
Abstract
Liver fibrosis affects over 100 million people in the world; it represents a multifactorial, fibro-inflammatory disorder characterized by exacerbated production of extracellular matrix with consequent aberration of hepatic tissue. The aetiology of this disease is very complex and seems to involve a broad spectrum of factors including the lifestyle, environment factors, genes and epigenetic changes. More evidences indicate that angiogenesis, a process consisting in the formation of new blood vessels from pre-existing vessels, plays a crucial role in the progression of liver fibrosis. Central to the pathogenesis of liver fibrosis is the hepatic stellate cells (HSCs) which represent a crossroad among inflammation, fibrosis and angiogenesis. Quiescent HSCs can be stimulated by a host of growth factors, pro-inflammatory mediators produced by damaged resident liver cell types, as well as by hypoxia, contributing to neoangiogenesis, which in turn can be a bridge between acute and chronic inflammation. As matter of fact, studies demonstrated that neutralization of vascular endothelial growth factor as well as other proangiogenic agents can attenuate the progression of liver fibrosis. With this review, our intent is to discuss the cause and the role of angiogenesis in liver fibrosis focusing on the current knowledge about the impact of anti-angiogenetic therapies in this pathology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genetic, environmental and lifestyle factors (e.g. alcohol abuse), mechanotransduction signal pathway and viral infections can contribute in onset and progression of liver fibrosis (LF) [1,2,3,4,5,6]. Histologically, this disorder can be classified as a chronic fibro-inflammatory condition characterized by an excessive deposition of extracellular matrix (ECM) proteins including collagen fibers (I, III, and IV) [7,8,9]. Clinically, portal hypertension can be a key feature in patients suffering from severe form of LF [10, 11]. Evidence from a number of studies demonstrates that angiogenesis, the formation of new blood vessels from pre-existing vasculature, plays a crucial role in the progression of this complex disease [12,13,14,15]. It is well known that inflammation and hypoxia are two elements that strongly promote neovascularization [16,17,18]. Interestingly, both phenomena can be considered as markers of LF [19, 20]; thus, it is reasonable that angiogenesis takes place during hepatic fibrogenesis [12, 13]. Consequently, it is conceivable that anti-angiogenic approaches could represent a useful tool in the treatment of LF. The present review will describe the general aspects of the pathogenesis of LF, focusing on the link between hepatic fibrogenesis and angiogenesis. Meanwhile, selective strategies targeting angiogenesis for the preservation of the hepatic tissue will be introduced.
The pathogenesis of liver fibrosis
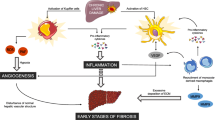
As aforementioned, histologically, fibrotic hepatic parenchyma is characterised by chronic inflammation and exacerbated production of ECM molecules with consequent abnormality in the liver tissue [7, 8]. The inflammatory foci are comprised of lymphocytes, plasma cells, monocytes/macrophages (LY6Chi phenotype) as well as granulocytes [21, 22]. All these inflammatory components indirectly participate in the process of fibrogenesis by producing soluble/paracrine signals including cytokines, chemotactic molecules, fibrogenic agents [23, 24]. Also, in the chronic hepatic injuries, cholangiocytes, hepatocytes, liver sinusoidal endothelial cells (LSECs) and non-sinusoidal endothelial cells (ECs), together with resident Kupffer cells secrete various sclerotic stimuli such as transforming growth factor- β (TGF-β, the “master mediator” of many fibrotic disorders), platelet-derived growth factor (PDGF), and epidermal growth factor (EGF) [25]. Figure 1 depicts the many cell types and molecular effectors involved in LF, leading to the activation of HSCs. Generally, the primary effectors of fibrogenesis are resident fibroblasts, myofibroblasts [26, 27], and their bone marrow-derived circulating precursors namely fibrocytes [28, 29]. In damaged liver, activated hepatic stellate cells (HSCs) are mainly responsible of fibrogenesis in at least 2 ways: on one hand they produce ECM, on the other hand impede ECM degradation by secreting proteases inhibitors including endogenous tissue inhibitors of metalloproteinases (TIMPs) [30, 31]. There is a lot of evidence showing that epithelial-mesenchymal-transition (EMT) has also a great importance in fibrotic lesions. Accordingly, hepatocytes as well as ECs/LSECs can undergo a process of epithelial-(endothelial)-mesenchymal transition (EMT) through autocrine/paracrine signals mediated, in part, by TGF-β [32]. Additionally, recent work discovered that pericytes, are also involved in the process of fibrogenesis [33,34,35]. In fact, these mural cells seem to have the capability to detach from basement membrane surrounding hepatic capillary to accumulate within the injured hepatic tissues, where they undergo phenotypic transformation into ECM-producing myofibroblasts [33,34,35]. As a consequence, a wide range of cells, growth factors and other stimuli are engaged in the liver fibrogenesis [33,34,35].

During liver injury, quiescent HSCs or other precursors (e.g. bone marrow-derived fibrocytes, portal fibroblasts, hepatocytes in the epithelial-mesenchymal transition) are activated by various cell types resident in the liver, including hepatocytes, cholangiocytes, sinusoidal and non-sinusoidal endothelial cells, pericytes, macrophages LY6Chi, Kupffer cells, as well as Th17 T cells and other lymphoid cells. All these cell types secrete pro-fibrogenic mediators that ultimately activate HSCs or other precursors that eventually transform into myofibroblasts and operate to deposit ECM. CCL CC chemokine ligands, DAMP danger associated molecular pattern, IL interleukin, NO nitric oxide, PDGF platelet-derived growth factor, ROS reactive oxygen species, TGF-β transforming growth factor-β, TNF-α tumor necrosis factor-α
The link between angiogenesis and liver fibrosis
Angiogenesis is a growth factor-dependent phenomenon taking place during all stage of the human development; during adult life, at least in healthy conditions, it happens only in certain circumstances, for example during pregnancy and menstrual cycle [36, 37]. By contrast, experimental and clinical evidences indicate that angiogenesis accelerates the progression of many disorders such as cancer growth and metastasis, rheumatoid arthritis, diabetic retinopathy and other complex diseases including LF [12, 38,39,40,41,42]. It is well known that inflammation and hypoxia are crucial elements in induction of neovascularization. As previously specified, hepatic tissue affected by fibrosis, shows permanent inflammation and low oxygen level, offering a prototypical microenvironment for neovascularization [43, 44]. In detail, accumulation of ECM in liver parenchyma is a main cause of hypoxia, which in turn, stabilizes the dimeric transcription factor “hypoxia-inducible factor” (HIF) [45]. HIF regulates the transcription of an array of genes including those controlling angiogenesis such VEGF, PDGF-B, matrix metalloproteinases (MMPs) as well as TIMPs [46,47,48,49]. As matter of fact, hypoxic areas co-localize with those of an increased microvessel density (MVD), fibrous septa and inflammatory foci [48, 50, 51]. In addition, hypoxia further stimulates the infiltration of inflammatory cells [52], which, in turn, contribute to angiogenesis and fibrotic phenomena [53]. In conclusion, in injured liver, hypoxia, angiogenesis, chronic inflammation and fibrosis drive each other following an activated loop, and synergistically exacerbate the severity of the LF (Fig. 2) [15, 16, 54].

Link among hypoxia, angiogenesis, inflammation and liver fibrosis. Hypoxia, angiogenesis, inflammation and fibrosis drive each other activating a pathological loop in liver
To incite neoangiogenesis, VEGF binds to its receptors VEGFRs stimulating the formation of new functional vessels (Fig. 3). By the signals generated by bound VEGFRs, VEGF is the leading regulator of ECs/LSECs activity during all steps of angiogenesis [55]. In LF, VEGF is, in part, produced by ECs/LSECs themselves suggesting an autocrine action of this signal pathway; but damaged hepatocytes and activated HSCs seem to be the principal sources of this relevant growth factor [56]. The latter evidence highlights the crucial role of HSCs in LF because they constitute a crossroad among inflammation, fibrosis and angiogenesis (Fig. 4) [12, 16, 57, 58]. PDGF-B, principally produced by ECs/LSECs, acts during vessel stabilization, orchestrating the formation/maturation of vascular tube and its coverage through the recruitment of PDGFRs positive pericytes/HSCs [57]. FGF, through an autocrine loop, is involved in LF angiogenesis not only inducing the activation of ECs/LSECs, but also, increasing HSC proliferation and recruitment [12]. Ang-2 is acutely released from activated ECs/LSECs, upon stimulation with inflammatory cytokines, proangiogenic factors, and hypoxia and competitively inhibits the binding of Ang-1 to Tie-2 [59] that, instead, serves to maintain survival and quiescence of endothelium [60]. Inflammatory cells also secrete a plethora of angiogenic factors (VEGF, PIGF, PDGF, FGF, Angiopoietins, TGF-β, etc.) [12, 61,62,63,64,65,66,67]. For example, both infiltrating macrophages and resident Kupffer cells, once activated, contribute to angiogenesis releasing reactive oxygen species (ROS), nitric oxide (NO), tumor necrosis factor- α (TNF-α) and other angiocrine molecules [12]. As above cited, in drastic circumstances, LF is complicated by portal hypertension (PHT) accompanied by severe hepatic structural disorder correlated to diffuse fibrosis [68]. Angiogenesis also participates in the pathogenesis of PHT, in part modulating HSCs activation and, on the other hand, provoking the formation of portal-veins collaterals [69]. Cellular molecules involved in promoting angiogenesis and their roles in LF are listed in Table 1 [57, 70,71,72,73,74,75].

The VEGF/VEGFR signalling axis, its contribution to angiogenesis and treatment modalities interfering with its activity. Binding of VEGF ligands to their cognate receptors (VEGFRs) leads to receptor dimerization and autophosphorylation triggering a down-stream intracellular phosphorylation cascade. Monoclonal antibodies target VEGFs, preventing its binding to VEGFRs, while monoclonal antibodies targeting VEGFRs prevent the binding of VEGFs, resulting in the inhibition of VEGFR signalling. The treatment of receptor tyrosine kinase inhibitors (RTK-Is) inhibits the activation of VEGF/VEGFR signalling

Schematic model of HSCs activation. Quiescent HSCs are activated, during lung injury, by a host of factors, including hypoxia, inflammatory stimuli and growth factors produced by liver cells, such as hepatocytes and endothelial cells. HSCs transform into myofibroblasts and contribute to angiogenesis, fibrosis and inflammation. Once activated, HSCs act as proangiogenic cells and may respond to stimuli such as hypoxia through the increase of VEGF, Ang-1, and their related receptors VEGFR-2 and Tie-2. Activated HSCs are the prime downstream effectors of excess ECM deposition and they also produce the fibrogenic cytokine TGF-β. Moreover, fibrolysis is compromised, e.g. by an increased synthesis of TIMPs and a decreased production of fibrolytic MMPs. Finally, activated HSCs contribute to inflammation in liver fibrosis by producing chemokines, including CC chemokines ligands (CCL2, CCL3, CCL5) and the CXC chemokines ligands (CXCL8, CXCl9, CXCL10, CXCL12). MCP-1 monocyte chemoattractant protein-1
Anti-angiogenesis approaches slow down LF
Considering the indisputable importance of neovascularization in LF progression, it is plausible that blocking angiogenesis may offer a method to attenuate the aberration of hepatic tissue or prevent more serious damage including cirrhosis [14, 76]. For this reason, some anti-angiogenic strategies including natural compounds are currently under investigation [77]. Since VEGF is the most efficient pro-angiogenic factor, generally most anti-angiogenic therapies have been focused on blocking the VEGF signal pathway [76, 78] (Fig. 3). Bevacizumab, a humanized monoclonal antibody neutralizing VEGF-A [72, 79, 80], in combination to other drugs, is currently used to treat different kinds of tumors [74, 81, 82]. It also shows a strong anti-fibrotic effect in human Tenon's fibrosis [83]. High VEGF-A levels in the aqueous humor of patients with nonneovascular glaucoma have been reported [84], and this increase may contribute to post-operative inflammation and fibrosis. Since Tenon’s fibroblasts have been shown to express VEGF-A receptors [84], these findings highlight once more the intimate relationship between angiogenesis and fibrosis, as it occurs in LF. An interesting study conducted by Huang et al. [85], showed that bevacizumab alleviates LF in vivo by neutralizing VEGF produced by hepatocytes and by blocking HSCs activation. The effects of VEGFRs neutralizing antibodies such as anti-VEGFR1 and anti-VEGFR2 have also been explored [86, 87] (Fig. 3). Results show that the use of anti-VEGFR-2 antibody results more effective than the anti-VEGFR-1 antibody when used alone [87], although combined treatment with both antibodies gave some tissue improvement in LF [87]. Additionally, the use of the multiple receptor tyrosine kinase inhibitors such as sorafenib and sunitinib blocking PDGFR-β and VEGFRs signaling pathways is under investigation [88] (Fig. 3). Sorafenib attenuates LF by reducing HSCs proliferation/activation and inducing their apoptosis both in vitro and in vivo [88, 89]. Sunitinib also decreases LF, switching off inflammation, HSCs activation and angiogenesis [90, 91]. PDGF-B and its signaling pathway and cyclooxygenase-2 are also involved in HSCs activation [92, 93]. Gao at al demonstrated that the use of celecoxib (a cyclooxygenase-2 inhibitor) shows similar effect in vivo as those obtained with sorafenib [94]. It is implicit that enhancing the expression and the activation of the ECM proteases can also contribute to the resolution of fibrosis [95, 96]. In line with this idea, it has been shown that the decrease of LF is associated with increased expression of MMPs (MMP-2 and -14) as well as decreased expression of TIMP-1 and -2 in hepatic tissue [30, 97].
Along with the use of immunotarget therapy above listed, recently, the regenerative potential of stem cells is being exploited in fibrotic diseases. Accordingly, the injection of bone marrow-derived mesenchymal stem cells (BMSCs) including endothelial progenitor’s cells (EPCs) seems to reduce the severity of LF by increasing the degradation of ECM by means of proteases/MMPs [98, 99]. In fact, experimental evidences showed that EPCs transplantation was shown to effectively promote the remodelling of damaged liver tissues in a dimethylnitrosamine (DMN) rat liver fibrosis model [100, 101].
Other approaches to slow down LF
Additionally, studies have demonstrated that LF may be prevented or reversed by bioactive food components and natural products, including fumagillin analogue (TNP-470), astaxanthin, curcumin, blueberry, silymarin, vitamins (C, D, E), resveratrol, quercetin, coffee and green tea extracts [102,103,104]. Generally, the anti-fibrotic effect of all these natural compounds seems to be mainly attributed not only to their antioxidant and anti-inflammatory features but also to their ability to revert the activated forms of HSCs in a more quiescent phenotype [102].
Current challenges and future directions
Inflammation, fibrosis and angiogenesis are strictly intertwined during the progression of chronic liver diseases (CLDs), including chronic viral hepatitis, PTH, non-alcoholic and alcoholic liver diseases. This brings to the notion that a wealth of cellular and molecular mechanisms are implicated in liver fibrosis and angiogenesis. Interactions among hepatocytes, HSCs, Kupffer cells, and endothelial cells have been described, with HSCs representing a crossroad at the interaction between inflammation, angiogenesis, and fibrosis. Angiogenic factors, including VEGF, PDGF, FGF, Ang-2, EGF, and various cytokines, are important mediators of angiogenesis in fibrosis associated with CLDs. Besides these factors, metabolic abnormalities, including adipokines, may dysregulate angiogenesis, and hence influence inflammation and fibrosis. Moreover, it has also been shown that endoplasmic reticulum stress and related unfolded protein response, and neuropilins are involved in liver angiogenesis and fibrosis [12]. Given the plethora of cellular and molecular mechanisms, a better appraisal of this complexity may be caught by three-dimensional (3D) models that can recapitulate liver architecture and interactions among different cell types [105, 106]. Indeed, one obstacle in the development of efficient therapies is the lack of robust and representative in vitro models of human liver fibrosis through which novel drugs can be tested. Currently used animal models are not useful for dissecting the relative role of each component since the predictive value for human physiologic responses in terms of pharmacokinetics and pharmacodynamics is sometimes poor. Moreover, they are not suitable for large scale screening of antifibrotic compounds. The main 3D models that are being used and implemented include cocultures of hepatocytes and HSCs, achieved by insert cultures, spheroids (presenting many cell types), or liver tissue cultures. More advanced techniques are bioprinting and microfabricated microfluidic devices to provide a constant flow of oxygen and fresh nutrients and remove the metabolic waste generated (as replacement of bile canaliculi). Finally, organotypic models, such as precision cut liver slices and decellularised 3D scaffolds, will offer more opportunities to test novel drugs in a context maintaining the intact hepatic architecture and cellular heterogeneity. Thus far, the main focus of the field has been on the maintenance of functional hepatocytes for prolonged culture periods; incorporation of non-parenchymal cells (such as endothelial cells, Kupffer cells, and HSCs) will allow the use of these culture systems for in vitro fibrosis studies [105]. In order to gain higher number of cells and make sustainable these models, stem cells are a suitable source of different cell types. Many stem cell types, including liver progenitor/stem cells, extra-hepatic biliary tree stem cells, embryonic stem cells as well as induced pluripotent stem cells (iPSCs) have been reported to generate hepatocyte-like cells [107] and cholangiocytes [108, 109], and more recently LSECs and HSCs [110]. Further studies should determine whether these cell types are fully functional and can reconstitute organotypic models. Another essential feature of these models will be the inclusion of stiffer materials mimicking the deposition of collagen that is a feature of liver fibrosis. Depending on the hardness of the substrates used, i.e. soft versus stiff, the quiescent phenotype of HSCs will be maintained, or they will transform into activated myofibroblasts [5, 106]. Recently, a novel 3D organotypic liver models comprised of hepatocytes, LSECs, HSCs, Kupffer cells, and the Space of Disse mimic demonstrated how a mechanical gradient resulted in transitioning phenotypes in hepatic cells and cause varying profiles of fibrotic markers [111]. Thus, mechanotransduction and biomechanics are parameters that should be envisioned as essential in constructing these models. These advanced in vitro models have been used for testing drug induced liver injury, determined by alcohol or medications, in the developmental phase of pharmaceuticals [105, 112] or in the evaluation of drugs already in clinical trials [113]. In addition to inhibitors of angiogenesis, that could result in unspecific effects, genetic tools may target profibrotic and proangiogenetic genes with an unprecedented precision. Small interfering RNAs and antisense oligonucleotides have been vehicled by nanocarriers (lipoplexes and nanoparticles) that are preferentially engulfed by nonparenchymal cells, prominently HSCs and myofibroblasts. The target genes to be downregulated include TGFβ-1, TGFβ receptors, osteopontin, integrins, and chemokine receptors [58]. Complex 3D and organotypic models are also essential in finding novel noninvasive markers of angiogenesis in liver fibrosis. Histological follow-up does not have the power to reliably detect antifibrotic drug effects in the short term. Validated serum markers would measure the activity of angiogenesis and fibrogenesis and therefore enable the selection of patients likely to respond to antiangiogenic and antifibrotic therapies, and to detect responders to these therapies. Finally, these models could capture the inter-individual genetic and environmental variations, increasing the pace towards the personalised medicine approach [58], and will be paramount to design more precise and real-to-life clinical trials.
Relevant conclusion
Anti-angiogenic therapy for hepatic fibrosis resolution has received increasing attention in recent years. However, it is not possible to overlook the fact that the LF is a multifactorial disorder, and angiogenesis in only one of the phenomena that favours its genesis and progression. Moreover, the limited preclinical/clinical studies impede to know in detail any counterproductive effects of antiangiogenic therapies in this aberrant circumstance. Consequently, further large randomized studies need to be conducted before deducing that anti-angiogenic approaches can be used in the treatment of liver fibrosis.
References
Ullah R, Rauf N, Nabi G et al (2019) Role of nutrition in the pathogenesis and prevention of non-alcoholic fatty liver disease: recent updates. Int J Biol Sci 15(2):265–276
Pelusi S, Cespiati A, Rametta R et al (2019) Prevalence and risk factors of significant fibrosis in patients with nonalcoholic fatty liver without steatohepatitis. Clin Gastroenterol Hepatol 17:2310–2319. https://doi.org/10.1016/j.cgh.2019.01.027
Zhang CY, Yuan WG, He P et al (2016) Liver fibrosis and hepatic stellate cells: etiology, pathological hallmarks and therapeutic targets. World J Gastroenterol 22(48):10512–10522
Zhou WC, Zhang QB, Qiao L (2014) Pathogenesis of liver cirrhosis. World J Gastroenterol 20(23):7312–7324
Liu L, You Z, Yu H et al (2017) Mechanotransduction-modulated fibrotic microniches reveal the contribution of angiogenesis in liver fibrosis. Nat Mater 16(12):1252–1261
Leandro G, Mangia A, Hui J et al (2006) Relationship between steatosis, inflammation, and fibrosis in chronic hepatitis C: a meta-analysis of individual patient data. Gastroenterology 130(6):1636–1642
Bonnans C, Chou J, Werb Z (2014) Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 15(12):786–801
Arriazu E, Ruiz de Galarreta M, Cubero FJ et al (2014) Extracellular matrix and liver disease. Antioxid Redox Signal 21(7):1078–1097
Iredale JP, Thompson A, Henderson NC (2013) Extracellular matrix degradation in liver fibrosis: biochemistry and regulation. Biochim Biophys Acta 1832(7):876–883
Gjeorgjievski M, Cappell MS (2016) Portal hypertensive gastropathy: a systematic review of the pathophysiology, clinical presentation, natural history and therapy. World J Hepatol 8(4):231–262
Iwakiri Y (2014) Pathophysiology of portal hypertension. Clin Liver Dis 18(2):281–291
Elpek G (2015) Angiogenesis and liver fibrosis. World J Hepatol 7(3):377–391
Bocca C, Novo E, Miglietta A (2015) Angiogenesis and fibrogenesis in chronic liver diseases. Cell Mol Gastroenterol Hepatol 1(5):477–488
Thomas H (2018) Liver: delineating the role of angiogenesis in liver fibrosis. Nat Rev Gastroenterol Hepatol 15(1):6
Zhang F, Lu Y, Zheng S (2015) Update on implications and mechanisms of angiogenesis in liver fibrosis. Hepatol Res 45(2):162–178
Paternostro C, David E, Novo E (2010) Hypoxia, angiogenesis and liver fibrogenesis in the progression of chronic liver diseases. World J Gastroenterol 16(3):281–288
Krock BL, Skuli N, Simon MC (2011) Hypoxia-induced angiogenesis: good and evil. Genes Cancer 2(12):1117–1133
Granger DN, Senchenkova E (2010) Angiogenesis. Inflammation and the microcirculation. Morgan & Claypool Life Sciences, San Rafael
Higashi T, Friedman SL, Hoshida Y (2017) Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev 121:27–42
Horvat T, Landesmann B, Lostia A (2017) Adverse outcome pathway development from protein alkylation to liver fibrosis. Arch Toxicol 91(4):1523–1543
Koyama Y, Brenner DA (2017) Liver inflammation and fibrosis. J Clin Invest 127(1):55–64
Tacke F, Zimmermann HW (2014) Macrophage heterogeneity in liver injury and fibrosis. J Hepatol 60(5):1090–1096
Wynn TA (2008) Cellular and molecular mechanisms of fibrosis. J Pathol 214(2):199–210. https://doi.org/10.1002/path.2277
Jiang JX, Török NJ (2013) Liver injury and the activation of the hepatic myofibroblasts. Curr Pathobiol Rep 1(3):215–223
Marrone G, Shah VH, Gracia-Sancho J (2016) Sinusoidal communication in liver fibrosis and regeneration. J Hepatol 65(3):608–617
Iwaisako K, Brenner DA, Kisseleva T (2012) What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J Gastroenterol Hepatol 27(Suppl 2):65–68
Xu J, Liu X, Koyama Y, Wang P et al (2014) The types of hepatic myofibroblasts contributing to liver fibrosis of different etiologies. Front Pharmacol 5:167
Lee SJ, Kim KH, Park KK (2014) Mechanisms of fibrogenesis in liver cirrhosis: the molecular aspects of epithelial-mesenchymal transition. World J Hepatol 6(4):207–216
Fausther M, Lavoie EG, Dranoff JA (2013) Contribution of myofibroblasts of different origins to liver fibrosis. Curr Pathobiol Rep 1(3):225–230
Gandhi CR, Pinzani M (2015) Stellate cells in health and disease. Elsevier, London, pp 107–124
Anjum S (2017) Frontiers in stem cell and regenerative medicine research. Bentham Science Publishers, Sharjah, pp 113–116
Choi SS, Diehl AM (2009) Epithelial-to-mesenchymal transitions in the liver. Hepatology 50(6):2007–2013
Lee JS, Semela D, Iredale J et al (2007) Sinusoidal remodeling and angiogenesis: a new function for the liver-specific pericyte? Hepatology 45(3):817–825
Greenhalgh SN, Iredale JP, Henderson NC (2013) Origins of fibrosis: pericytes take centre stage. F1000Prime Rep 5:37
Geevarghese A, Herman IM (2014) Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Transl Res 163(4):296–306
Salajegheh A (2016) Introduction to angiogenesis in normal physiology, disease and malignancy. Angiogenesis in health, disease and malignancy. Springer, Cham, pp 1–9
Iruela-Arispe ML, Zovein A (2017) Fetal and neonatal physiology, 5th edn. Elsevier, Amsterdam, pp 85–89
Keith B, Simon MC (2015) Tumor angiogenesis. In: Mendelsohn J (ed) The molecular basis of cancer, 4th edn. Elsevier, Philadelphia, pp 257–268
Bielenberg DR, Zetter BR (2015) The contribution of angiogenesis to the process of metastasis. Cancer J 21(4):267–273
Pezzella F, Harris AL, Tavassoli M (2015) Blood vessels and cancer much more than just angiogenesis. Cell Death Discov 1:15064
Elshabrawy HA, Chen Z, Volin MV (2015) The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 18(4):433–448
Crawford TN, Alfaro DV, Kerrison JB et al (2009) Diabetic retinopathy and angiogenesis. Curr Diabetes Rev 5(1):8–13
Watnick RS (2012) The role of the tumor microenvironment in regulating angiogenesis. Cold Spring Harb Perspect Med 2(12):a006676
Landskron G, De la Fuente M, Thuwajit P et al (2014) Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014:149185. https://doi.org/10.1155/2014/149185
Ju C, Colgan SP, Eltzschig HK (2016) Hypoxia-inducible factors as molecular targets for liver diseases. J Mol Med (Berl) 94(6):613–627
LaGory EL, Giaccia AJ (2016) The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol 18(4):356–365
Hamik A, Wang B, Jain MK et al (2006) Transcriptional regulators of angiogenesis. Arterioscler Thromb Vasc Biol 26(9):1936–1947
Ramakrishnan S, Anand V, Roy S (2014) Vascular endothelial growth factor signaling in hypoxia and inflammation. J Neuroimmune Pharmacol 9(2):142–160
Shin DH, Dier U, Melendez JA et al (2015) Regulation of MMP-1 expression in response to hypoxia is dependent on the intracellular redox status of metastatic bladder cancer cells. Biochim Biophys Acta 1852(12):2593–2602
Fu Y, Peng H, Zhang X et al (2016) Assessment of fibrotic tissue and microvascular architecture by in-line phase-contrast imaging in a mouse model of liver fibrosis. Eur Radiol 26(9):2947–2955
Chouaib S, Messai Y, Couve S et al (2012) Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front Immunol 3:21
Nath B, Szabo G (2012) Hypoxia and hypoxia inducible factors: diverse roles in liver diseases. Hepatology 55(2):622–633
Seki E, Schwabe RF (2015) Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology 61(3):1066–1079
Coulon S, Heindryckx F, Geerts A et al (2011) Angiogenesis in chronic liver disease and its complications. Liver Int 31(2):146–162
Walter TJ, Cast AE, Huppert KA et al (2014) Epithelial VEGF signaling is required in the mouse liver for proper sinusoid endothelial cell identity and hepatocyte zonation in vivo. Am J Physiol Gastrointest Liver Physiol 306(10):849–862
Corpechot C, Barbu V, Wendum D et al (2002) Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology 35(5):1010–1021
Ying HZ, Chen Q, Zhang WY et al (2017) PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (review). Mol Med Rep 16(6):7879–7889
Schuppan D, Ashfaq-Khan M, Yang AT et al (2018) Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol 68–69:435–451
Hakanpaa L, Sipila T, Leppanen VM et al (2015) Endothelial destabilization by angiopoietin-2 via integrin β1 activation. Nat Commun 6:5962
Fukuhara S, Sako K, Noda K et al (2010) Angiopoietin-1/Tie2 receptor signaling in vascular quiescence and angiogenesis. Histol Histopathol 25(3):387–396
Kong X, Horiguchi N, Mori M et al (2012) Cytokines and STATs in liver fibrosis. Front Physiol 3:69
Dooley S, ten Dijke P (2012) TGF-β in progression of liver disease. Cell Tissue Res 347(1):245–256
Friedman SL (2008) Mechanisms of hepatic fibrogenesis. Gastroenterology 134(6):1655–1669
Lee UE, Friedman SL (2011) Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol 25(2):195–206
Elpek G (2014) Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: an update. World J Gastroenterol 20(23):7260–7276
Cong M, Iwaisako K, Jiang C et al (2012) Cell signals influencing hepatic fibrosis. Int J Hepatol 2012:158547. https://doi.org/10.1155/2012/158547
Kostallari E, Shah VH (2016) Angiocrine signaling in the hepatic sinusoids in health and disease. Am J Physiol Gastrointest Liver Physiol 311(2):246–251
Bandali MF, Mirakhur A, Lee EW et al (2017) Portal hypertension: Imaging of portosystemic collateral pathways and associated image-guided therapy. World J Gastroenterol 23(10):1735–1746
Fernandez M (2015) Molecular pathophysiology of portal hypertension. Hepatology 61(4):1406–1415
Novo E, Cannito S, Zamara E et al (2007) Proangiogenic cytokines as hypoxia-dependent factors stimulating migration of human hepatic stellate cells. Am J Pathol 170(6):1942–1953
Dewidar B, Meyer C, Dooley S et al (2019) TGF-β in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells 8(11):1419
Schumacher JD, Guo GJ (2016) Regulation of hepatic stellate cells and fibrogenesis by fibroblast growth factors. Biomed Res Int 2016:8323747. https://doi.org/10.1155/2016/8323747
Poisson J, Lemoinne S, Boulanger C et al (2017) Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol 66(1):212–227
Yang C, Zeisberg M, Mosterman B (2003) Liver fibrosis: insights into migration of hepatic stellate cells in response to extracellular matrix and growth factors . Gastroenterology 124(1):147–159
Reeves HL, Scott L, Friedman SL (2002) Activation of hepatic stellate cells–a key issue in liver fibrosis. Front Biosci 7:d808-826
Ding Q, Tian XG, Li Y et al (2015) Carvedilol may attenuate liver cirrhosis by inhibiting angiogenesis through the VEGF-Src-ERK signaling pathway. World J Gastroenterol 21(32):9566–9576
Domitrović R, Potočnjak I (2016) A comprehensive overview of hepatoprotective natural compounds: mechanism of action and clinical perspectives. Arch Toxicol 90(1):39–79
Park S, Kim JW, Kim JH et al (2015) Differential roles of angiogenesis in the induction of fibrogenesis and the resolution of fibrosis in liver. Biol Pharm Bull 38(7):980–985
Wang Y, Fei D, Vanderlaan M (2004) Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis 7(4):335–345
Ferrara N, Hillan KJ, Novotny W (2005) Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun 333(2):328–335
Kong DH, Kim MR, Jang JH et al (2017) A review of anti-angiogenic targets for monoclonal antibody cancer therapy. Int J Mol Sci 18(8):1786
Ilic I, Jankovic S, Ilic M (2016) Bevacizumab combined with chemotherapy improves survival for patients with metastatic colorectal cancer: evidence from meta analysis. PLoS One 11(8):e0161912
O’Neill EC, Qin Q, Van Bergen NJ et al (2010) Antifibrotic activity of bevacizumab on human Tenon’s fibroblasts in vitro. Invest Ophthalmol Vis Sci 51(12):6524–6532
Li Z, Van Bergen T, Van de Veire S et al (2009) Inhibition of vascular endothelial growth factor reduces scar formation after glaucoma filtration surgery. Invest Ophthalmol Vis Sci 50:5217–5225
Huang Y, Feng H, Kan T et al (2013) Bevacizumab attenuates hepatic fibrosis in rats by inhibiting activation of hepatic stellate cells. PLoS ONE 8(8):e73492
Falcon BL, Chintharlapalli S, Uhlik MT et al (2016) Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharmacol Ther 164:204–225
Yoshiji H, Kuriyama S, Yoshii J et al (2003) Vascular endothelial growth factor and receptor interaction is a prerequisite for murine hepatic fibrogenesis. Gut 52(9):1347–1354
Qu K, Huang Z, Lin T et al (2016) New insight into the anti-liver fibrosis effect of multitargeted tyrosine kinase inhibitors: from molecular target to clinical trials. Front Pharmacol 6:300
Hao H, Zhang D, Shi J et al (2016) Sorafenib induces autophagic cell death and apoptosis in hepatic stellate cell through the JNK and Akt signaling pathways. Anticancer Drugs 27(3):192–203
Wang Y, Gao J, Zhang D et al (2010) New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J Hepatol 53(1):132–144
Su TH, Shiau CW, Jao P et al (2015) Sorafenib and its derivative SC-1 exhibit antifibrotic effects through signal transducer and activator of transcription 3 inhibition. Proc Natl Acad Sci USA 112(23):7243–7248
Lu YY, Gao JH, Zhao C et al (2018) Cyclooxygenase-2 up-regulates hepatic somatostatin receptor 2 expression. Sci Rep 8:11033
Adachi T, Togashi H, Suzuki A et al (2005) NAD(P)H oxidase plays a crucial role in PDGF-induced proliferation of hepatic stellate cells. Hepatology 41(6):1272–1281
Gao JH, Wen SL, Yang WJ et al (2013) Celecoxib ameliorates portal hypertension of the cirrhotic rats through the dual inhibitory effects on the intrahepatic fibrosis and angiogenesis. PLos One 8(7):e69309
Giannandrea M, Parks WC (2014) Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech 7(2):193–203
Roeb E (2018) Matrix metalloproteinases and liver fibrosis (translational aspects). Matrix Biol 68–69:463–473
Okazaki I, Ninomiya Y, Kyuichi T et al (2003) Extracellular matrix and the liver: approach to gene therapy. Elsevier Science, San Diego, pp 115–126
Liu F, Liu ZD, Wu N et al (2013) In vitro interactions between rat bone marrow-derived endothelial progenitor cells and hepatic stellate cells: interaction between EPCs and HSCs. In Vitro Cell Dev Biol Anim 49(7):537–547
Lan L, Liu R, Qin LY et al (2018) Transplantation of bone marrow-derived endothelial progenitor cells and hepatocyte stem cells from liver fibrosis rats ameliorates liver fibrosis. World J Gastroenterol 24(2):237–247
Nakamura T, Torimura T, Iwamoto H et al (2012) Prevention of liver fibrosis and liver reconstitution of DMN-treated rat liver by transplanted EPCs. Eur J Clin Invest 42(7):717–728
Acton QA (2013) Liver fibrosis: new insights for the healthcare professional. ScholarlyBrief, Atlanta, pp 54–57
Salomone F, Godos J, Zelber-Sagi S (2016) Natural antioxidants for non-alcoholic fatty liver disease: molecular targets and clinical perspectives. Liver Int 36(1):5–20
Casas-Grajales S, Muriel P (2015) Antioxidants in liver health. World J Gastrointest Pharmacol Ther 6(3):59–72
Hernández-Ortega LD, Alcántar-Díaz BE, Ruiz-Corro LA et al (2012) Quercetin improves hepatic fibrosis reducing hepatic stellate cells and regulating pro-fibrogenic/anti-fibrogenic molecules balance. J Gastroenterol Hepatol 27(12):1865–1872
van Grunsven LA (2017) 3D in vitro models of liver fibrosis. Adv Drug Deliv Rev 121:133–146
Mazza G, Al-Akkad W, Rombouts K (2017) Engineering in vitro models of hepatofibrogenesis. Adv Drug Deliv Rev 121:147–157
Zhang Z, Liu J, Liu Y et al (2013) Generation, characterization and potential therapeutic applications of mature and functional hepatocytes from stem cells. J Cell Physiol 228:298–305
Ogawa M, Ogawa S, Bear CE et al (2015) Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat Biotechnol 33:853–861
Sampaziotis F, Cardoso de Brito M, Madrigal P et al (2015) Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat Biotechnol 33:845–852
Koui Y, Kido T, Ito T et al (2017) An in vitro human liver model by iPSC-derived parenchymal and non-parenchymal cells. Stem Cell Reports 9(2):490–498
Orbach SM, Ford AJ, Saverot S-E et al (2018) Multi-cellular transitional organotypic models to investigate liver fibrosis. Acta Biomater 82:79–92
Lauschke VM, Shafagh RZ, Hendriks DFG et al (2019) 3D primary hepatocyte culture systems for analyses of liver diseases, drug metabolism, and toxicity: emerging culture paradigms and applications. Biotechnol J 14(7):e1800347
Mukherjee S, Zhelnin L, Sanfiz A et al (2019) Development and validation of an in vitro 3D model of NASH with severe fibrotic phenotype. Am J Transl Res 11(3):1531–1540
Author information
Authors and Affiliations
Contributions
MD wrote and supervised manuscript; DGS assisted in the final preparation of the manuscript; ZM assisted manuscript preparation; CM wrote “Current Challenges and Future Directions” and supervised the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zadorozhna, M., Di Gioia, S., Conese, M. et al. Neovascularization is a key feature of liver fibrosis progression: anti-angiogenesis as an innovative way of liver fibrosis treatment. Mol Biol Rep 47, 2279–2288 (2020). https://doi.org/10.1007/s11033-020-05290-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05290-0