third column summarizes the role of mitochondrial function in redox homeostasis in PE placentas. The mitochondrial ETC produces a significant amount of ROS (e.g., superoxide anion and hydrogen peroxide) as adverse byproducts of ATP generation [15, 60]. In normal gestation, there is an appropriate balance between ROS and lipid peroxidation (e.g., fatty acids lipid peroxidation products: malondialdehyde and 4-hydroxy-2-nonenal) and antioxidants, including antioxidant enzymes (e.g., superoxide dismutase, catalase (CAT), or glutathione peroxidase (GPX) and small molecule antioxidants (glutathione, thioredoxin, or tocopherol) [47, 61]. Preeclampsia is known to cause an imbalance between ROS and antioxidant scavengers, or a redox imbalance [15, 47, 61]. Preeclampsia placentas mildly damaged with oxidative stress also increase compensatory mechanisms, such as increased expression of peroxiredoxin 2 (PRDX2), GPX, and CAT, to protect against ROS-induced mitochondrial injury [44, 62]. A series of cellular insults (e.g., elevated levels of ROS) accelerate mitophagy which eliminates dysfunctional or redundant mitochondria to maintain cellular homeostasis [63]. However, excessive ROS cause severe mitochondrial dysfunction by further disrupting the normal function of the ETC machinery, ultimately leading to cell death through apoptosis [9].
Recently, mitochondrial transcription factor A (TFAM) has attracted attention as a gene that may be a link to tie the ROS signaling and mitochondrial dysfunction [64]. TFAM is essential for the regulation of mtDNA transcription and replication [22, 64]. Impairment of TFAM function has been shown to cause mtDNA depletion, deficient OXPHOS, and abnormal mitochondrial structure [22, 64]. Reactive oxygen species can disrupt mtDNA stability and induce mitochondrial dysfunction by reducing TFAM expression [64]. Indeed, mRNA and protein expression levels of the TFAM gene were significantly downregulated in the eoPE placentas compared to the control [45]. This suggests that TFAM was depleted by oxidative stress and further caused cellular damage through mitochondrial dysfunction. For example, in a mouse myocardial infarction model, overexpression of human TFAM has been reported to increase the amount of mtDNA and could effectively ameliorate mitochondrial dysfunction [65]. Therefore, timely removal of excess ROS or upregulation of TFAM expression may lead to survival of trophoblast cells in PE placentas. In addition to ROS and antioxidant levels, real-time quantification of TFAM expression and mtDNA content may help better assess mitochondrial function in PE placentas.
Mitochondrial dynamics
Several studies have evaluated the ultrastructural changes of mitochondria in PE placentas using transmission electron microscopy [9, 66] (Fig. 1, fourth column). Ultrastructural analysis of trophoblasts and pMSCs of PE placentas revealed marked mitochondrial swelling with loss of cristae, vacuolation and damage, and excessive fragmentation [25, 29, 66]. These findings were more pronounced in trophoblast cells of eoPE placentas compared to loPE placentas [9, 29]. Mitochondrial morphology reflects mitochondrial bioenergetics and dynamics, i.e., fission and fusion, in PE [26]. Mitochondrial fusion/fission dynamics seem differentially regulated in normal and PE placentas, particularly in eoPE and loPE [9]. However, as research progresses, conflicting results have been obtained regarding some molecules (e.g., MFN1, MFN2, OPA1, DRP1, TFAM, voltage-dependent anion-selective channel protein 1 (VDAC1), nuclear respiratory factor 1 (NRF1)) associated with mitochondrial function in PE [9, 10, 25, 45, 48]. Yu et al. [67] showed that MFN2 expression was significantly decreased in PE placentas compared to normal placentas. Moreover, other authors [10, 25] found that OPA1 expression was decreased in eoPE. Indeed, OPA1 mRNA and protein expression was decreased in PE rat model induced by 11β-HSD2 inhibitor carbenoxolone (CBX) [36]. Additionally, the expression of MFN1, MFN2, and OPA1 has been reported to be downregulated in loPE placentas [10, 67]. Furthermore, mitochondrial fragmentation in cytotrophoblasts [25] or mesenchymal cells [29] of PE placentas was demonstrated to be caused by activation of DRP1. However, it has also been reported that DRP1 expression is not upregulated in eoPE placentas [10, 45]. Broadly summarized, these literatures have shown that the mitochondrial dynamic balance in PE placentas may be tilted toward fission due to decreased MFN1, MFN2, and OPA1 expression and increased or unchanged DRP1 expression/activation.
In the placenta during early pregnancy, increased vascular resistance and uteroplacental hypoperfusion often cause hypoxia [4]. It is well known in the oncology field that mitochondrial fission is induced by hypoxia [38]. HIF-1, a central oxygen-threshold sensor in the human placenta, is significantly expressed in the first trimester of pregnancy and highly upregulated in PE placentas [68]. Indeed, genetic and pharmacological overexpression of HIF1A gene in mice resemble the phenotype of the human PE [69, 70]. HIF-1 is significantly upregulated in PE placentas and plays crucial roles in endothelial cell dysfunction and PE pathogenesis through activation of anti-angiogenic and inhibition of proangiogenic factors [68]. Furthermore, HIF-1 is known to upregulate the expression and posttranslational modification of DRP1 [29, 71]. Therefore, hypoxia may promote mitochondrial fission via HIF-1-dependent upregulation of DRP1 expression in the placenta [29]. It is recognized that there are molecular and functional similarities between the human placentas from early pregnancy (less than 10 weeks) and from PE pregnancies, reflecting the unique environment with low-oxygen conditions [72]. Mitochondria before 10 weeks of gestation are morphologically small and globular (i.e., mitochondrial fission), whereas mitochondria after 10 weeks of gestation are more elongated and interconnected (i.e., mitochondrial fusion) [29, 72]. This may be because at around 10–12 weeks of gestation, the conversion of the narrow arteries into distended uteroplacental arteries causes a rise in oxygen tension and suppresses the HIF-1-induced DRP1 expression [72]. Collectively, hypoxia may induce mitochondrial fission in PE placentas via upregulating DRP1 gene expression in a HIF1-dependent manner. However, there is no direct evidence that upregulation of DRP1 expression or downregulation of OPA1 expression plays essential roles in various pathways that regulate PE development and progression.
Furthermore, ceramide, which is produced by hydrolysis of sphingomyelin present in cell membranes, is known as a factor that induces mitochondrial fission [25, 73]. HIF-1 increases the production of ceramide by promoting the expression of the ceramide synthase sialidase 3 [73]. Ceramide is known to increase the expression of DRP1 through upregulation of the Bcl-2 member BCL2 family apoptosis regulator BOK (BOK) [25]. Indeed, ceramide promoted mitochondrial fission in human choriocarcinoma JEG3 cells, primary isolated cytotrophoblasts, and PE trophoblast cells [25]. Therefore, enhanced mitochondrial fission in trophoblast cells of PE placentas may be caused by the HIF-1/ceramide/DRP1-dependent signaling pathways.
On the other hand, several papers revealed that MFN [74] and OPA1 [45], which are involved in mitochondrial fusion, are upregulated in PE placentas [22, 48]. Bartho et al. [22] found that the expression of MFN1 is clearly increased in preterm PE placentas compared to control preterm placentas. Additionally, serum levels of MFN2 are found to be significantly higher in the PE group than in the control group, particularly highest in the eoPE group [74]. High maternal serum MFN2 levels may also be a consequence of a compensatory mechanism against hypoxia caused by PE [9]. However, placental and serum MFN1 and MFN2 were not measured simultaneously in the same case, so it is unclear whether serum MFN originates from the placenta. Furthermore, Holland et al. showed that OPA1 was increased in eoPE placentas [48], and OPA1 and MFN1 were increased, and FIS1 was decreased in loPE placentas [46]. MFN expression was elevated in loPE but not in eoPE [48]. OPA1-dependent fusion protects mitochondria from mitophagy during nutrient starvation and may enhance quality control [75]. These data suggest that the expression of mitochondrial fusion genes can affect disease severity and progression.
Finally, cellular senescence/aging is a process that leads to a state of irreversible growth arrest but remains metabolically active [76]. Placental senescence/aging is demonstrated by increased ROS accumulation, p53 signaling, and endoplasmic reticulum stress, and decreased mitochondrial density, respiratory capacity, and ATP production [21, 40]. Focusing on mitochondrial dynamics, the morphology of mitochondria in normal placental syncytiotrophoblasts shows small round punctate structures, suggesting increased fission and a senescent state [77]. Placental senescence/aging is controlled by downregulation of mitochondrial expression of MFN1 and MFN2 [22, 78]. Thus, downregulation of MFN expression in normal placenta indicates physiological aging towards the end of pregnancy. In this context, mitochondrial dynamics may be positively (e.g., cell survival through quality control mechanisms [79]) or negatively (e.g., cell death through apoptosis mechanisms [21]) regulated in PE placentas, depending on the severity of tissue damage. Indeed, genes related to mitochondrial senescence/aging (e.g., MFN1, TFAM, and translocase of outer mitochondrial membrane 20 (TOMM20)) are increased in preterm PE placentas compared to gestation-matched control placentas [22]. MFN1 and OPA1 shift cellular metabolism from glycolysis toward mitochondrial respiration during senescence/aging and facilitate cell survival through upregulating ATP generation [80]. This may be a compensatory mechanism to maintain or restore an adequate function in PE placentas. Ultimately, decreased expression of MFN1 and OPA1 may result in further decline in mitochondrial function and rapidly cause irreversible placental damage possibly via disruption of the compensation mechanism [21]. Depending on disease severity, there are likely at least two stages of PE: compensated and decompensated.
Collectively, previous studies have yielded contradictory results regarding MFN1, MFN2, OPA1, and DRP1 expression in PE placentas. Altered mitochondrial dynamics in PE placentas can influence the choice between maintaining mitochondrial quality or inducing further impairment. Altered mitochondrial fusion/fission dynamics could explain the phenotypic variation of PE.
Changes in mtDNA content
Mitochondrial DNA replication controlled by mitochondria dynamics plays an important role in mitochondrial biogenesis and vice versa [7] (Fig. 1, second column from the right). Loss of mitochondrial fusion has been shown to cause defects in mtDNA replication [7]. Cell-free DNA secreted from normal placenta enters maternal circulation, and their concentration increases with gestational age [81]. There are some reports that have measured mtDNA content in the placenta and blood of patients with PE. Reports of mtDNA content in patients with PE were inconsistent, showing increasing [82] or decreasing mtDNA content [36, 81, 83]. Circulating cell-free mitochondrial DNA (ccf-mtDNA) concentrations [82] and placental mtDNA copy number [45] have been reported to be significantly higher in eoPE patients than in age matched controls. Since OPA1 regulates mtDNA stability and increases mtDNA copy number, ccf-mtDNA may be increased in PE patients with elevated placental OPA1 expression. Mitochondrial DNA can induce inflammatory cytokines associated with innate immunity in a Toll-like receptor 9 (TLR9)-dependent manner [82]. In fact, TLR9 activity is significantly increased in PE patients than in control patients [82]. On the other hand, Cushen et al. showed that ccf-mtDNA concentrations were significantly decreased in patients with PE compared with healthy controls [81]. Indeed, the TFAM gene and protein are significantly reduced in some PE placentas, suggesting that mitochondrial function and mtDNA replication are severely impaired [15, 45]. When extensive cell death occurs, the release of mtDNA fragments into the maternal circulation increases, but may decrease over time. Therefore, changes in mtDNA content in blood and placenta might be influenced by the severity as well as the time course of mitochondrial dysfunction in PE.
Changes in the expression of molecules related to mitophagy
Autophagy is an evolutionarily conserved recycling process in eukaryotes to maintain cellular homeostasis against various conditions such as nutritional deficiencies or stress [19]. Mitophagy is an important mitochondrial quality control mechanism, eliminates mutated mtDNA, limits production of damaging ROS, ensures mitochondrial homeostasis, and contributes to energy production [21] (Fig. 1, rightmost column). The Ser/Thr kinase phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) accumulates on the surface of dysfunctional mitochondria, activates the E3 ubiquitin ligase Parkin, and jointly mediates mitophagy [84]. Damaged mitochondria generated by fission are selectively removed by mitophagy [9, 84]. Many mitochondrial cargo receptors (MCRs), including BCL2 interacting protein 3 (BNIP3), BNIP3L (NIX), FUN14 domain containing 1 (FUNDC1), Bcl2-L-13, FKBP prolyl isomerase 8 (FKBP8), FANC-C, Prohibitin-2 (PHB-2), Metaxin-1, and Cardiolipin, are required to target mitochondria to autophagosomes [56]. For example, BNIP3 activates mitophagy and prevents cellular damage [56]. There is increasing evidence that autophagy or mitophagy is essential for placental development and is involved in establishing and maintaining pregnancy [19]. As an adaptive response to stresses such as hypoxia and nutrient deprivation during early pregnancy, EVT utilize the mitophagy mechanism to obtain the energy needed for cell migration [19]. Deficient mitophagy induces mitochondrial dysfunction, impaired mitochondrial quality control, and apoptosis [79].
Abnormalities in placental mitophagy have recently become a hot topic in PE [14]. Although activation of mitophagy is one of the defence lines against oxidative stress during PE, mitophagy is thought to be altered due to changes in the expression of specific genes related to mitochondrial dynamics (e.g., MFN1, MFN2, OPA1, and DRP1) [22, 78], mitochondrial biogenesis (e.g., peroxisome proliferator-activated receptor-gamma co-activator-1alpha (PGC-1α) and sirtuin 3 (SIRT3)) [15], mitochondrial aging (e.g., TFAM, TOMM20, and VDAC1) [15, 22, 45, 85], autophagy related markers (e.g., transcription factor EB (TFEB), lysosomal associated membrane protein 1 (LAMP1), LAMP2, autophagy marker light chain protein 3 (LC3A, also known as microtubule associated protein 1 light chain 3 alpha), cathepsin D (CTSD), Beclin 1, sequestosome 1 (p62/SQSTM1), and DNA damage regulated autophagy modulator 1 (DRAM1)) [15, 18, 19, 86, 87], and MCR molecules (e.g., BNIP3 and BNIP3L) [18, 44, 78, 88, 89]. Therefore, it is important to explore the expression of genes and proteins related to mitophagy in order to clarify its mechanistic involvement in the basic pathophysiology of PE. Several reports have shown that the key mitophagy machinery is significantly impaired in PE placentas compared to normal placentas [18, 86, 90]. . The expression of MCR molecules (e.g., BNIP3) induced by stress such as hypoxia and nutritional deprivation is suppressed in PE placentas compared to normal placentas [18]. Downregulation of BNIP3 expression reduces mitophagy and potentiate cell death [18, 56]. Additionally, the DRAM1 gene encodes a lysosomal membrane protein that is required for the induction of autophagy [47]. The HIF-1α-induced experimental animal data revealed decreased DRAM1 expression, widespread mitochondrial dysfunction, and significantly decreased mitophagy [47]. Thus, defects in mitophagy in PE placentas can trigger irreversible cell death through the induction of apoptosis [91]. Conversely, there are some reports that activation of autophagy was observed more frequently in PE placentas than in normal placentas [19, 25, 47, 79]. Yildirim et al. summarized the role of BNIP3, DRAM1, and FUNDC1, mediators of receptor-mediated mitophagy, in the progression of preeclampsia, suggesting that mitophagy-related pathways may be associated with the pathophysiology of preeclampsia [92]. Several mitophagy-related proteins were found to be overexpressed in PE placentas. For example, p62/SQSTM1, a stress-inducible cellular protein that interacts with the autophagy machinery, accumulates in EVT of PE placentas [19]. Indeed, the number of autophagic vacuoles or LC3 dots, a marker to monitor autophagy [15, 19, 79], the expression of mitophagy markers (e.g., p62/SQSTM1 and Beclin 1) [18, 19, 79, 87, 90], and PINK1/Parkin-mediated mitophagy [23, 82] were more pronounced in PE placentas than in normal placentas. These researchers believe that autophagy or mitophagy activity is increased in PE to selectively remove the accumulation of dysfunctional mitochondria. Recent review summarizes our current understanding of the roles of mitochondrial quality control in PE [93].
As shown here, existing studies reported inconsistent findings, with some reports demonstrating that mitophagy is activated [19, 25, 79] or suppressed [18, 19, 86, 90] in PE placenta. To provide possible reasons for this inconsistency, we re-evaluated the expression levels of key regulators of mitophagy by subdividing PE into early- and late-onset phenotypes (Table 2). The expression of proteins associated with mitochondrial aging (e.g., VDAC1, TFAM), glycolysis (e.g., HK1 (hexokinase 1)), biogenesis (e.g., PGC-1α and PGC-1β), and autophagy and mitophagy (e.g., LC3A) was significantly elevated in loPE placentas compared to normal placentas [15]. VDAC1 has many biological functions, including reprogramming energy metabolism, maintaining mitochondrial function, mediating Ca2+ transport, controlling ROS release, and regulating PINK1/Parkin-mediated mitophagy and apoptosis [94, 95]. Hexokinase converts glucose to glucose-6-phosphate, induces activation of glycolysis, and can partially compensate for an energy deficit due to reduction in oxidative metabolism [15]. Additionally, the reduced expression of genes related to glycolysis or gluconeogenesis (e.g., enolase 2 (ENO2), phosphoglycerate kinase 1 (PGK1), and HK2) was obvious in the decidua of eoPE placentas [96]. Furthermore, BNIP3 can protect against cell injury via mitophagy, which removes damaged mitochondria. BNIP3 has been shown to be upregulated in eoPE placentas [44], suggesting that mitophagy is activated and maintain mitochondrial quality control and homeostasis even in eoPE placentas. However, not only defective mitophagy but also extensive mitophagy results in depletion of cell ATP pool, extensive cell damage, and apoptosis. In addition, Tong et al. showed increased BNIP3 expression in PE patients compared to that in controls [96], while Zhou et al. reported a decrease [18]. Knockdown of BNIP3 promotes ROS accumulation, autophagy impairment, mitochondrial damage, and apoptosis [18]. PE placentas with decreased BNIP3 expression may be more susceptible to apoptosis due to impaired mitophagy. Furthermore, Bcl-2 expression is upregulated in the uterine myometrium and placenta of the patients with eoPE, leading to increased antioxidant capacity [15]. Bcl-2 significantly inhibits the mitophagy process through inhibition of Beclin1 activity [15, 97]. Beclin 1 is a Bcl-2-interacting protein that promotes mitophagy. These results suggest that quality control and cellular repair mechanisms may already be compromised in some eoPE. From the above, part of the eoPE placenta still retains the ability to maintain cell survival, while others have already impaired sufficient homeostatic compensation mechanisms [48].
Finally, we examine the occurrence of apoptosis in early- and late-onset PE. Mitophagy is critical to regulate apoptosis resistance, and impairment in the mitophagy pathway induces apoptosis [18]. When a variety of compensatory mechanisms break down, a variety of stimuli, including hypoxia, oxidative stress, and acidosis may trigger mitophagy impairment and ultimately result in apoptosis in placenta [9]. Apoptosis of trophoblast cells is significantly increased in PE compared to normal placenta [98]. Trophoblasts in eoPE placentas alter the ratio of pro- and anti-apoptotic molecules toward apoptosis properties (i.e., an increase in the pro-apoptotic Bcl-2-associated X protein (BAX) and a decrease in the anti-apoptotic BCL2) [99, 100]. A further increase in apoptosis (i.e., increased BAX/BCL2 ratio) was observed in eoPE placentas compared to controls [9]. This suggests that the earlier the onset of PE, the more severe the apoptosis. Therefore, key differences in the gene-expression patterns that affect mitophagy processes in PE placentas may be used as indicators to distinguish between eoPE and loPE, or surrogate markers to determine disease severity. Collectively, some compensatory mechanisms capable of counterbalancing apoptosis may be activated to maintain mitochondrial homeostasis in loPE, while such mechanisms are already disrupted in eoPE, which potentially may cause apoptosis. Altered protein expression associated with mitophagy and apoptosis in early- and late-onset PE may depend on compensatory mechanisms of mitochondrial function.
Understanding the pathogenesis of PE through animal models
Several experimental animal models have been established by pharmacological (carbenoxolone (CBX) [101], HIF-1α [47, 70], or N(ω)-nitro-L-arginine methyl ester (L-NAME) [102]) or surgical (reduced uterine perfusion pressure (RUPP) models [103]) manipulation to better understand the complex pathogenesis of PE [104]. A literature search was conducted to determine whether these models reflect various causes of PE, particularly mitochondrial dysfunction, in humans. Carbenoxolone is a competitive inhibitor of 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) and L-NAME is a vasoconstrictor. 11β-HSD2 converts cortisol into inactive 11-keto metabolites and compromised enzyme activity causes sodium retention and hypertension. Indeed, in cultured human EVT, CBX-induced 11β-HSD2 suppression leads to mitochondrial dysfunction and disrupts mtDNA stability through downregulating OPA1 expression [36]. Abnormal mitochondrial function, such as impaired OXPHOS, carbon metabolism, and glutathione metabolism, aberrant sphingolipid pathway, and accumulation of mutated mtDNA, has important roles in the pathogenesis of PE in the CBX model [36]. Moreover, L-NAME inhibits nitric oxide synthase and suppresses endothelium-dependent vasodilation. A decrease in the level of mitochondrial membrane potential, a hallmark of mitochondrial dysfunction, was demonstrated in the placenta of the L-NAME-induced PE model [105]. Additionally, mitochondrial dysfunction, including high levels of oxidative stress and apoptosis, decreased mitophagy, and significantly decreased DRAM1 expression, were found in the placentas of HIF-1α-induced PE model [47, 70]. DRAM1 is known to induce autophagy related 5 (ATG5)-independent autophagy, so decreased DRAM1 expression can induce mitochondrial dysfunction. All of these animal models cause secondary mitochondrial dysfunction, but the regulation of DRAM1 gene expression may be a model for understanding how mitophagy directly affects mitochondrial function and oxidative stress in PE. Collectively, mitochondrial abnormalities that frequently occur in the placentas of PE patients are also found in PE-like animal models. With the aid of animal models, we are beginning to comprehend the molecular mechanisms underlying mitochondrial dysfunction that controls the development of PE. However, it remains controversial whether mitochondrial abnormalities are the cause of PE or the result of compensatory mechanisms.
Therapeutic potential of targeting mitochondrial dynamics and mitophagy
Currently, there is no fundamental treatment for PE, and the most common approach to clinical management is to terminate the pregnancy, which often induces preterm birth [1]. Correct diagnosis and appropriate treatment of PE require accurate understanding of the disease status in real time. In this review, we highlighted that the complex mechanisms underlying PE include impaired energy metabolism, redox imbalance, dysregulation of mitochondrial biogenesis and dynamics, accumulation of mutated mtDNA, dysfunction of autophagy and mitophagy, senescence, and inadequate or complete apoptosis. This suggests that controlling mitochondrial function, which is a hallmark of PE, may prevent disease progression. First, since the elevated level of ROS triggers mitochondrial dysfunction, antioxidants may be ideal candidates for the development of therapeutics [15, 36, 37, 106]. Indeed, interventional studies showed that coenzyme Q10, a key molecule in the mitochondrial ETC, may be protective against PE development, at least to some extent [107]. However, although it is true that oxidative damage is involved in the pathophysiology of PE, current antioxidant treatments that focus solely on reducing oxidative stress have been reported to be ineffective [108]. Additionally, the therapeutic potential of MitoQ, a potent mitochondria-targeting antioxidant, was evaluated using a PE animal model [109]. Animal studies showed that MitoQ protects against PE during late pregnancy, but increases the risk of PE when administered in early pregnancy. High levels of ROS cause oxidative damage, which aids in the development of PE, but moderate ROS levels are essential for ensuring normal placental development [110]. Thus, MitoQ could be administered depending on a placental oxidative stress severity. However, it is difficult to identify multiple and diverse factors which must be balanced in an ever-changing redox status in PE placentas. These factors include ROS generation, ETC activity, and metabolic shifts toward glycolysis or OXPHOS. For example, some EVT are known to favor OXPHOS over glycolysis and increase ROS generation through upregulation of OPA1 expression [35, 36]. However, in other EVT with elevated DRP1 expression, ROS production is reduced due to a metabolic shift towards increased glycolysis [29, 37]. Antioxidant therapy may be required for cell populations that overexpress OPA1. Accurately assessing mitochondrial dynamics might help identify patients who can benefit from antioxidant therapy.
Second, there is growing interest in potential modulators of mitochondrial mitophagy as preclinical methods to prevent PE. Optimal mitophagy promotes trophoblast cell survival, but its dysfunction leads to apoptosis [19, 79]. There is a strong desire to develop new drugs that can control the expression of target proteins involved in the regulation of mitochondrial dynamics and mitophagy (e.g., BNIP3, TFAM, DRAM1, MFN1, MFN2, OPA1, and DRP1) (see the subsection 3.2.5). For example, preclinical studies are providing attractive candidates for modulating mitochondrial dynamics, such as the quinazolinone derivative known as the Drp1 inhibitor mitochondrial division inhibitor-1 (Mdivi-1) [29]. In the field of cancer, Mdivi-1 appears to be a promising therapeutic strategy for the treatment of a variety of cancers [111]. Additionally, upregulation of DRAM1 expression induces autophagy in the placenta of PE mice [47]. Therefore, pharmacological modulators of mitochondrial dynamics and mitophagy hold great potential in advancing the fundamental research and clinical translation of PE therapy. However, it is currently difficult to monitor mitochondrial function in real time. Furthermore, the lack of molecular markers that can diagnose mitophagy activation or inhibition with high accuracy remains a major barrier to PE research and treatment. Therefore, preclinical research on the regulation of mitochondrial dynamics in PE is in its infancy.
Discussion
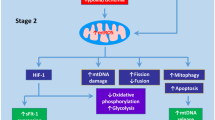
In this review, we summarize evidence of spatial and temporal changes in mitochondrial function (i.e., mitochondrial biogenesis, dynamics, and mitophagy) in normal cytotrophoblasts, syncytiotrophoblasts, and EVT, and discuss for the first time the possibility that disruption of the mitochondrial compensatory mechanism for these changes may be linked to the etiology of PE. First, placental mitochondria have a wide range of biological functions (i.e., mitochondrial flexibility, adaptation, and compensation) with respect to coordinating trophoblast cell function to support energy production for proper growth of the fetus. Indeed, syncytiotrophoblasts have distinct mitochondrial morphology and function compared with that in cytotrophoblasts (Fig. 2). This is because genes and proteins related to mitochondrial biogenesis and dynamics and energy metabolism are differentially expressed in cytotrophoblasts and syncytiotrophoblasts (Table 1). Mitochondria appear to constantly adapt to intrinsic and extrinsic stresses across gestation, fine-tuning key processes such as mitochondrial biogenetics, dynamics, mitophagy, and apoptosis in a timely manner [77, 112]. Therefore, understanding the spatial and temporal changes in mitochondrial dynamics, biogenesis, and mitophagy in the normal placenta is important to elucidate the pathogenesis of PE.

Regulation of mitochondrial dynamics and mitophagy in normal and preeclamptic placentas. Cytotrophoblast cells differentiate into syncytiotrophoblasts and EVT, and these cells cope with the ever-changing environments (e.g., nutrient and oxygen supply) (Area surrounded by solid line). Mitochondria have evolved specific strategies such as biogenesis and dynamics that significantly impact efficient energy production, cell survival, and death. The present review suggests that PE may be associated with decompensation of placental function (Area surrounded by dotted line). PE may be characterized by two phases: a long compensatory phase and a progressive decompensated phase. Impaired mitophagy due to mitochondrial dysfunction may be one of the most important determinants in compensated and uncompensated PE. Morphological and biochemical assessment of mitophagy in preeclamptic placentas is useful for risk stratification and management. Assessment of compensation or decompensation may further provide clues for predicting disease severity. In contrast to loPE where mitophagy function is compensated, eoPE is associated with decompensation of or defects in mitophagy, which leads to apoptosis. However, too little data has been accumulated to define loPE and eoPE as compensated and decompensated, respectively
Cytotrophoblasts differentiate into syncytiotrophoblasts and EVT, and these cells cope with the ever-changing environments (e.g., nutrient and oxygen supply) (Area surrounded by solid line). Mitochondria have evolved specific strategies such as biogenesis and dynamics that significantly impact efficient energy production, cell survival, and death. The present review suggests that PE may be associated with decompensation of placental function (Area surrounded by dotted line). Preeclampsia may be characterized by two phases: a long compensatory phase and a progressive decompensated phase. Impaired mitophagy due to mitochondrial dysfunction may be one of the most important determinants in compensated and uncompensated PE. Morphological and biochemical assessment of mitophagy in preeclamptic placentas is useful for risk stratification and management. Assessment of compensation or decompensation may further provide clues for predicting disease severity. In contrast to loPE where mitophagy function is compensated, eoPE is associated with decompensation of or defects in mitophagy, which leads to apoptosis. However, too little data has been accumulated to define loPE and eoPE as compensated and decompensated, respectively.
Second, many researchers believe that impaired redox homeostasis may adversely affect vascular endothelial cell function [106], causes systemic vasoconstriction and inflammation, and contributes to multiorgan dysfunction, leading to the development and progression of PE [4, 10]. At present, the molecular basis of PE is still unresolved, and the boundary between the two forms, loPE and eoPE, remains unclear, except for a spectrum of onset time and severity of presentation [1, 4, 9]. It has been demonstrated that in PE placentas, hypoxia-mediated mitochondrial fission induces regulation of ROS generation and activation of mitophagy machinery and triggers increased mitochondrial fragmentation and placental tissue damage over time [21, 25, 45, 106]. In PE, the degree of oxidative stress is greater than in normal pregnancy, and mitochondrial function is partially or almost completely impaired, depending on the severity of the disease [10]. Given that mitochondria undergo their dynamics and autophagy to exert the proper function when trophoblast cells experience oxidative stress [87], the development of PE may be associated with mitochondrial dysfunction [36]. Recent studies have increasingly revealed aberrant regulation of mitochondrial function in PE placentas (Fig. 2). The disruption of homeostatic balance in mitochondrial dynamics may lead to different phenotypes of PE, depending on its severity [25, 113]. Recent advances in high-throughput technologies using gene microarray and proteomic analysis have identified several molecular mechanisms relevant to mitochondrial function within and between clinical phenotypes (see the subsection 3.2.5). Although the pathogenesis of loPE and eoPE cannot be simply considered to be activation and impairment of mitophagy, respectively, mitochondrial dynamics and their function appear to be different in eoPE and loPE placentas [25, 90]. For example, OPA1 [48] and TFAM [45] are downregulated in eoPE placentas, suggesting that mitochondria are highly fragmented. The autophagy and mitophagy mechanisms could already be disrupted in such placentas. On the other hand, increased expression of MFN1 [48] and MFN2 [74] in the loPE placenta leads to increased ROS signaling, mitochondrial dynamics and biogenesis, and mitophagy, which in turn maintains mitochondrial health even after the presence of the molecular- and organelle-level dysfunction. Conversely, persistent downregulation of MFN1 and MFN2 expression leads to loss of placental function, so pregnancy termination may be recommended even in patients diagnosed with loPE. Thus, compensatory reserve for mitochondrial dysfunction may determine disease severity. However, the current model is simplistic, since the early/late onset classification does not reflect the pathophysiology of PE and it is unlikely that the timing of the clinical manifestations only relies on the mitochondrial adaptations to environmental stress, especially hypoxia.
Finally, we discuss why contradictory results were obtained regarding some markers of mitochondrial biogenesis (e.g., PDK, PDH, HK, or PGC-1), dynamics (e.g., MFN1, MFN2, OPA1, or DRP1), and mitophagy (e.g., TFAM, or DRAM1) in PE placentas (see the subsections 3.2.3 and 3.2.5). First, mitochondrial fission is augmented in both first trimester placenta under physiological hypoxia and PE placenta under pathological hypoxia [29]. However, there is currently no way to differentiate between these two events (i.e., physiological or pathological). Second, the internal structure of the placenta is complex, so the results vary from one part of the placenta to another. The expression of genes and proteins related to mitochondrial function in PE placental tissue may depend on the proportion of cytotrophoblasts and syncytiotrophoblasts in the sample. We know that the former is basically involved in mitochondrial fusion, and the latter in fission (Table 1). Finally, adaptive or compensatory mechanisms enable trophoblast cells to survive even when mitochondrial function is temporarily impaired. For example, redox imbalance and impaired energy metabolism reduce the mitochondrial OXPHOS capacity, alter mitochondrial morphology and dynamics, and lead to the accumulation of mutated mtDNA and then dysfunction of mitophagy (Fig. 2). However, the flexibility of trophoblast cells allows the upregulation of compensatory pathways (e.g., increased antioxidant activity or increased mitochondrial repair genes) to repair salvageable organelles [48]. Therefore, endogenous repair mechanisms may restore placental function by fine-tuning mitochondrial dynamics through both the spatial and temporal variations; however, over time, irreversible mitochondrial damage may also worsen placental function via impaired mitophagy. Disruption of compensatory mechanisms in mitochondrial dynamics may lead to apoptotic trophoblast cell death.
In conclusion, this review suggests that mitochondrial dysfunction and disruption of its compensatory mechanisms have been implicated in the pathogenesis of PE, albeit with inconsistent results. Poor mitochondrial adaptation and compensation is believed to lead to development of pathologies of pregnancy, including PE. Understanding the pathogenesis of PE requires the identification of informative candidate molecules related to mitochondrial function.
Future perspectives
Trophoblasts have a variety of survival strategies to sense oxygen and nutrient levels and adjust energy metabolic pathways in their ever-changing environments within the placenta. Monitoring the quality of trophoblast cells requires real-time biochemical quantification of genes or proteins related to mitochondrial biogenesis, dynamics, and mitophagy in individual placentas. However, a comprehensive assessment of mitochondrial function in each placenta is virtually impossible due to measurement difficulties. To resolve this issue, quantification of circulating cell-free DNA (ccf-DNA) and exosomes may be practical. Ccf-DNA and exosomes are essential molecular mediators that reflect the characteristics of producer cells. They are considered to be involved in regulating numerous physiological and pathological processes, including mitochondrial function in trophoblast cells. Liquid biopsy is a novel sampling method based on recent biotechnology to overcome some inherent limitations [114]. Gene expression analysis verified by reverse transcription-polymerase chain reaction (RT-PCR) assay can be performed across a custom gene panel. The custom panel includes candidate genes potentially related to mitochondrial dynamics, biogenesis, and mitophagy (e.g., PDK, PDH, HK, PGC-1, MFN1, MFN2, OPA1, DRP1, TFAM, or DRAM1). For example, decreased expression of genes such as MFN1, MFN2, OPA1, and TFAM may help identify patients who would benefit from pregnancy termination. Such panel-based testing can transform risk stratification and management of the PE patients. Research focusing on mitochondrial function will gain more clinical importance as an effective treatment strategy for PE. Furthermore, understanding how the mitochondrial morphology and function influence cell fate decisions of trophoblast stem cells is an important question in normal placentation biology.
Data availability
No new data were created.
References
Tranquilli AL, Dekker G, Magee L, Roberts J, Sibai BM, Steyn W, Zeeman GG, Brown MA (2014) The classification, diagnosis and management of the hypertensive disorders of pregnancy: a revised statement from the ISSHP. Pregnancy Hypertens 4(2):97–104. https://doi.org/10.1016/j.preghy.2014.02.001
Staff AC (2019) The two-stage placental model of preeclampsia: an update. J Reprod Immunol 134–135:1–10. https://doi.org/10.1016/j.jri.2019.07.004
Tomimatsu T, Mimura K, Endo M, Kumasawa K, Kimura T (2017) Pathophysiology of preeclampsia: an angiogenic imbalance and long-lasting systemic vascular dysfunction. Hypertens Res 40(4):305–310. https://doi.org/10.1038/hr.2016.152
Garrido-Gomez T, Dominguez F, Quiñonero A, Diaz-Gimeno P, Kapidzic M, Gormley M, Ona K, Padilla-Iserte P, McMaster M, Genbacev O, Perales A, Fisher SJ, Simón C (2017) Defective decidualization during and after severe preeclampsia reveals a possible maternal contribution to the etiology. Proc Natl Acad Sci U S A 114(40):E8468–E8477. https://doi.org/10.1073/pnas.1706546114
Redman CWG, Staff AC, Roberts JM (2022) Syncytiotrophoblast stress in preeclampsia: the convergence point for multiple pathways. Am J Obstet Gynecol 226(2S):S907–S927. https://doi.org/10.1016/j.ajog.2020.09.047
Archibald JM (2015) Endosymbiosis and eukaryotic cell evolution. Curr Biol 25R911–25R921. https://doi.org/10.1016/j.cub.2015.07.055
Kondadi AK, Anand R, Reichert AS (2019) Functional interplay between Cristae Biogenesis, Mitochondrial Dynamics and mitochondrial DNA Integrity. Int J Mol Sci 204311. https://doi.org/10.3390/ijms20174311
Ma Y, Wang L, Jia R (2020) The role of mitochondrial dynamics in human cancers. Am J Cancer Res 10:1278–1293 eCollection 2020
Marín R, Chiarello DI, Abad C, Rojas D, Toledo F, Sobrevia L (2020) Oxidative stress and mitochondrial dysfunction in early-onset and late-onset preeclampsia. Biochim Biophys Acta Mol Basis Dis 1866(12):165961. https://doi.org/10.1016/j.bbadis.2020.165961
Zhou X, Han TL, Chen H, Baker PN, Qi H, Zhang H (2017) Impaired mitochondrial fusion, autophagy, biogenesis and dysregulated lipid metabolism is associated with preeclampsia. Exp Cell Res 359(1):195–204. https://doi.org/10.1016/j.yexcr.2017.07.029
Annesley SJ, Fisher PR (2019) Mitochondria in Health and Disease. Cells 8(7):680. https://doi.org/10.3390/cells8070680
Furui T, Kurauchi O, Tanaka M, Mizutani S, Ozawa T, Tomoda Y (1994) Decrease in cytochrome c oxidase and cytochrome oxidase subunit I messenger RNA levels in preeclamptic pregnancies. Obstet Gynecol 84(2):283–288
Oh SY, Choi SJ, Kim KH, Cho EY, Kim JH, Roh CR (2008) Autophagy-related proteins, LC3 and Beclin-1, in placentas from pregnancies complicated by preeclampsia. Reprod Sci 15(9):912–920. https://doi.org/10.1177/1933719108319159
Kalkat M, Garcia J, Ebrahimi J, Melland-Smith M, Todros T, Post M, Caniggia I (2013) Placental autophagy regulation by the BOK-MCL1 rheostat. Autophagy 9(12):2140–2153. https://doi.org/10.4161/auto.26452
Vishnyakova PA, Volodina MA, Tarasova NV, Marey MV, Kan NE, Khodzhaeva ZS, Vysokikh MY, Sukhikh GT (2017) Alterations in antioxidant system, mitochondrial biogenesis and autophagy in preeclamptic myometrium. BBA Clin 8:35–42. https://doi.org/10.1016/j.bbacli.2017.06.002
Xu P, Zheng Y, Liao J, Hu M, Yang Y, Zhang B, Kilby MD, Fu H, Liu Y, Zhang F, Xiong L, Liu X, Jin H, Wu Y, Huang J, Han T, Wen L, Gao R, Fu Y, Fan X, Qi H, Baker PN, Tong C (2023) AMPK regulates homeostasis of invasion and viability in trophoblasts by redirecting glucose metabolism: implications for pre-eclampsia. Cell Prolif 56(2):e13358. https://doi.org/10.1111/cpr.13358
Jahan F, Vasam G, Green AE, Bainbridge SA, Menzies KJ, Jahan F (2023) Vasam GPlacental mitochondrial function and dysfunction in Preeclampsia. Int J Mol Sci 24(4):4177. https://doi.org/10.3390/ijms24044177
Zhou X, Zhao X, Zhou W, Qi H, Zhang H, Han TL, Baker P (2021) Impaired placental mitophagy and oxidative stress are associated with dysregulated BNIP3 in preeclampsia. Sci Rep 11(1):20469. https://doi.org/10.1038/s41598-021-99837-1
Nakashima A, Aoki A, Kusabiraki T, Cheng SB, Sharma S, Saito S (2017) Autophagy regulation in preeclampsia: pros and cons. J Reprod Immunol 123:17–23. https://doi.org/10.1016/j.jri.2017.08.006
Fisher JJ, McKeating DR, Cuffe JS, Bianco-Miotto T, Holland OJ, Perkins AV (2019) Proteomic Analysis of Placental Mitochondria following trophoblast differentiation. Front Physiol 10:1536. https://doi.org/10.3389/fphys.2019.01536
Manna S, McCarthy C, McCarthy FP (2019) Placental ageing in adverse pregnancy outcomes: Telomere Shortening, Cell Senescence, and mitochondrial dysfunction. Oxid Med Cell Longev 2019:3095383. https://doi.org/10.1155/2019/3095383
Bartho LA, McKeating DR, Hannan NJ, Kaitu’u-Lino TJ, Perkins AV (2022) Transcriptional profiles of genes related to mitochondrial aging in placental pathologies. Mol Hum Reprod 29(9):gaac026. https://doi.org/10.1093/molehr/gaac026
Martínez F, Kiriakidou M, Strauss JF 3rd (1997) Structural and functional changes in mitochondria associated with trophoblast differentiation: methods to isolate enriched preparations of syncytiotrophoblast mitochondria. Endocrinology 138(5):2172–2183. https://doi.org/10.1210/endo.138.5.5133
Scorrano L (2007) Multiple functions of mitochondria-shaping proteins. Novartis Found Symp 287:47–55. https://doi.org/10.1002/9780470725207.ch4. discussion 55 – 9
Ausman J, Abbade J, Ermini L, Farrell A, Tagliaferro A, Post M, Caniggia I (2018) Ceramide-induced BOK promotes mitochondrial fission in preeclampsia. Cell Death Dis 9(3):298. https://doi.org/10.1038/s41419-018-0360-0
Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11(12):872–884. https://doi.org/10.1038/nrm3013
Yu SB, Pekkurnaz G (2018) Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. J Mol Biol 430(21):3922–3941. https://doi.org/10.1016/j.jmb.2018.07.027
Abdullah MO, Zeng RX, Margerum CL, Papadopoli D, Monnin C, Punter KB, Chu C, Al-Rofaidi M, Al-Tannak NF, Berardi D, Rattray Z, Rattray NJW, Abraham SA, Eskelinen EL, Watson DG, Avizonis D, Topisirovic I, Chan EYW (2022) Mitochondrial hyperfusion via metabolic sensing of regulatory amino acids. Cell Rep 40(7):111198. https://doi.org/10.1016/j.celrep.2022.111198
Gillmore T, Farrell A, Alahari S, Sallais J, Kurt M, Park C, Ausman J, Litvack M, Post M, Caniggia I (2022) Dichotomy in hypoxia-induced mitochondrial fission in placental mesenchymal cells during development and preeclampsia: consequences for trophoblast mitochondrial homeostasis. Cell Death Dis 13(2):191. https://doi.org/10.1038/s41419-022-04641-y
Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y (2013) Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32(40):4814–4824. https://doi.org/10.1038/onc.2012.494
Bustamante J, Ramírez-Vélez R, Czerniczyniec A, Cicerchia D, Aguilar de Plata AC, Lores-Arnaiz S (2014) Oxygen metabolism in human placenta mitochondria. J Bioenerg Biomembr 46(6):459–469. https://doi.org/10.1007/s10863-014-9572-x
Adebayo M, Singh S, Singh AP, Dasgupta S (2021) Mitochondrial fusion and fission: the fine-tune balance for cellular homeostasis. FASEB J 35(6):e21620. https://doi.org/10.1096/fj.202100067R
De Castillo losR, Zarco-Zavala D, Olvera-Sanchez M, Pardo S, Juarez JP, Martinez O, Mendoza-Hernandez F, García-Trejo G, Flores-Herrera JJ (2011) Atypical cristae morphology of human syncytiotrophoblast mitochondria: role for complex V. J Biol Chem 286(27):23911–23919. https://doi.org/10.1074/jbc.M111.252056
Kolahi KS, Valent AM, Thornburg KL, Cytotrophoblast (2017) Not Syncytiotrophoblast, dominates glycolysis and oxidative phosphorylation in Human Term Placenta. Sci Rep 7:42941. https://doi.org/10.1038/srep42941
Walker OS, Gurm H, Sharma R, Verma N, May LL, Raha S (2021) Delta-9-tetrahydrocannabinol inhibits invasion of HTR8/SVneo human extravillous trophoblast cells and negatively impacts mitochondrial function. Sci Rep 11(1):4029. https://doi.org/10.1038/s41598-021-83563-9
Long J, Huang Y, Tang Z, Shan Y, Feng D, Wang W, Liu J, Huang Y, Gu H, Guo D, Yao R, Ni X (2022) Mitochondria targeted antioxidant significantly alleviates Preeclampsia caused by 11β-HSD2 dysfunction via OPA1 and MtDNA maintenance. Antioxid (Basel) 11(8):1505. https://doi.org/10.3390/antiox11081505
Vangrieken P, Al-Nasiry S, Bast A, Leermakers PA, Tulen CBM, Janssen GMJ, Kaminski I, Geomini I, Lemmens T, Schiffers PMH, van Schooten FJ, Remels AHV (2021) Hypoxia-induced mitochondrial abnormalities in cells of the placenta. PLoS ONE 16(1):e0245155. https://doi.org/10.1371/journal.pone.0245155
Kim DY, Jung SY, Kim YJ, Kang S, Park JH, Ji ST, Jang WB, Lamichane S, Lamichane BD, Chae YC, Lee D, Chung JS, Kwon SM (2018) Hypoxia-dependent mitochondrial fission regulates endothelial progenitor cell migration, invasion, and tube formation. Korean J Physiol Pharmacol 22(2):203–213. https://doi.org/10.4196/kjpp.2018.22.2.203
Arimoto-Ishida E, Sakata M, Sawada K, Nakayama M, Nishimoto F, Mabuchi S, Takeda T, Yamamoto T, Isobe A, Okamoto Y, Lengyel E, Suehara N, Morishige K, Kimura T (2009) Up-regulation of alpha5-integrin by E-cadherin loss in hypoxia and its key role in the migration of extravillous trophoblast cells during early implantation. Endocrinology 150(9):4306–4315. https://doi.org/10.1210/en.2008-1662
Cox LS, Redman C (2017) The role of cellular senescence in ageing of the placenta. Placenta 52:139–145. https://doi.org/10.1016/j.placenta.2017.01.116
Hu X-Q, Zhang L (2022) Mitochondrial dysfunction in the pathogenesis of Preeclampsia. Curr Hypertens Rep 24(6):157–172. https://doi.org/10.1007/s11906-022-01184-7Epub 2022 Mar 7
Correia Y, Scheel J, Gupta S, Wang K (2021) Placental mitochondrial function as a driver of angiogenesis and placental dysfunction. Biol Chem 402(8):887–909. https://doi.org/10.1515/hsz-2021-0121
Smith AN, Wang X, Thomas DG, Tatum RE, Booz GW, Cunningham MW (2021) The role of mitochondrial dysfunction in Preeclampsia: causative factor or collateral damage? Am J Hypertens 34(5):442–452. https://doi.org/10.1093/ajh/hpab003
Xu Z, Jin X, Cai W, Zhou M, Shao P, Yang Z, Fu R, Cao J, Liu Y, Yu F, Fan R, Zhang Y, Zou S, Zhou X, Yang N, Chen X, Li Y (2018) Proteomics Analysis Reveals Abnormal Electron Transport and excessive oxidative stress cause mitochondrial dysfunction in placental tissues of early-onset Preeclampsia. Proteom Clin Appl 12(5):e1700165. https://doi.org/10.1002/prca.201700165
Vishnyakova PA, Volodina MA, Tarasova NV, Marey MV, Tsvirkun DV, Vavina OV, Khodzhaeva ZS, Kan NE, Menon R, Vysokikh MY, Sukhikh GT (2016) Mitochondrial role in adaptive response to stress conditions in preeclampsia. Sci Rep 6:32410. https://doi.org/10.1038/srep32410
He L, Wang Z, Sun Y (2004) Reduced amount of cytochrome c oxidase subunit I messenger RNA in placentas from pregnancies complicated by preeclampsia. Acta Obstet Gynecol Scand 83(2):144–148. https://doi.org/10.1111/j.0001-6349.2004.00345.x
Chen G, Lin Y, Chen L, Zeng F, Zhang L, Huang Y, Huang P, Liao L, Yu Y (2020) Role of DRAM1 in mitophagy contributes to preeclampsia regulation in mice. Mol Med Rep 22(3):1847–1858. https://doi.org/10.3892/mmr.2020.11269
Holland OJ, Cuffe JSM, Dekker Nitert M, Callaway L, Kwan Cheung KA, Radenkovic F, Perkins AV (2018) Placental mitochondrial adaptations in preeclampsia associated with progression to term delivery. Cell Death Dis 9(12):1150. https://doi.org/10.1038/s41419-018-1190-9
Beyramzadeh M, Dikmen ZG, Erturk NK, Tuncer ZS, Akbiyik F (2017) Placental respiratory chain complex activities in high risk pregnancies. J Matern Fetal Neonatal Med 30(24):2911–2917. https://doi.org/10.1080/14767058.2016.1268594
Chhimpa N, Singh N, Puri N, Kayath HP (2023) The Novel role of mitochondrial citrate synthase and citrate in the pathophysiology of Alzheimer’s Disease. J Alzheimers Dis 94(s1):S453–S472. https://doi.org/10.3233/JAD-220514
Liu X, Zuo R, Bao Y, Qu X, Sun K, Ying H (2017) Down-regulation of PDK4 is critical for the switch of Carbohydrate catabolism during syncytialization of human placental trophoblasts. Sci Rep 7(1):8474. https://doi.org/10.1038/s41598-017-09163-8
Dewi S, Triatmono VR, Rasyada Ralas PR, Veraldi V, Alfian M, Iswanti I, Prijanti FC (2022) Increasing of LDH Specific Activity and PEPCK Level play a role on activation of Gluconeogenesis Pathway in early onset Pre-eeclampsia Placenta. Rep Biochem Mol Biol 11(2):320–326. https://doi.org/10.52547/rbmb.11.2.320
Waker CA, Albers RE, Pye RL, Doliboa SR, Wyatt CN, Brown TL, Mayes DA (2017) AMPK Knockdown in placental labyrinthine progenitor cells results in restriction of critical Energy resources and terminal differentiation failure. Stem Cells Dev 26(11):808–817. https://doi.org/10.1089/scd.2016.0252
Herzig S, Shaw RJ (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19(2):121–135. https://doi.org/10.1038/nrm.2017.95
Kumagai A, Itakura A, Koya D, Kanasaki K (2018) AMP-Activated protein (AMPK) in pathophysiology of pregnancy complications. Int J Mol Sci 19(10):3076. https://doi.org/10.3390/ijms19103076
Poole LP, Macleod KF (2021) Mitophagy in tumorigenesis and metastasis. Cell Mol Life Sci 78(8):3817–3851. https://doi.org/10.1007/s00018-021-03774-1
Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ (2016) Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351(6270):275–281. https://doi.org/10.1126/science.aab4138
Shin EK, Kang HY, Yang H, Jung EM, Jeung EB (2016) The regulation of fatty acid oxidation in human preeclampsia. Reprod Sci 23(10):1422–1433. https://doi.org/10.1177/1933719116641759
Bartha JL, Visiedo F, Fernández-Deudero A, Bugatto F, Perdomo G (2012) Decreased mitochondrial fatty acid oxidation in placentas from women with preeclampsia. Placenta 33(2):132–134. https://doi.org/10.1016/j.placenta.2011.11.027
Wang Y, Walsh SW (2001 Feb-Mar) Increased superoxide generation is associated with decreased superoxide dismutase activity and mRNA expression in placental trophoblast cells in pre-eclampsia. Placenta 22(2–3):206–212. https://doi.org/10.1053/plac.2000.0608
Aouache R, Biquard L, Vaiman D, Miralles F (2018) Oxidative stress in Preeclampsia and placental diseases. Int J Mol Sci 19:1496. https://doi.org/10.3390/ijms19051496
Taravati A, Tohidi F (2018) Comprehensive analysis of oxidative stress markers and antioxidants status in preeclampsia. Taiwan J Obstet Gynecol 57(6):779–790. https://doi.org/10.1016/j.tjog.2018.10.002
Li J, Dong X, Liu JY, Gao L, Zhang WW, Huang YC, Wang Y, Wang H, Wei W, Xu DX (2024) FUNDC1-mediated mitophagy triggered by mitochondrial ROS is partially involved in 1-nitropyrene-evoked placental progesterone synthesis inhibition and intrauterine growth retardation in mice. Sci Total Environ 908:168383. https://doi.org/10.1016/j.scitotenv.2023.168383
Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y, Yang G, Chen Y, Cheng J, Lu Y, Liu J (2021) Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 11(4):1845–1863. https://doi.org/10.7150/thno.50905
Yan W, Zhang H, Liu P, Wang H, Liu J, Gao C, Liu Y, Lian K, Yang L, Sun L, Guo Y, Zhang L, Dong L, Lau WB, Gao E, Gao F, Xiong L, Wang H, Qu Y, Tao L (2013) Impaired mitochondrial biogenesis due to dysfunctional adiponectin-AMPK-PGC-1α signaling contributing to increased vulnerability in diabetic heart. Basic Res Cardiol 108(3):329. https://doi.org/10.1007/s00395-013-0329-1
Zsengellér ZK, Rajakumar A, Hunter JT, Salahuddin S, Rana S, Stillman IE, Ananth Karumanchi S (2016) Trophoblast mitochondrial function is impaired in preeclampsia and correlates negatively with the expression of soluble fms-like tyrosine kinase 1. Pregnancy Hypertens 6(4):313–319. https://doi.org/10.1016/j.preghy.2016.06.004
Yu J, Guo X, Chen R, Feng L (2016) Downregulation of Mitofusin 2 in Placenta is related to Preeclampsia. Biomed Res Int 2016:6323086. https://doi.org/10.1155/2016/6323086
Soleymanlou N, Jurisica I, Nevo O, Ietta F, Zhang X, Zamudio S, Post M, Caniggia I (2005) Molecular evidence of placental hypoxia in preeclampsia. J Clin Endocrinol Metab 90(7):4299–4308. https://doi.org/10.1210/jc.2005-0078
Sallais J, Park C, Alahari S, Porter T, Liu R, Kurt M, Farrell A, Post M, Caniggia I (2022) HIF1 inhibitor acriflavine rescues early-onset preeclampsia phenotype in mice lacking placental prolyl hydroxylase domain protein 2. JCI Insight 7(23):e158908. https://doi.org/10.1172/jci.insight.158908
Albers RE, Kaufman MR, Natale BV, Keoni C, Kulkarni-Datar K, Min S, Williams CR, Natale DRC, Brown TL (2019) Trophoblast-specific expression of Hif-1α results in Preeclampsia-Like symptoms and fetal growth restriction. Sci Rep 9(1):2742. https://doi.org/10.1038/s41598-019-39426-5
Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL (2012) Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110(11):1484–1497. https://doi.org/10.1161/CIRCRESAHA.111.263848
Caniggia I, Winter J, Lye SJ, Post M (2000 Mar-Apr) Oxygen and placental development during the first trimester: implications for the pathophysiology of pre-eclampsia. Placenta 21:25–30 Suppl A:S. https://doi.org/10.1053/plac.1999.0522
Xia QS, Lu FE, Wu F, Huang ZY, Dong H, Xu LJ, Gong J (2020) New role for ceramide in hypoxia and insulin resistance. World J Gastroenterol 26(18):2177–2186. https://doi.org/10.3748/wjg.v26.i18.2177
Aydogan Mathyk B, Temel Yuksel I, Tayyar A, Aslan Cetin B, Tayyar AT, Koroglu N (2020) Maternal serum mitofusin-2 levels in patients with preeclampsia: the possible role of mitochondrial dysfunction in preeclampsia. J Matern Fetal Neonatal Med 33(11):1861–1866. https://doi.org/10.1080/14767058.2018.1532497
Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J (2011) Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A 108(25):10190–10195. https://doi.org/10.1073/pnas.1107402108
Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M, Sollott SJ, Zorov DB (2018) Mitochondrial membrane potential. Anal Biochem 552:50–59. https://doi.org/10.1016/j.ab.2017.07.009
Fisher JJ, Bartho LA, Perkins AV, Holland OJ (2020) Placental mitochondria and reactive oxygen species in the physiology and pathophysiology of pregnancy. Clin Exp Pharmacol Physiol 47:176–184. https://doi.org/10.1111/1440-1681.13172
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160(2):189–200. https://doi.org/10.1083/jcb.200211046
Akcora Yildiz D, Irtegun Kandemir S, Agacayak E, Deveci E (2017) Evaluation of protein levels of autophagy markers (Beclin 1 and SQSTM1/p62) and phosphorylation of cyclin E in the placenta of women with preeclampsia. Cell Mol Biol (Noisy-le-grand) 63(12):51–55. https://doi.org/10.14715/cmb/2017.63.12.12
Son JM, Sarsour EH, Kakkerla Balaraju A, Fussell J, Kalen AL, Wagner BA, Buettner GR, Goswami PC (2017) Mitofusin 1 and optic atrophy 1 shift metabolism to mitochondrial respiration during aging. Aging Cell 16(5):1136–1145. https://doi.org/10.1111/acel.12649
Cushen SC, Ricci CA, Bradshaw JL, Silzer T, Blessing A, Sun J, Zhou Z, Scroggins SM, Santillan MK, Santillan DA, Phillips NR, Goulopoulou S (2022) Reduced maternal circulating cell-free mitochondrial DNA is Associated with the development of Preeclampsia. J Am Heart Assoc 11(2):e021726. https://doi.org/10.1161/JAHA.121.021726
Williamson RD, McCarthy FP, Kenny LC, McCarthy CM (2019) Activation of a TLR9 mediated innate immune response in preeclampsia. Sci Rep 9(1):5920. https://doi.org/10.1038/s41598-019-42551-w
Qiu C, Hevner K, Enquobahrie DA, Williams MA (2012) A case-control study of maternal blood mitochondrial DNA copy number and preeclampsia risk. Int J Mol Epidemiol Genet 3(3):237–244
Ding WX, Yin XM (2012) Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 393(7):547–564. https://doi.org/10.1515/hsz-2012-0119
Kang D, Kim SH, Hamasaki N Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion 2007 Feb-Apr ;7(1–2):39–44. https://doi.org/10.1016/j.mito.2006.11.017
Nakashima A, Cheng SB, Ikawa M, Yoshimori T, Huber WJ, Menon R, Huang Z, Fierce J, Padbury JF, Sadovsky Y, Saito S, Sharma S (2020) Evidence for lysosomal biogenesis proteome defect and impaired autophagy in preeclampsia. Autophagy 16(10):1771–1785. https://doi.org/10.1080/15548627.2019.1707494
Ivankovic D, Chau KY, Schapira AH, Gegg ME (2016) Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem 136(2):388–402. https://doi.org/10.1111/jnc.13412
Zhang L, Han L, Ma R, Hou X, Yu Y, Sun S, Xu Y, Schedl T, Moley KH, Wang Q (2015) Sirt3 prevents maternal obesity-associated oxidative stress and meiotic defects in mouse oocytes. Cell Cycle 14(18):2959–2968. https://doi.org/10.1080/15384101.2015.1026517
Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao WT, Ma HK, Jiang MD, Xu TT, Xu J, Shen Y, Song B, Gao PJ, Han WQ, Zhang W (2020) HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol 36:101671. https://doi.org/10.1016/j.redox.2020.101671
Ribeiro VR, Romao-Veiga M, Nunes PR, Peracoli JC, Peracoli MTS (2022) Increase of autophagy marker p62 in the placenta from pregnant women with preeclampsia. Hum Immunol 83(5):447–452. https://doi.org/10.1016/j.humimm.2022.02.005
Tekola-Ayele F, Workalemahu T, Gorfu G, Shrestha D, Tycko B, Wapner R, Zhang C, Louis GMB (2019) Sex differences in the associations of placental epigenetic aging with fetal growth. Aging 11(15):5412–5432. https://doi.org/10.18632/aging.102124
Yildirim RM, Ergun Y, Murat Basar M (2022) Mitochondrial dysfunction, Mitophagy and their correlation with Perinatal complications: Preeclampsia and Low Birth Weight. Biomedicines 10(10):2539. https://doi.org/10.3390/biomedicines10102539
Peng X, Hou R, Yang Y, Luo Z, Cao Y (2022) Current Studies of Mitochondrial Quality Control in the Preeclampsia. Front Cardiovasc Med. Feb 28:9:836111. https://doi.org/10.3389/fcvm.2022.836111. eCollection 2022
Hu H, Guo L, Overholser J, Wang X (2022) Mitochondrial VDAC1: a potential therapeutic target of inflammation-related diseases and Clinical opportunities. Cells 11(19):3174. https://doi.org/10.3390/cells11193174
Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12(2):119–131. https://doi.org/10.1038/ncb2012
Tong J, Zhao W, Lv H, Li WP, Chen ZJ, Zhang C (2018) Transcriptomic profiling in human decidua of severe Preeclampsia detected by RNA sequencing. J Cell Biochem 119(1):607–615. https://doi.org/10.1002/jcb.26221
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell 122(6):927–939. https://doi.org/10.1016/j.cell.2005.07.002
Sharp AN, Heazell AE, Crocker IP, Mor G (2010) Placental apoptosis in health and disease. Am J Reprod Immunol 64(3):159–169. https://doi.org/10.1111/j.1600-0897.2010.00837.x
Kasture V, Kale A, Randhir K, Sundrani D, Joshi S (2019) Effect of maternal omega-3 fatty acids and vitamin E supplementation on placental apoptotic markers in rat model of early and late onset preeclampsia. Life Sci 239:117038. https://doi.org/10.1016/j.lfs.2019.117038
Zhao L, Ma R, Zhang L, Yuan X, Wu J, He L, Liu G, Du R (2019) Inhibition of HIF-1a-mediated TLR4 activation decreases apoptosis and promotes angiogenesis of placental microvascular endothelial cells during severe pre-eclampsia pathogenesis. Placenta 83:8–16. https://doi.org/10.1016/j.placenta.2019.06.375
Wang G, Huang Y, Hu T, Zhang B, Tang Z, Yao R, Huang Y, Fan X, Ni X (2020) Contribution of placental 11β-HSD2 to the pathogenesis of preeclampsia. FASEB J 34(11):15379–15399. https://doi.org/10.1096/fj.202001003RR
Molnár M, Sütö T, Tóth T, Hertelendy F (1994) Prolonged blockade of nitric oxide synthesis in gravid rats produces sustained hypertension, proteinuria, thrombocytopenia, and intrauterine growth retardation. Am J Obstet Gynecol 170(5 pt 1):1458–1466. https://doi.org/10.1016/s0002-9378(94)70179-2
Li J, LaMarca B, Reckelhoff JF (2012) A model of preeclampsia in rats: the reduced uterine perfusion pressure (RUPP) model. Am J Physiol Heart Circ Physiol 303:H1–H8. https://doi.org/10.1152/ajpheart.00117.2012
Gatford KL, Andraweera PH, Roberts CT, Care AS (2020) Animal models of preeclampsia. Hypertension 75:1363–1381. https://doi.org/10.1161/hypertensionaha.119.14598
Xu X, Pan J-R, Zhang Y-Z (2019) CoQ10 alleviate preeclampsia symptoms by enhancing the function of mitochondria in the placenta of pregnant rats with preeclampsia. Hypertens Pregnancy 38(4):217–222. https://doi.org/10.1080/10641955.2019.1649420
Raijmakers MTM, Dechend R, Poston L (2004) Oxidative stress and preeclampsia: rationale for antioxidant clinical trials. Hypertension 44(4):374–380. https://doi.org/10.1161/01.HYP.0000141085.98320.01
Teran E, Hernandez I, Nieto B, Tavara R, Ocampo JE, Calle A (2009) Coenzyme Q10 supplementation during pregnancy reduces the risk of pre-eclampsia. Int J Gynaecol Obstet 105(1):43–45. https://doi.org/10.1016/j.ijgo.2008.11.033
Salles AM, Galvao TF, Silva MT, Motta LCD, Pereira MG (2012) Antioxidants for preventing preeclampsia: a systematic review. ScientificWorldJournal 2012:243476. https://doi.org/10.1100/2012/243476
Yang Y, Xu P, Zhu F, Liao J, Wu Y, Hu M, Fu H, Qiao J, Lin L, Huang B, Jin H, Liu X, Zheng Y, Wen L, Saffery R, Kilby MD, Yan J, Kenny LC, Qi H, Tong C, Baker PN (2021) The potent antioxidant MitoQ protects against Preeclampsia during Late Gestation but increases the risk of Preeclampsia when administered in early pregnancy. Antioxid Redox Signal 34(2):118–136. https://doi.org/10.1089/ars.2019.7891
Yang Y, Jin H, Qiu Y, Liu Y, Wen L, Fu Y, Qi H, Baker PN, Tong C (2022) Reactive oxygen species are essential for placental angiogenesis during early Gestation. Oxid Med Cell Longev 2022:4290922. https://doi.org/10.1155/2022/4290922
Wang J, Hansen K, Edwards R, Van Houten B, Qian W (2015) Mitochondrial division inhibitor 1 (mdivi-1) enhances death receptor-mediated apoptosis in human ovarian cancer cells. Biochem Biophys Res Commun 456(1):7–12. https://doi.org/10.1016/j.bbrc.2014.11.010
Lu M, Sferruzzi-Perri AN (2021) Placental mitochondrial function in response to gestational exposures. Placenta 104:124–137. https://doi.org/10.1016/j.placenta.2020.11.012
Chan DC (2020) Mitochondrial dynamics and its involvement in Disease. Annu Rev Pathol 15:235–259. https://doi.org/10.1146/annurev-pathmechdis-012419-032711
Nikanjam M, Kato S, Kurzrock R (2022) Liquid biopsy: current technology and clinical applications. J Hematol Oncol 15(1):131. https://doi.org/10.1186/s13045-022-01351-y
Acknowledgements
Figures were created by Toyomi Kobayashi (Ms.Clinic MayOne, Nara, Japan; https://www.mscl-mayone.com/; accessed on 9 October 31, 2023).
Author information
Authors and Affiliations
Contributions
Conception and design, H.K. Acquisition of data, S.M., C.Y., and S.I. Analysis and Interpretation of data, H.S. Drafting of the manuscript, H.K. Critical revision of the manuscript for important intellectual content, S.M., C.Y., H.S., and S.I. Statistical analysis, H.S. Administrative technical or material support, H.K. Supervision, S.I. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
The submitted paper is a review article and has not been approved by the Institutional Review Board and the Research and Ethical Committee of Nara Medical University Graduate School of Medicine, Kashihara, Japan.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kobayashi, H., Yoshimoto, C., Matsubara, S. et al. An integral role of mitochondrial function in the pathophysiology of preeclampsia. Mol Biol Rep 51, 330 (2024). https://doi.org/10.1007/s11033-024-09285-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09285-z