
Abstract
Background
Diploid cells of Saccharomyces cerevisiae undergo either pseudohyphal differentiation or sporulation in response to depletion of carbon and nitrogen sources. Distinct signaling pathways regulate filamentation and sporulation in response to nutrient limitation. How these pathways are coordinated for implementing distinct cell fate decisions in response to similar nutritional cues is an enigma. Although the role of trehalose pathway in sporulation has been extensively studied, it’s possible role in pseudohyphal differentiation has been unexplored.
Methods and results
Briefly, tps1 and tps2 mutants were tested for their ability to form pseudohyphae independently as well as in the background of GPR1 and RAS2 mutations. Here, we demonstrate that disruption of TPS1 but not TPS2 inhibits pseudohyphae formation. Interestingly, deletion of GPR1 suppresses the above defect. Further genetic analysis revealed that TPS1 and TPS2 exert opposing effects in triggering filamentation.
Conclusion
We provide new insights into the role of an otherwise well-known pathway of trehalose biosynthesis in pseudohyphal differentiation. Based on additional data we propose that downstream signaling, mediated by cAMP may be modulated by nutrient mediated differential regulation of RAS2 by TPS1 and TPS2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Organisms have evolved a plethora of developmental and differentiation mechanisms to overcome nutritional deprivation. This involves extensive rewiring of signaling mechanisms to be able to meet the changing metabolic requirements of the cell. Yeast, Saccharomyces cerevisiae, undergoes both metabolic as well as morphologic adaptation in order to overcome nutrient deprivation [1, 2]. Thus, diploid cells of S. cerevisiae have the potential to either sporulate or achieve pseudohyphal differentiation in response to carbon and/or nitrogen depletion [3,4,5,6]. Despite intense investigations, it is still unclear as to how these two developmental processes of pseudohyphae formation and sporulation emerge in response to the common trigger of low nitrogen and low glucose [4, 7, 8]. Interestingly, the cAMP-PKA pathway is involved in both acetate mediated spore formation [9, 10] as well as glucose mediated pseudohyphal differentiation [1, 11].
Signaling for pseudohyphal transition in response to low ammonium occurs via MEP2, an ammonium transporter [12]. It was demonstrated that MEP2 is a transceptor i.e. in addition to signaling, the transport function of Mep2p was necessary for filamentation response [13]. NPR1, a TORC1 effector kinase, positively regulates MEP2 under conditions of poor nitrogen availability to trigger pseudohyphae formation [14, 15]. Although it has been demonstrated that MEP2 signals via cAMP [12], the underlying mechanisms have not been fully elucidated. [13]. Of relevance here is the observation that the filamentation defect of a mep2 mutant is overcome upon addition of cAMP or by constitutively active alleles of GPA2 or RAS2 [12].
Pseudohyphal differentiation was originally thought to occur only in response to low ammonium and abundant glucose [5]. In contrast, Iyer et al. [4] demonstrated that in addition to signaling from low ammonium, low glucose signaling was also essential for filamentation response. Glucose mediated signaling for filamentation occurs through GPR1-GPA2 axis [7, 16]. GPA2 relays the signal to cAMP via adenylate cyclase [7] as does RAS2 [17]. The activation of either RAS2 or GPA2 can elicit the transcriptional changes required for glucose mediated increase of cAMP [2]. It was previously reported that KRH1/2 interfere with GPR1-GPA2 coupling thereby inhibiting downstream signaling via cAMP [18]. Iyer and Bhat [19] demonstrated that KRH1 and KRH2 are non-redundant and uncovered distinct roles for these two kelch proteins in inducing pseudohyphae by using low glucose. Thus, this study highlighted the significance of glucose limiting condition in filamentation. This was consistent with the observation that FLO11, a key gene regulating pseudohyphal differentiation [20], is glucose repressed [21]. Further, SNF1, a gene that is required to alleviate glucose repression [22], is also necessary for formation of pseudohyphae [23]. Thus, it is evident that glucose limitation is also a key component of pseudohyphal differentiation process just as it is for spore formation. This compelled us to revisit the possibility of new players in regulating pseudohyphal differentiation in response to depleting ammonium when glucose is limiting.
It has been observed that components of the glucose regulated trehalose biosynthetic pathway [24] are essential for sporulation [25]. The trehalose pathway involves two enzymes namely TPS1 (trehalose phosphate synthase) and TPS2 (trehalose phosphate phosphatase). TPS1 catalyzes the formation of trehalose-6-phosphate (Tre6P) from UDP-Glucose and glucose-6-phosphate [26]. Observations in Candida albicans had indicated that disruption of TPS1 resulted in a decrease in virulence of the strain as well as an inability to form hyphae [27]. Disruption of TPS2 caused a reduction in virulence without affecting hyphae formation [28]. Further, it was demonstrated that TPS2 and GPR1 functioned synergistically in trehalose metabolism as well as virulence [29]. The TPS genes have been shown not only to regulate differentiation in C. albicans but also to regulate several processes in plants ranging from cell morphology to architecture of inflorescence and other developmental processes [30].
Based on the above and the observation that low glucose is pivotal in pseudohyphae formation as well as in restoring the glycolytic imbalance in a tps1 mutant, we hypothesized that the trehalose biosynthetic pathway could be involved in filamentation response. Here, we show that tps1 but not tps2 mutant is defective in pseudohyphae formation. The use of SLALD (Synthetic low ammonium low dextrose) medium [4] in addition to SLAD (Synthetic low ammonium dextrose) medium [12] enabled the dissection of the independent roles of TPS1 and TPS2. Our results demonstrate that TPS1 and TPS2 may regulate RAS2 differentially depending upon the availability of nutrients, to signal filamentation via cAMP/PKA pathway.
Materials and methods
Media and strains
The strains used in this study are isogenic derivatives of ∑1278b strain (as listed in Online Resource 1). The strains were constructed using standard methods [31]. Genes were disrupted (primers listed in Online Resource 2) using marker based polymerase chain reaction (PCR) methods [32]. For all mutants used in this study, the entire coding region was replaced by the disruption cassette. Double disruptants were generated by mating the individual mutants followed by sporulation and segregation of haploids. The haploid strains were diploidized using mating type switching induced by HO plasmid.
Pseudohyphal growth assay
Synthetic low ammonium dextrose (SLAD) medium (50 µM ammonium sulphate and 2% glucose) or Synthetic low ammonium low dextrose (SLALD) medium (50 µM ammonium sulphate and 0.05% glucose) were used to score pseudohyphal growth [3, 4, 33]. Cells were spread for single colonies and incubated for 6 days at 30 °C unless mentioned otherwise. Images of colonies were captured at × 10 magnification using a Nikon Coolpix 8400 camera attached to a Nikon TS 100 microscope. Three independent colony images which are representative of at least three experimental repetitions are shown.
Spotting assay
The glucose growth phenotype was determined using the spotting assay. Cells were grown to 0.5 OD600, washed twice with sterile distilled water and five-fold serial dilutions were spotted on yeast extract peptone dextrose (YPD) agar (1% yeast extract, 2% peptone and 2% glucose) or yeast extract peptone galactose (YPGal) agar (1% yeast extract, 2% peptone and 2% galactose). YPGal medium served as a control. Images were captured after 2 days of incubation.
Western blot analysis
Crude cell extracts were prepared as described by [34]. Briefly, cells were grown to OD600 of 3–4 in YPD medium and then harvested. For analysis in SLALD medium, cells grown to 3–4 OD600 were collected by centrifugation, washed twice with sterile distilled water, re-suspended in SLALD and transferred to SLALD (one-fifth volume) and incubated for an additional 8 h before being harvested. 200 mg cells were lysed with 0.2 g of glass beads after adding lysis buffer as described [34]. Centrifugation at 8000 rpm for 5 min yielded the crude protein extract used for western blot analysis. 20 µg protein was loaded. The blots were developed with antibodies against yeast Ras2p from Santacruz Biotechnology Inc. For the loading control, Glucose 6-phosphate dehydrogenase (G6PDH), antibodies from Sigma-Aldrich were used. Both the anti-mouse pAb (for Ras2p) as well as the anti-rabbit pAb (for G6PDH) were alkaline phosphatase conjugates obtained from Sigma-Aldrich. Quantification of western blot data was carried out using ImageJ analysis [35]. The final normalized values i.e. the ratio of the net bands (after background subtraction) namely the net protein band to the net loading control band are represented in the graph. The data represented is an average of three experimental repetitions with the error bars indicating the standard deviation. Variation in protein loaded is less than 10% based on quantification of G6PDH band intensity (see Online Resource 3).
Results
Glucose growth defect of the tps1 mutant is both strain as well as ploidy dependent
It is well established that mutation in TPS1 causes a growth defect on fermentable carbon sources [34, 36]. This phenotype was observed in a haploid i.e. tps1::TRP1 of the W303 strain background [34]. In the study by van Heerden et al. [36], the diploid strain of S288c i.e. tps1::G418/tps1::G418 was used (personal communication). Suzuki et al. [37], reported that transposon insertion mutation at the TPS1 locus i.e. tps1::mTn3 was lethal in the ∑1278 strain background. Contrary to this finding, we observed that the tps1 mutant in the ∑1278 background i.e. tps1::G418 was viable. In an attempt to determine the glucose growth phenotype of the ∑1278 tps::G418 mutants, we analyzed growth on glucose as well as galactose media. Galactose medium was used as a control condition. We observed that the tps2 mutant did not exhibit any growth defect. Although the tps1Δ haploid strain in the ∑1278 background had a growth defect (Fig. 1a), the diploid strain was able to grow on glucose (Fig. 1b). Thus it is evident that glucose growth phenotype of the tps1 mutant strains varies with different lineages as well as the ploidy status of the cell. Surprisingly, we observed that the diploid tps1 mutant exhibited a growth defect on galactose. While it is difficult to explain this observation, we surmise that this may be a deleterious effect caused because glucose-6-phosphate formed from galactose accumulates above a threshold in the cell, in the absence of TPS1.

Effect of carbon source on tps mutants. Glucose growth phenotype of a haploid and b diploid TPS mutants. Three dilutions (fivefold) were spotted. Effect of zygosity on pseudohyphal growth phenotype. Colony images of c homozygous and d heterozygous TPS mutants are shown
TPS1 is required for filamentation response
Trehalose synthesis in yeast occurs in response to adverse environmental conditions, including nutritional stress [38, 39]. Since filamentation occurs in response to nutrient limitation, we hypothesized that TPS1 and/or TPS2 may be involved in regulating this response. Therefore, independent colonies of tps1 and tps2 as well as the tps1tps2 double mutant were analyzed for filamentation in SLAD as well as SLALD media (Fig. 1c). As expected, tps1 was defective in pseudohyphae formation. In contrast, the tps2 mutant had no filamentation defect indicating that TPS1 but not TPS2 is required for this response. However, the tps1tps2 double mutant formed pseudohyphae only in SLALD but not SLAD medium. A possible explanation for this would be that TPS2 is required to alleviate glucose mediated repression of filamentation. In SLALD medium, this requirement is of no consequence as the glucose concentration is such that repression does not occur. Further, strains heterozygous for the TPS1 as well as the TPS2 loci exhibit different phenotypes as compared to the strains homozygous for the same loci (Fig. 1d), indicating that the effective concentrations of intermediates of the trehalose synthesis pathway may play a role.
The next step was to determine the pathway through which TPS1 acts. Studies in C. albicans have demonstrated that TPS enzymatic activity was higher in the gpr1Δ strain [40]. To determine if there was any genetic interaction between GPR1 and TPS1 or TPS2, in S. cerevisiae, pseudohyphal growth of gpr1tps1 and gpr1tps2 mutants was monitored. Both the haploid as well as the diploid strains of the gpr1tps1 double mutant exhibited a growth defect on high glucose (data not shown). It is possible that in the absence of both TPS1 as well as GPR1, in an ammonium deficient environment, the cell experiences severe effects of perceived absence of glucose, resulting in the growth defect. In SLALD medium, however, the filamentation defect of tps1 mutant was overcome upon disruption of GPR1 (Fig. 2a, right panel and Fig. 2b), indicating that mutation in GPR1 was epistatic over mutation in TPS1, in signaling for pseudohyphae. This effect is probably mediated via the cAMP-PKA pathway as extraneous addition of cAMP overcomes the filamentation defect of the tps1 mutant in SLALD medium (Fig. 3, left panel). Further, our observations indicate that mutation in TPS2 is epistatic over mutation in GPR1 in SLAD medium (Fig. 2a, compare top and bottom panels). This is in contrast to that observed in C. albicans where mutation in TPS2 and GPR1 result in a synergistic effect in suppression of filamentation [29]. The only plausible explanation for this phenotype is that accumulation of Tre6P probably triggers filamentation through some unknown mechanism. However, this is counter-intuitive given that pseudohyphae formation is favoured by low glucose conditions [4] while Tre6P promotes glucose repression [41].

Effect of GPR1 disruption on filamentation in the background of TPS mutations. a Pseudohyphae formation at the end of 6 days of incubation. b Filamentation response of the gpr1tps1 double mutant at the end of 6 or 8 days of incubation on SLALD medium. The photographs at the end of 8 days are not of the same colonies as that shown at the end of 6 days

a Effect of extraneous addition of 1 mM cAMP on pseudohyphal differentiation of ras2 disruption in the background of tps mutations. b Effect of RAS2 disruption on pseudohyphae formation in the background of TPS mutations
TPS1 and TPS2 have a differential role in regulating RAS2 depending upon nutrient availability
It has been observed that tps1 mutation results in activation of RAS2 [34]. However, the effect of tps2 mutation on RAS2 is not known. In order to determine whether the effect on RAS2 is limited only to TPS1 or does TPS2 also have a role and whether this effect led to regulation of pseudohyphal growth, filamentation was monitored in both ras2tps1 as well as ras2tps2 double mutants. Interestingly, disruption of tps2 but not tps1 restored pseudohyphae formation in the ras2 mutant (Fig. 3b). Further, pseudohyphae formation in the ras2tps2 double mutant was enhanced in SLALD as compared to SLAD medium (Fig. 3b, compare left and right panels). Therefore, expression of Ras2p was monitored under nutrient complete (YPD medium) as well as nutrient limiting (SLALD medium) conditions in the diploid strains of tps1Δ and tps2Δ. Diploid strains were used for this experiment to ensure that observations are a more accurate reflection of the role played by TPS genes, as pseudohyphal differentiation is exhibited only by diploid cells. Contrary to earlier reports in the haploid tps1 mutant where RAS2 is activated in glucose causing the growth defect [34], we observed that Ras2p expression was reduced in the diploid tps1 mutant in YPD medium (Fig. 4a and c). This meant one of two possibilities; either that regulation of Ras2p by TPS1 resulted in opposite effects in the haploid versus the diploid cell or that Ras2p is upregulated upon TPS1 disruption in W303 strain [34] while it is downregulated in the ∑1278 strain background (this study). Whatever the reason, our observations indicate that this mutant is unable to form pseudohyphae. Since, Ras2p expression is reduced in high glucose in a diploid tps1 mutant, the defect in pseudohyphae formation is probably due to lower levels of Ras2p. Further, our data indicates that in YPD medium, Ras2p expression is upregulated in a tps2 mutant. This suggests that opposing effects exerted by TPS and TPS2 may be co-ordinated to effect signaling mediated through RAS2. In SLALD medium, however, Ras2p expression is decreased in tps2 but not tps1 mutant (Fig. 4b and c). In a tps1 mutant, even though Ras2p is expressed in SLALD medium, pseudohyphae formation is inhibited suggesting that TPS1 also regulates pseudohyphal growth in a RAS2 independent manner. To summarize, our observations indicate that TPS1 and TPS2 possibly regulate RAS2 differentially under conditions of nutrient abundance or depletion, to trigger pseudohyphal differentiation.

Expression of Ras2p in the TPS mutants. a In nutrient rich YPD medium. b In nutrient deficient SLALD medium. c Western blot quantification data. The bars represent the ratio of the normalized values i.e. background-subtracted Ras2p to background-subtracted G6PDH, error bars show standard deviation (n = 3)
To determine whether exogenous cAMP could rescue the defect caused by reduction in Ras2p levels mediated through TPS1, effect of extraneous addition of cAMP was monitored in the tps1, tps1ras2 as well as the ras2 mutant as a control (Fig. 3a). As discussed in the earlier section, the pseudohyphal defect of the tps1 mutant was rescued on addition of cAMP in SLALD but not SLAD medium (Fig. 3a, left panel). The filamentation defect of the ras2 mutant was restored on exogenous cAMP addition in SLAD medium only (Fig. 3a, middle panel) while the ras2tps1 double mutant remained defective (Fig. 3a, Right panel). Surprisingly, addition of cAMP did not rescue the filamentation defect of either the ras2 mutant or the ras2tps1 double mutant on SLALD medium (Fig. 3a, middle and right panels). These results indicate the possibility that signaling from glucose is necessary in addition to that from cAMP in the absence of RAS2.
Discussion
TPS1/2 regulate dimorphic transition and thereby virulence in Candida albicans [28] as well as Magnaporthe grisea [42]. There is evidence to show that TPS genes regulate multiple processes involved in growth and development in plants as well [30, 43]. In S. cerevisiae, although there is a large body of data available on the deleterious effects of TPS1 or TPS2 mutations [36, 44,45,46], how these effects are generated is unclear. Gibney et al. [47] demonstrated that phenotypes of the tps1 mutant could not be reversed by simply increasing intracellular concentration of trehalose. This meant that the phenotypes were not due to the depletion of intracellular trehalose concentration per se. It is possible that the enzymes of trehalose pathway exert a more complex metabolic effect on the physiology of the cell [47, 48].
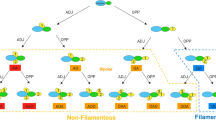
Our observations imply that components of the trehalose biosynthetic pathway may determine whether the cell goes into pseudohyphal differentiation or sporulation in response to nutritional stress by regulation of cAMP and thereby downstream signaling. This is in accordance with an earlier observation that intracellular concentration of trehalose correlated with pseudohyphae formation [4]. Based on our results, we propose a model (Fig. 5) wherein, the trehalose biosynthetic pathway regulates pseudohyphal differentiation in multiple ways. We propose that TPS1 functions in two ways. On one hand it acts by activation of RAS2. On the other hand, it positively regulates cAMP in a RAS2 independent manner. This is supported by our observation that the tps1 mutant is unable to form pseudohyphae in SLALD medium (Fig. 1c) although expression of RAS2 is not reduced in SLALD medium in this strain (Fig. 4). This RAS2 independent effect of TPS1 could be mediated via GPR1 or through a hitherto unidentified mechanism.

Schematic illustration of signaling and possible metabolic effect mediated by the trehalose biosynthetic pathway components. Extracellular glucose availability determines regulation of RAS2 by TPS1 and TPS2 which in turn regulates cAMP effects. In addition, TPS1 exerts an independent effect probably mediated through GPR1 while TPS2 possibly alleviates glucose repression by directing the metabolic flux from Tre6P to trehalose. Bold lines represent interactions based on our observations, dotted lines represent proposed interactions, compound dotted lines represent proposed glucose mediated signaling via TPS2. Key: → Activation ⊣ Inhibition
TPS2 however, appears to play a predominant and more complicated role. While our observations suggest that TPS2 is not involved in pseudohyphae formation, mutation in tps2 overcomes the filamentation defect of tps1 mutant in SLALD (Fig. 1c, bottom right), that of gpr1 mutant in SLAD (Fig. 2a, bottom left) and that of ras2 mutant in SLAD as well as SLALD media (Fig. 3b). According to our model, it is likely that TPS2 exerts a regulatory effect by suppressing RAS2 in addition to exerting a metabolic or regulatory effect mediated through Tre6P. It has been reported that Tre6P promotes glucose repression by suppressing genes required for gluconeogenesis [41]. We propose that this is the basis for the filamentation response of the tps1tps2 mutant (Fig. 1c, compare bottom left and right panels). In this double mutant, it is possible that Tre6P accumulates because of tps2 mutation and prevents pseudohyphae formation in SLAD medium. However, in SLALD medium, the glucose concentration is below that required for glucose repression and the tps1tps2 double mutant is able to overcome the filamentation defect by virtue of the general effect of alleviation of glucose repression. Our observation that filamentation defect of the gpr1 mutant is overcome upon tps2 mutation (Fig. 2a, compare top and bottom of left panel) supports the idea that Tre6P exerts a positive regulatory effect on pseudohyphae formation. However, this argument is counter-intuitive as Tre6P is known to promote glucose repression which is a condition that inhibits filamentation and probably not likely to occur. Thus, the role of Tre6P if any, is not clear. The positive effect of tps2 mutation in the background of ras2 mutations is possibly due to fall in trehalose levels. This is based on our earlier observation that lower trehalose level in the cell correlates with pseudohyphae formation [4]. Thus, our data suggests that both Tps1p and Tps2p could be bi-functional proteins exerting both a regulatory as well as a metabolic effect.
Based on the results of western blot, we further hypothesize that TPS1 and TPS2 coordinate to regulate the expression of Ras2p based on glucose availability (Fig. 5) and thereby affect the downstream concentration of cAMP, to trigger filamentation. That glucose is a key nutrient in this signaling is strengthened by our observations on SLALD medium supplemented with cAMP (Fig. 3a, middle panel), where exogenous cAMP addition is able to rescue filamentation in SLAD but not SLALD medium. This data suggests that signaling from glucose is essential in addition to that from cAMP in the absence of RAS2, to trigger pseudohyphae formation. It is possible that this signal is transmitted through TPS1 either via GPR1-GPA2 axis or directly via glucose. All things considered, our data clearly implies that RAS2 signaling in the context of pseudohyphal differentiation needs further evaluation. This idea is supported by the existence of conflicting reports on the filamentation response of the ras2 mutant [49,50,51]. Monitoring the effect of perturbations in ammonium and glucose sensing pathways on RAS2 expression in the tps mutants could shed some light on the possible mechanism mediated through the trehalose biosynthetic pathway.
To summarize, the results of this study shed light on the metabolic basis of cellular differentiation effected via TPS1/2. However, it is still unclear whether the observed effects are mediated through trehalose, the metabolic intermediates or through the TPS1/2 encoded proteins per se or a combination thereof. One way of addressing this would be to isolate missense mutations in TPS1 that specifically knock off the enzyme function without affecting pseudohyphae formation. Understanding the mechanistic basis of this metabolic effect is fundamental for elucidating the mechanism of fungal virulence. The wider and more significant implication of the study is in understanding the metabolic basis of differentiation in response to nutrient limitation as the nutrient dependent TOR, SNF1 and PKA mediated signalling pathways are highly conserved amongst eukaryotes.
References
Gagiano M, Bauer FF, Pretorius IS (2002) The sensing of nutritional status and the relationship to filamentous growth in Saccharomyces cerevisiae. FEMS Yeast Res 2:433–470. https://doi.org/10.1016/S1567-1356(02)00133-2
Gancedo JM (2008) The early steps of glucose signalling in yeast. FEMS Microbiol Rev 32:673–704
Gimeno CJ, Styles CA, Fink GR (1992) Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: regulation by starvation and RAS. Cell 68:1077–1090
Iyer RS, Das M, Bhat PJ (2008) Pseudohyphal differentiation defect due to mutations in GPCR and ammonium signaling is suppressed by low glucose concentration: a possible integrated role for carbon and nitrogen limitation. Curr Gen 54:71–81. https://doi.org/10.1007/s00294-008-0202-1
Lengeler KB, Davidson RC, Souza CD, Harashima T, Shen W, Wang P, Pan X, Waug M, Heitman J (2000) Signal transduction cascades regulating fungal development and virulence. MMBR 64:746–785
Madhani HD (2000) Interplay of intrinsic and extrinsic signals in yeast differentiation. PNAS 97:13461–13463
Cullen PJ, Sprague GF (2012) The regulation of filamentous growth in yeast. Genetics 190:23–49. https://doi.org/10.1534/genetics.111.127456
Honigberg SM, Purnapatre K (2003) Signal pathway integration in the switch from the mitotic cell cycle to meiosis in yeast. J Cell Sci 116:2137–2147. https://doi.org/10.1242/jcs.00460
Jungbluth M, Taxis C (2012) Acetate regulation of spore formation is under the control of the Ras/Cyclic AMP/protein kinase A pathway and carbon dioxide in Saccharomyces cerevisiae. Eukaryot Cell 11:1021–1032. https://doi.org/10.1128/EC.05240-11
Weidberg H, Moretto F, Spedale G, Amon A, van Werven FJ (2016) Nutrient control of yeast gametogenesis is mediated by TORC1, PKA and energy availability. PLoS Genet 12:1–26. https://doi.org/10.1371/journal.pgen.1006075
van De Velde S, Thevelein JM (2008) Cyclic AMP-protein kinase A and Snf1 signaling mechanisms underlie the superior potency of sucrose for induction of filamentation in Saccharomyces cerevisiae. Eukaryot Cell 7:286–293. https://doi.org/10.1128/EC.00276-07
Lorenz MC, Heitman J (1998) The MEP2 ammonium permease regulates pseudohyphal differentiation in Saccharomyces cerevisiae. EMBO J 17:1236–1247. https://doi.org/10.1093/emboj/17.5.1236
Rutherford JC, Chua G, Hughes T, Cardenas ME, Heitman J (2008) A Mep2-dependent transcriptional pofile links permease function to gene expression during pesudohyphal growth in Saccharomyces cerevisiae. Mol Biol Cell 19:3028–3039
Boeckstaens M, André B, Marini AM (2007) The yeast ammonium transport protein Mep2 and its positive regulator, the Npr1 kinase, play an important role in normal and pseudohyphal growth on various nitrogen media through retrieval of excreted ammonium. Mol Microbiol 64:534–546
Boeckstaens M, Llinares E, Van Vooren P, Marini AM (2014) The TORC1 effector kinase Npr1 fine tunes the inherent activity of the Mep2 ammonium transport protein. Nat Comm 5:3101. https://doi.org/10.1038/ncomms4101
Xue Y, Hirsc JP (1998) GPR1 encodes a putative G protein-coupled receptor that associates with the Gpa2p G α subunit and functions in a Ras-independent pathway. EMBO J 17:1996–2007
Santangelo GM (2006) GLucose signaling in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 70:253–282
Harashima T, Heitman J (2005) Gα subunit Gpa2 recruits kelch repeat subunits that inhibit receptor-G protein coupling during cAMP induced dimorphic transitions in Saccharomyces cerevisiae. Mol Biol Cell 16:4557–4571
Iyer RS, Bhat PJ (2017) KRH1 and KRH2 are functionally non-redundant in signaling for pseudohyphal differentiation in Saccharomyces cerevisiae. Curr Gen 63:851–859. https://doi.org/10.1007/s00294-017-0684-9
Rupp S, Summers E, Lo HJ, Madhani H, Fink G (1999) MAP kinase and cAMP filamentation signaling pathways converge on the unusually large promoter of the yeast FLO11 gene. EMBO J 18:1257–1269
Kuchin S, Vyas VK, Carlson M (2002) Snf1 protein kinase and the repressors Nrg1 and Nrg2 regulate FLO11, haploid invasive growth, and diploid pseudohyphal differentiation. Mol Cell Biol 22:3994–4000
Carlson M, Osmond BC, Neigeborn L, Botstein D (1984) A suppressor of SNF1 mutations causes constitutive high-level invertase synthesis in yeast. Genetics 107:19–32
Kuchin S, Vyas VK, Carlson M (2003) Role of the yeast Snf1 protein kinase in invasive growth. Biochem Soc Trans 31:175–177. https://doi.org/10.1042/bst0310175
Apweiler E, Sameith K, Margaritis T, Brabers N, van de Pasch L, Bakker LV, van Leenen D, Holstege FCP, Kemmeren P (2012) Yeast glucose pathways converge on the transcriptional regulation of trehalose biosynthesis. BMC Genomics 13:239
De Silva-Udawatta MN, Cannon JF (2001) Roles of trehalose phosphate synthase in yeast glycogen metabolism and sporulation. Mol Microbiol 40:1345–1356. https://doi.org/10.1046/j.1365-2958.2001.02477.x
Thevelein JM, Hohmann S (1995) Trehalose synthase: guard to the gate of glycolysis in yeast? TIBS 20:3–9
Zaragoza O, Blazquez MA, Gancedo C (1998) Disruption of the Candida albicans TPS1 gene encoding trehalose-6-phosphate synthase impairs formation of hyphae and decreases infectivity. J Bact 180:3809–3815
van Dijck P, De Rop L, Szlufcik K, Van Ael E, Thevelein JM (2002) Disruption of the Candida albicans TPS2 gene encoding trehalose-6-phosphate phosphatase decreases infectivity without affecting hypha formation. Infect Immun 70:1772–1782. https://doi.org/10.1128/IAI.70.4.1772-1782.2002
Maidan MM, De Rop L, Relloso M, Diez-Orejas R, Thevelein JM, Van Dijck P (2008) Combined inactivation of the Candida albicans GPR1 and TPS2 genes results in avirulence in a mouse model for systemic infection. Infect Immun 76:1686–1694. https://doi.org/10.1128/IAI.01497-07
Chary SN, Hicks GR, Yoon GC, Carter D, Raikhel NV (2008) Trehalose-6-phosphate synthase/phosphatase regulates cell shape and plant architecture in arabidopsis. Plant Physiol 146:97–107. https://doi.org/10.1104/pp.107.107441
Adams A, Gottschling DE, Kaiser CA, Stearns T (1997) Methods in yeast genetics cold spring harbor. Cold Spring Harbor Laboratory, New York, pp 1–157
Wach A, Brachat A, Pohlmann R, Philippsen P (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10:1793–1808
Lorenz MC, Heitman J (1997) Yeast pseudohyphal growth is regulated by GPA2, a G protein alpha homolog. EMBO J 16:7008–7018. https://doi.org/10.1093/emboj/16.23.7008
Peeters K, Van Leemputte F, Fischer B, Bonini BM, Quezada H, Tsytlonok M, Haesen D, Vanthienen W, Bernardes N, Gonzalez-Blas CB, Janssens V, Tompa P, Versées W, Thevelein JM (2017) Fructose-1,6-bisphosphate couples glycolytic flux to activation of Ras. Nat Commun 8:922. https://doi.org/10.1038/s41467-017-01019-z
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675
van Heerden JH, Wortel MT, Bruggeman FJ, Heijnen JJ, Bollen YJM, Planqué R, Hulshof J, O’Toole TG, Wahl SA, Teusink B (2014) Lost in transition: start-up of glycolysis yields subpopulations of nongrowing cells. Science 343:1245114. https://doi.org/10.1126/science.1245114
Suzuki C, Hori Y, Kashiwagi Y (2003) Screening and characterization of transposon-insertion mutants in a pseudohyphal strain of Saccharomyces cerevisiae. Yeast 20:407–415
Eleutherio E, Panek A, De Mesquita JF, Trevisol E, Magalhães R (2015) Revisiting yeast trehalose metabolism. Curr Gen 61:263–274. https://doi.org/10.1007/s00294-014-0450-1
Lillie SH, Pringle JR (1980) Reserve carbohydrate metabolism in Saccharomyces cerevisiae: responses to nutrient limitation. J Bact 143:1384–1394
Serneels J, Tournu H, DijckP V (2012) Tight control of trehalose content is required for efficient heat-induced cell e.longation in Candida albicans. J Biol Chem 287:36873–36882
Vicente RL, Spina L, Gómez JPL, Dejean S, Parrou JL, François JM (2018) Trehalose-6-phosphate promotes fermentation and glucose repression in Saccharomyces cerevisiae. Microb Cell 5:444–459. https://doi.org/10.15698/mic2018.10.651
Wilson RA, Jenkinson JM, Gibson RP, Littlechild JA, Wang ZY, Talbot NJ (2007) Tps1 regulates the pentose phosphate pathway, nitrogen metabolism and fungal virulence. EMBO J 26:3673–3685
Satoh-Nagasawa N, Nagasawa N, Malcomber S, Sakai H, Jackson D (2006) A trehalose metabolic enzyme controls inflorescence architecture in maize. Nature 11:227–230
Bell W, Klaassen P, Ohnacker M, Boller T, Herweijer M, Schoppink P, Van der zee P, Wiemken A (1992) Characterization of the 56-kDa subunit of yeast trehalose-6-phosphate synthase and cloning of its gene reveal its identity with the product of CIF1, a regulator of carbon catabolite inactivation. Eur J Biochem 209:951–959
Deroover S, Ghillebert R, Broeckx T, Winderickx J, Rolland F (2016) Trehalose-6-phosphate synthesis controls yeast gluconeogenesis downstream and independent of SNF1. FEMS Yeast Res 16:1–15. https://doi.org/10.1093/femsyr/fow036
Se Virgilio C, Burckert N, Bell W, Jeno P, Boller T, Wiemken A (1993) Disruption of TPS2, the gene encoding the 100-kDa subunit of the trehalose-6-phosphate synthase/phosphatase complex in Saccharomyces cerevisiae, causes accumulation of trehalose-6-phosphate and loss of trehalose-6-phosphate phosphatase activity. Eur J Biochem 212:315–323
Gibney PA, Schieler A, Chen JC, Rabinowitz JD, Botstein D (2015) Characterizing the in vivo role of trehalosein Saccharomyces cerevisiae using the AGT1 transporter. PNAS 112:6116–6121
Tomova AA, Kujumdzieva AV, Petrova VY (2019) Carbon source influences Saccharomyces cerevisiae yeast cell survival strategies: quiescence or sporulation. Biotechnol Biotechnol Equip 33:1464–1470. https://doi.org/10.1080/13102818.2019.1674188
Kubler E, Mosch HU, Rupp S, Lisanti MP (1997) Gpa2p, a G-protein α-subunit, regulates growth and pseudohyphal development in Saccharomyces cerevisiae via a cAMP-dependent mechanism. J Biol Chem 272:20321–20323
Mosch HU, Kubler E, Krappman S, Fink GR, Braus GH (1999) Crosstalk between the Ras2p-controlled mitogen activated protein kinase and cAMP pathways during invasive growth of Saccharomyces cerevisiae. Mol Biol Cell 10:1325–1335
Ryan O, Shapiro RS, Kurat CF et al (2012) Global gene deletion analysis exploring yeast filamentous growth. Science 337:1353–1356
Lorenz MC, Pan X, Harashima T, Cardenas ME, Xue Y, Hirsch JP, Heitman J (2000) The G protein-coupled receptor Gpr1 is a nutrient sensor that regulates pseudohyphal differentiation in Saccharomyces cerevisiae. Genetics 154:609–622
Acknowledgements
This work was supported by funding awarded to Dr. Revathi Iyer, by the Department of Science and Technology, India, under the DST WOS-A scheme (SR/WOS-A/LS-58/2017). We thank Prof. R. Patkar, for graciously permitting the use of his laboratory facilities to study the effect of cAMP on the the ras2 mutant.
Funding
This work was supported by funding awarded to Dr. Revathi Iyer, by the Department of Science and Technology, India, under the DST WOS-A scheme (Grant No. SR/WOS-A/LS-58/2017).
Author information
Authors and Affiliations
Contributions
Both authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Dr. RI. The first draft of the manuscript was written by Dr. RI. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Iyer, R., Bhat, P.J. Trehalose biosynthetic pathway regulates filamentation response in Saccharomyces cerevisiae. Mol Biol Rep 49, 9387–9396 (2022). https://doi.org/10.1007/s11033-022-07792-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07792-5